MEK1 Inhibitor Combined with Irradiation Reduces Migration of Breast Cancer Cells Including miR-221 and ZEB1 EMT Marker Expression

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
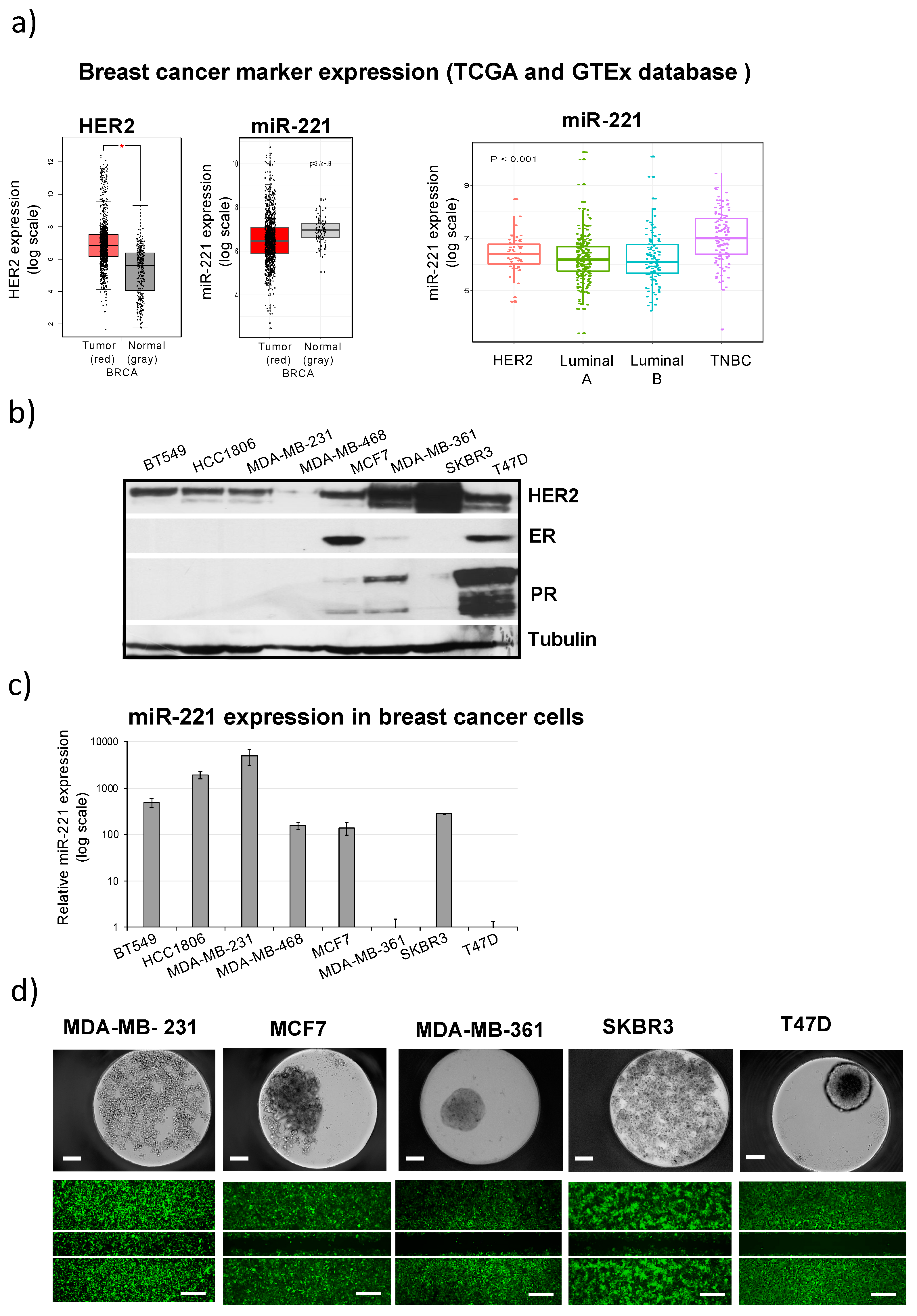
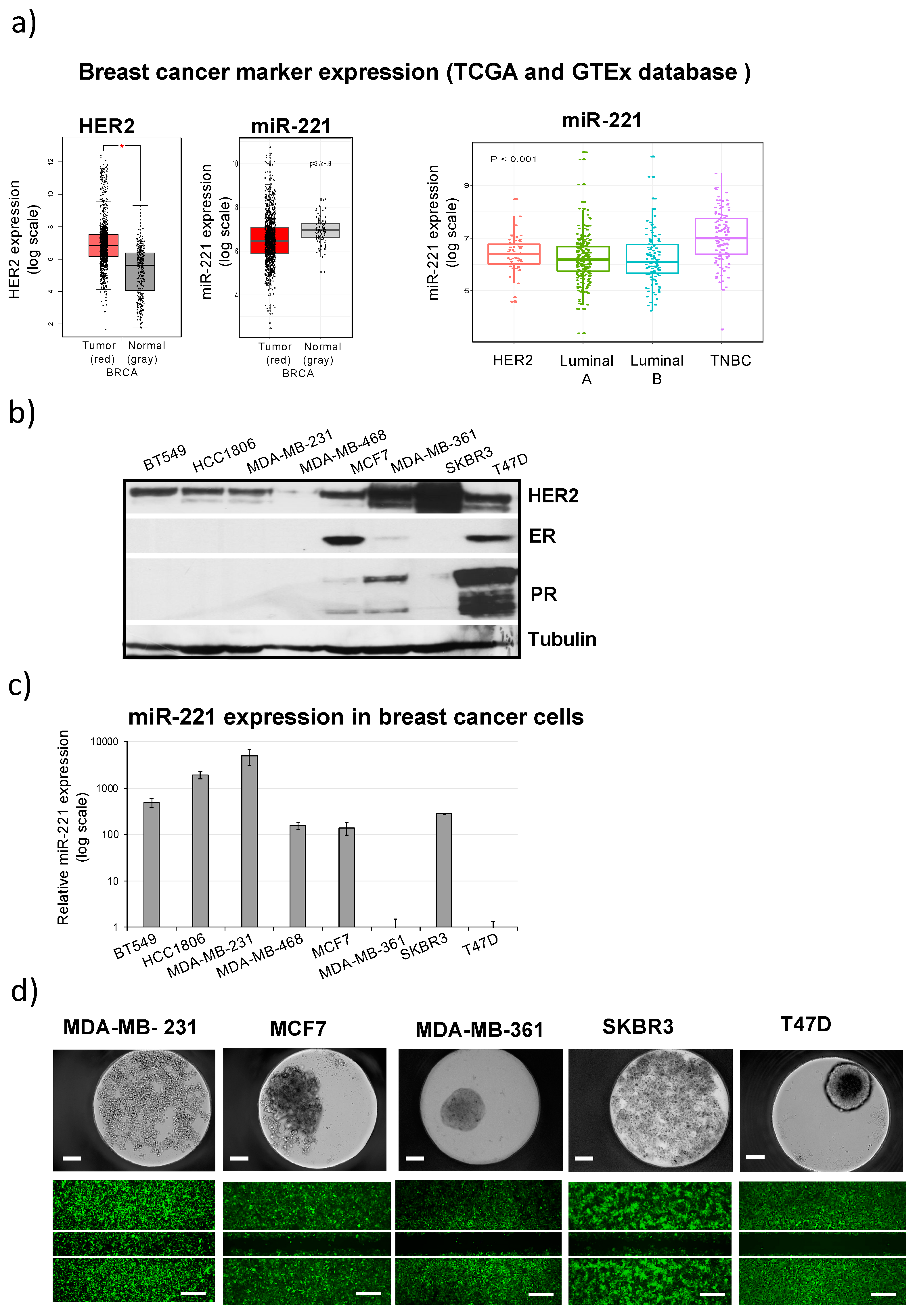
2.1. Differential miR-221 Expression in Breast Cancer Subgroups and Cell Lines
2.2. High miR-221 Expression Levels in Breast Cancer Cells Correlate with Low Attachment, Low Adhesion and High Migration Activity
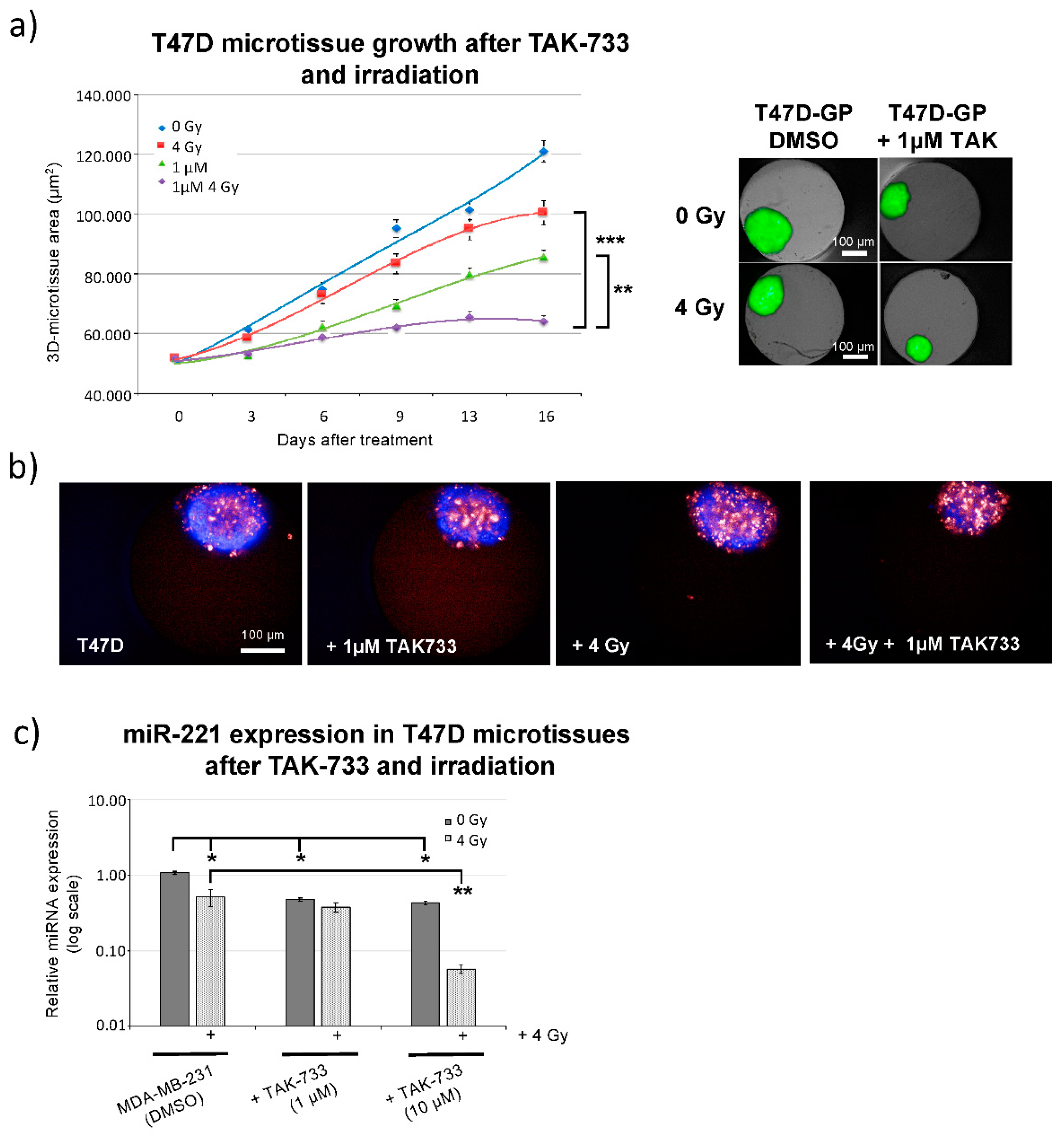
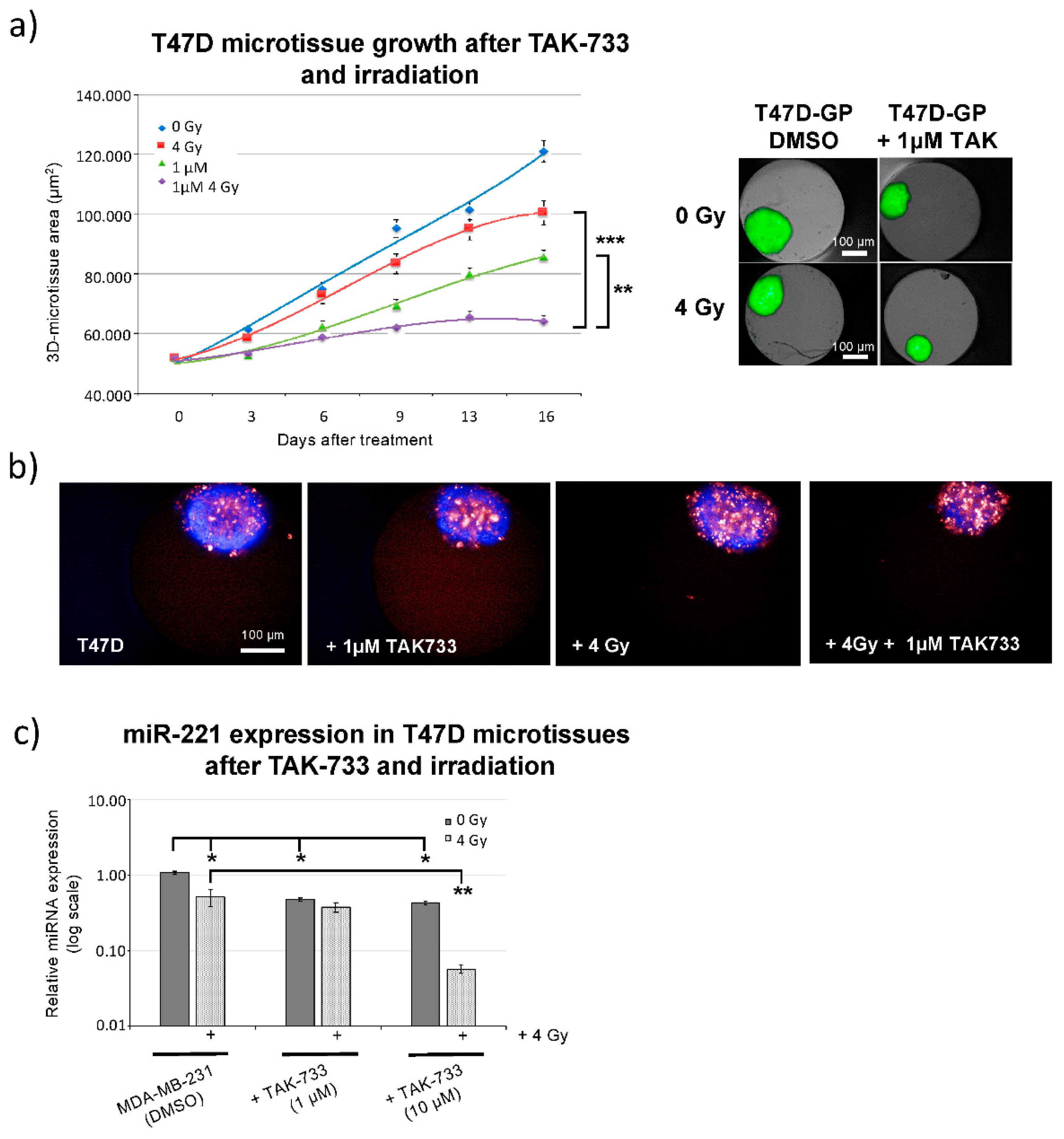
2.3. Breast Cancer 3D-Spheroid Analysis Using MEK1 Inhibitor (TAK-733) in Combination with Irradiation
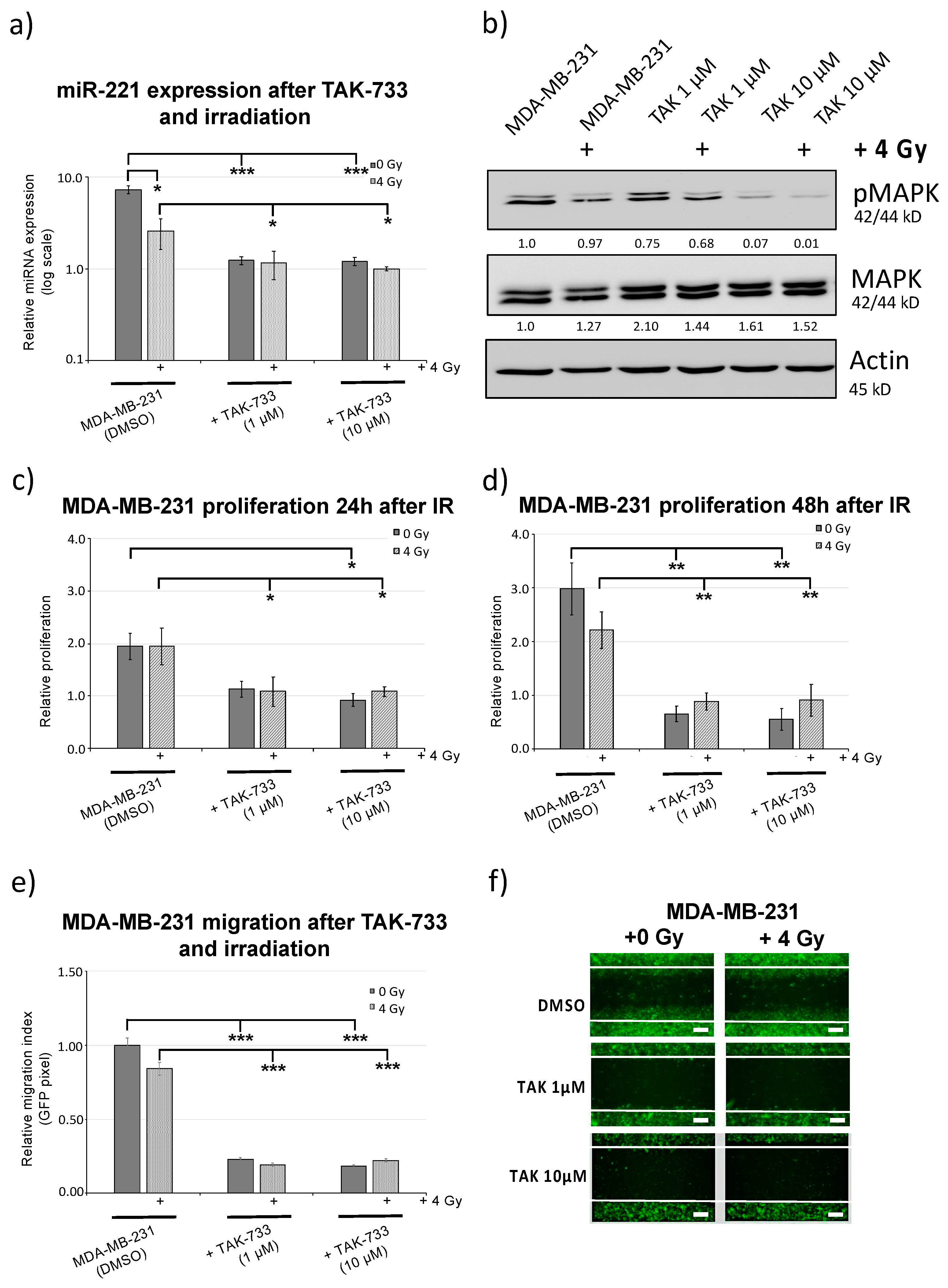
2.4. MEK1 Inhibitor (TAK-733) Impedes Breast Cancer Cell Migration in Combination with Irradiation
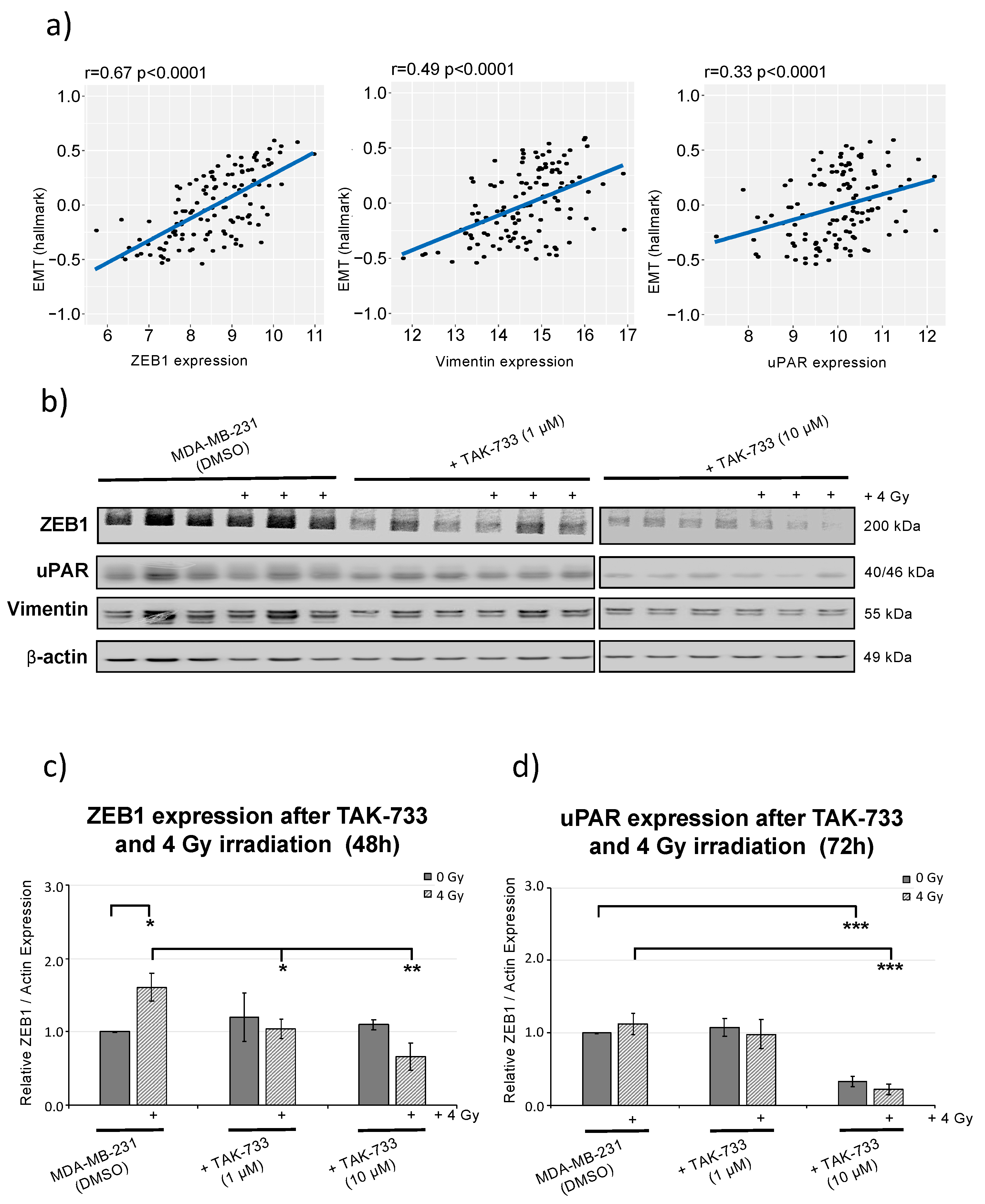
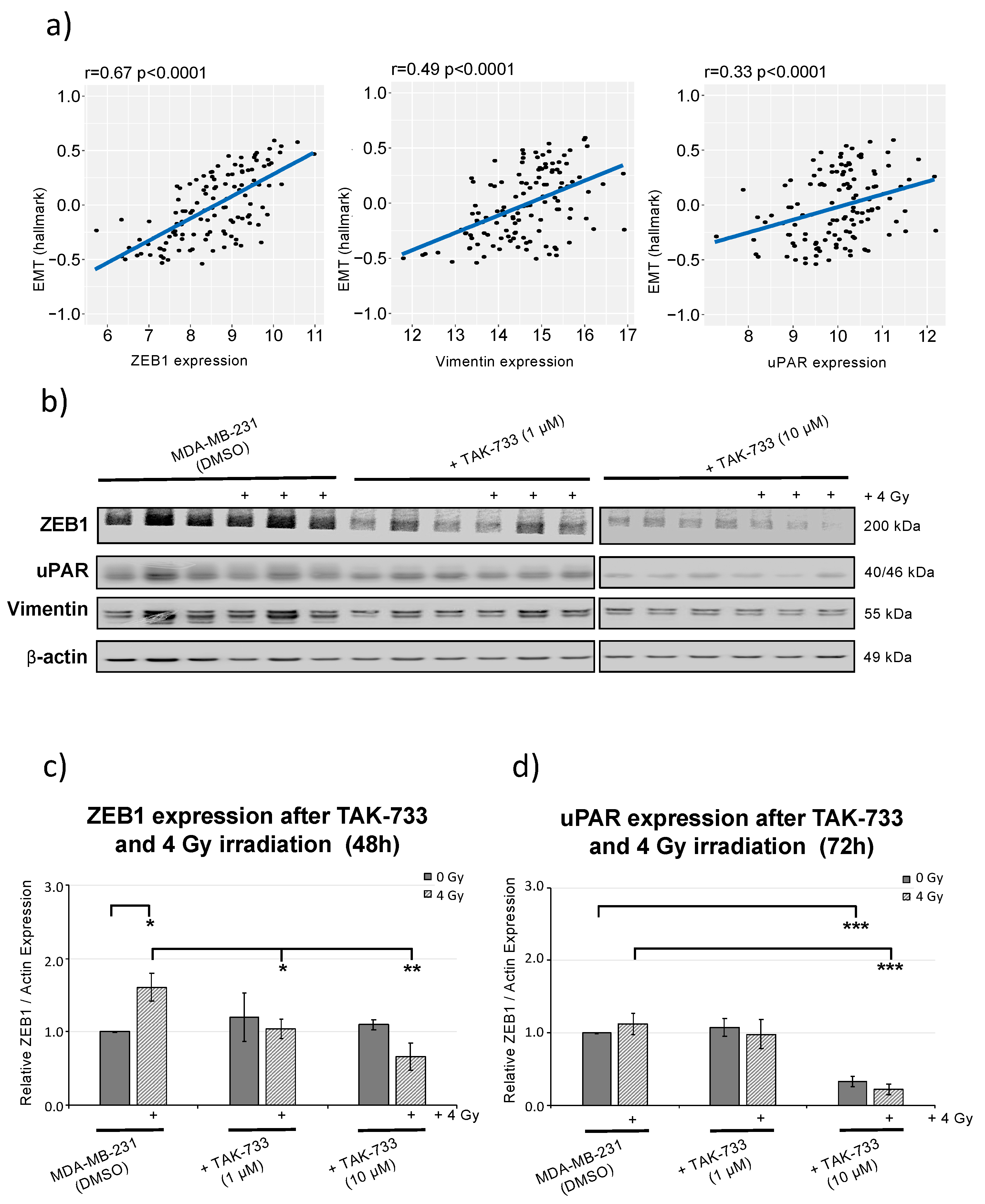
2.5. MEK1 Inhibitor (TAK-733) Combined with Irradiation Inhibits ZEB1 (EMT Marker) Activity
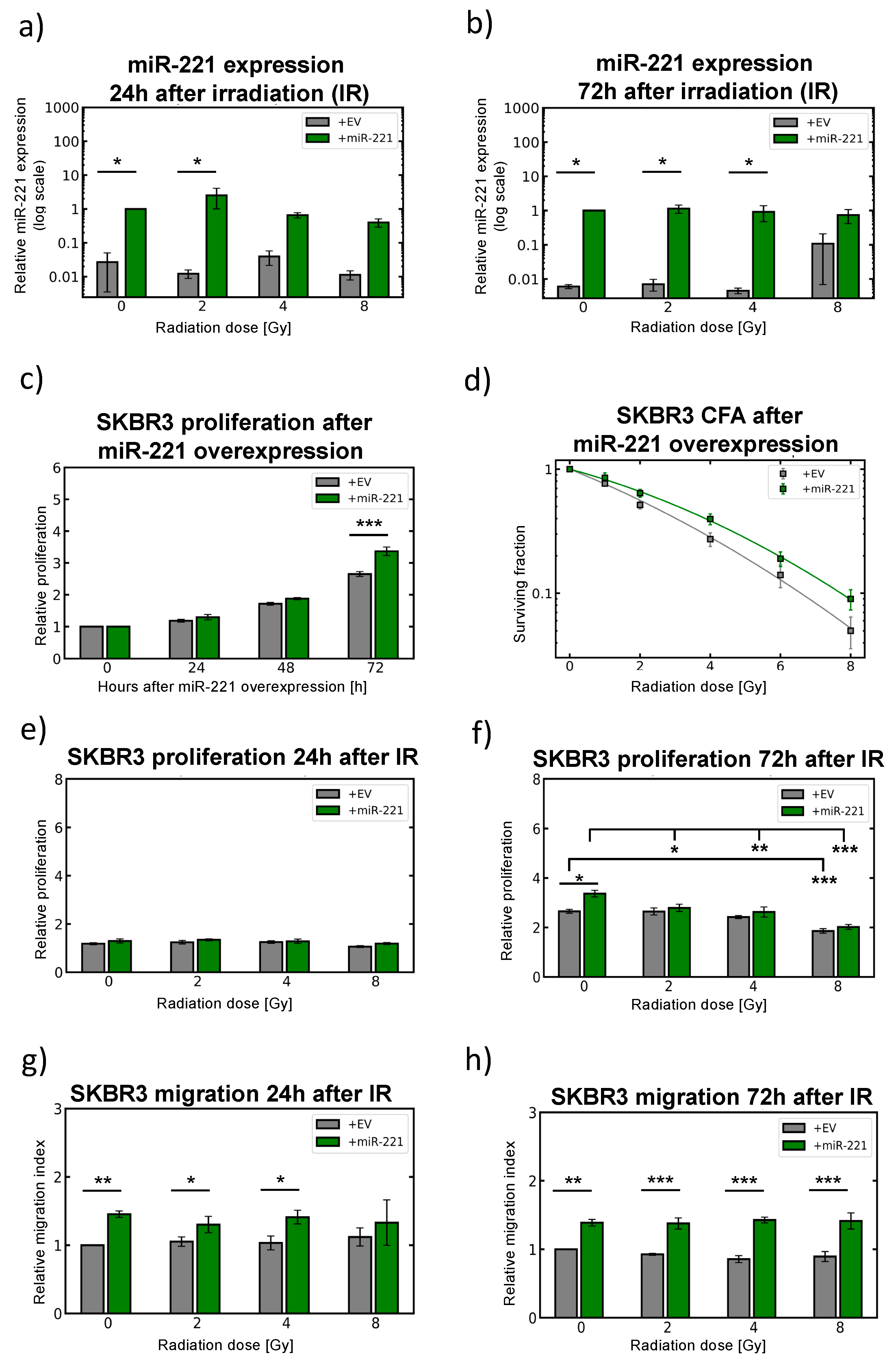
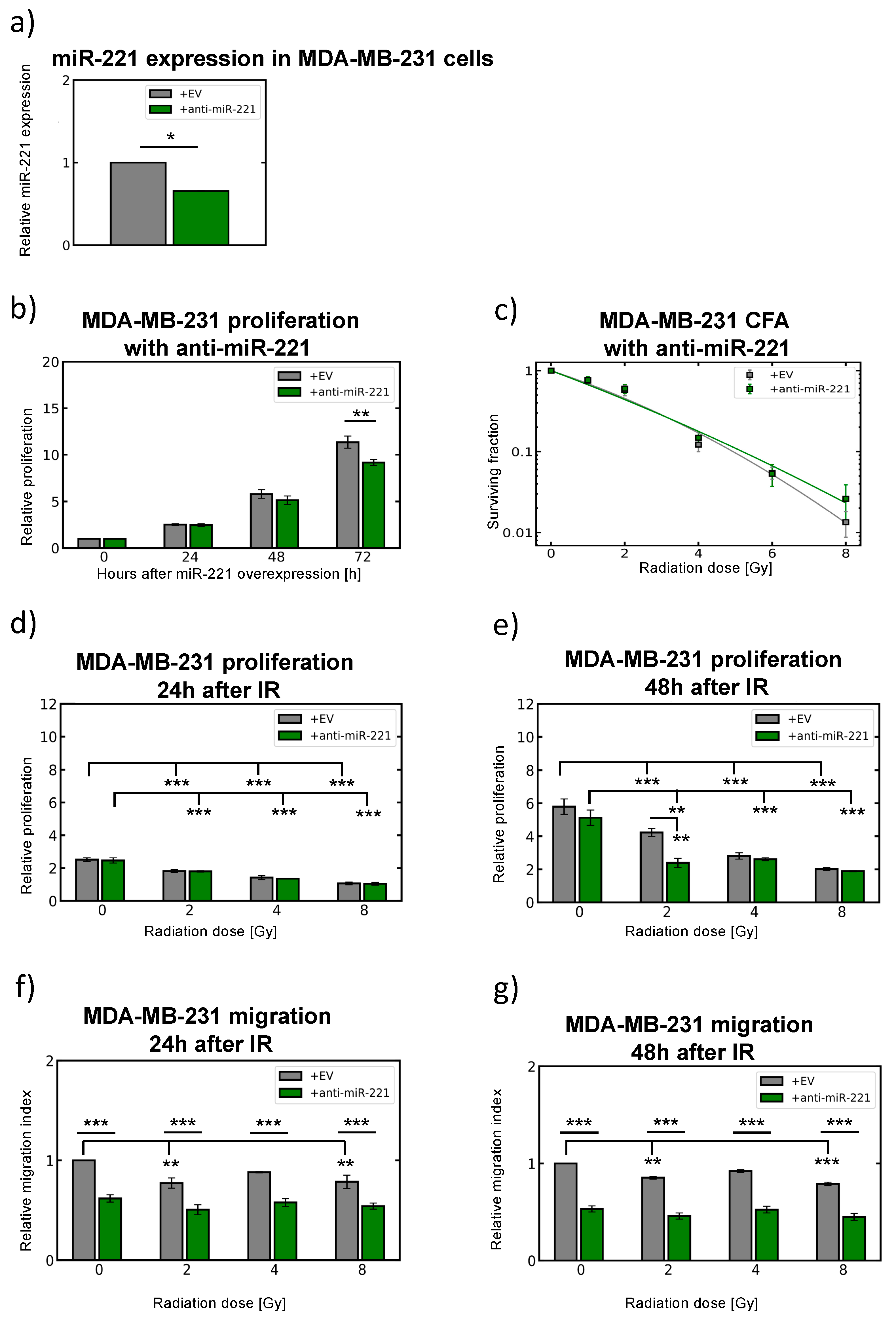
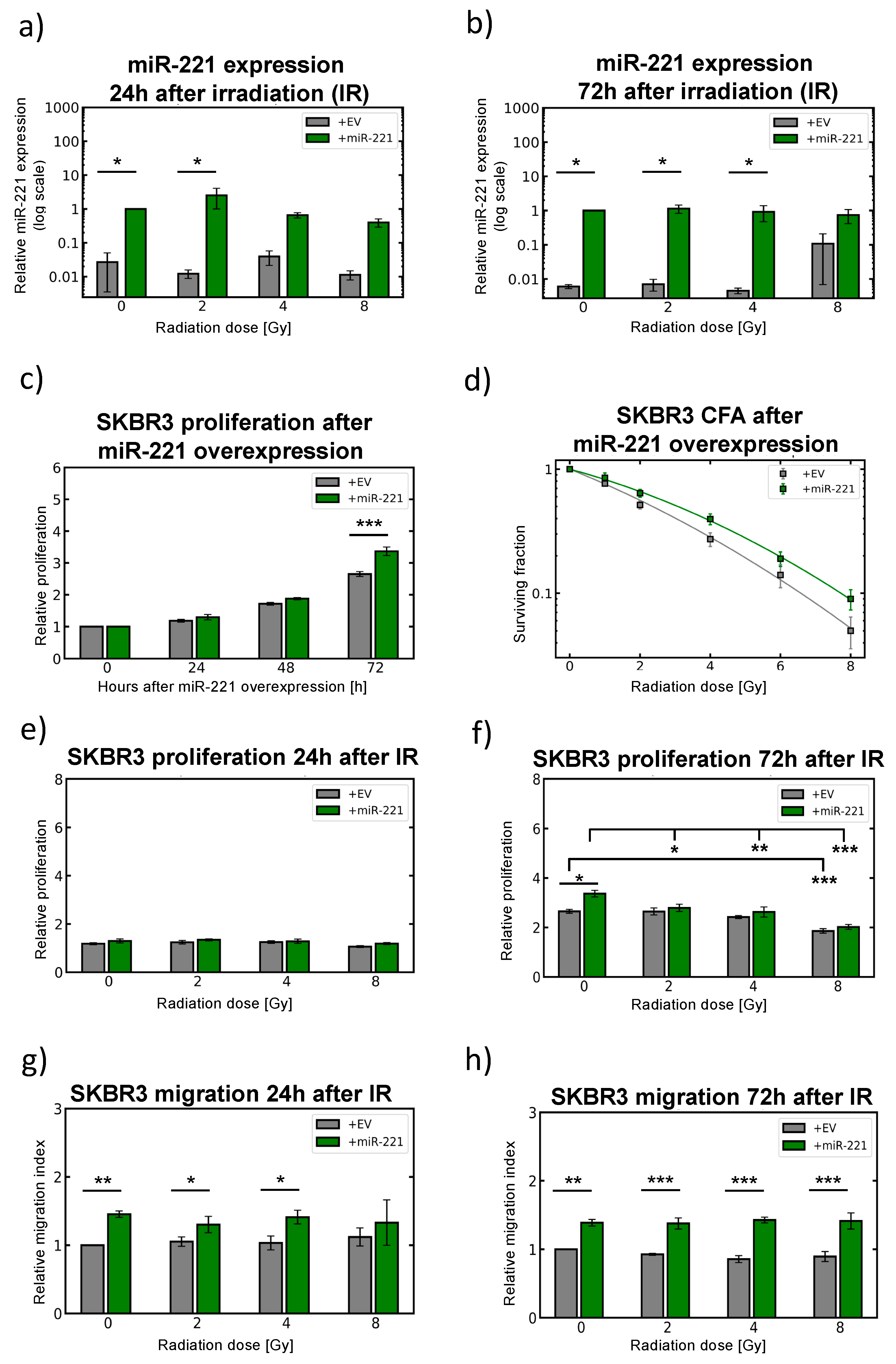
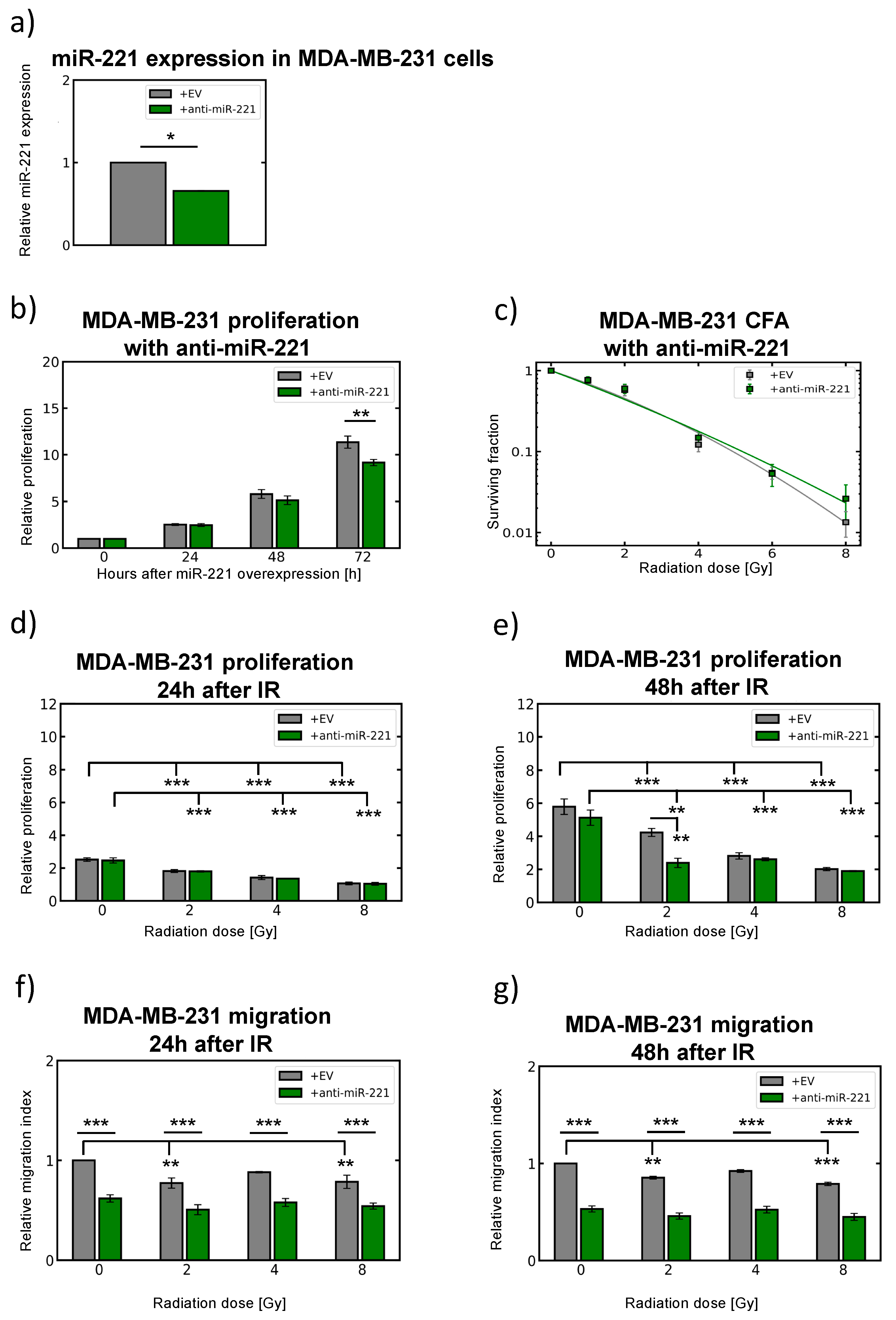
2.6. Modulation of miR-221 Expression Alters Migration Capacity
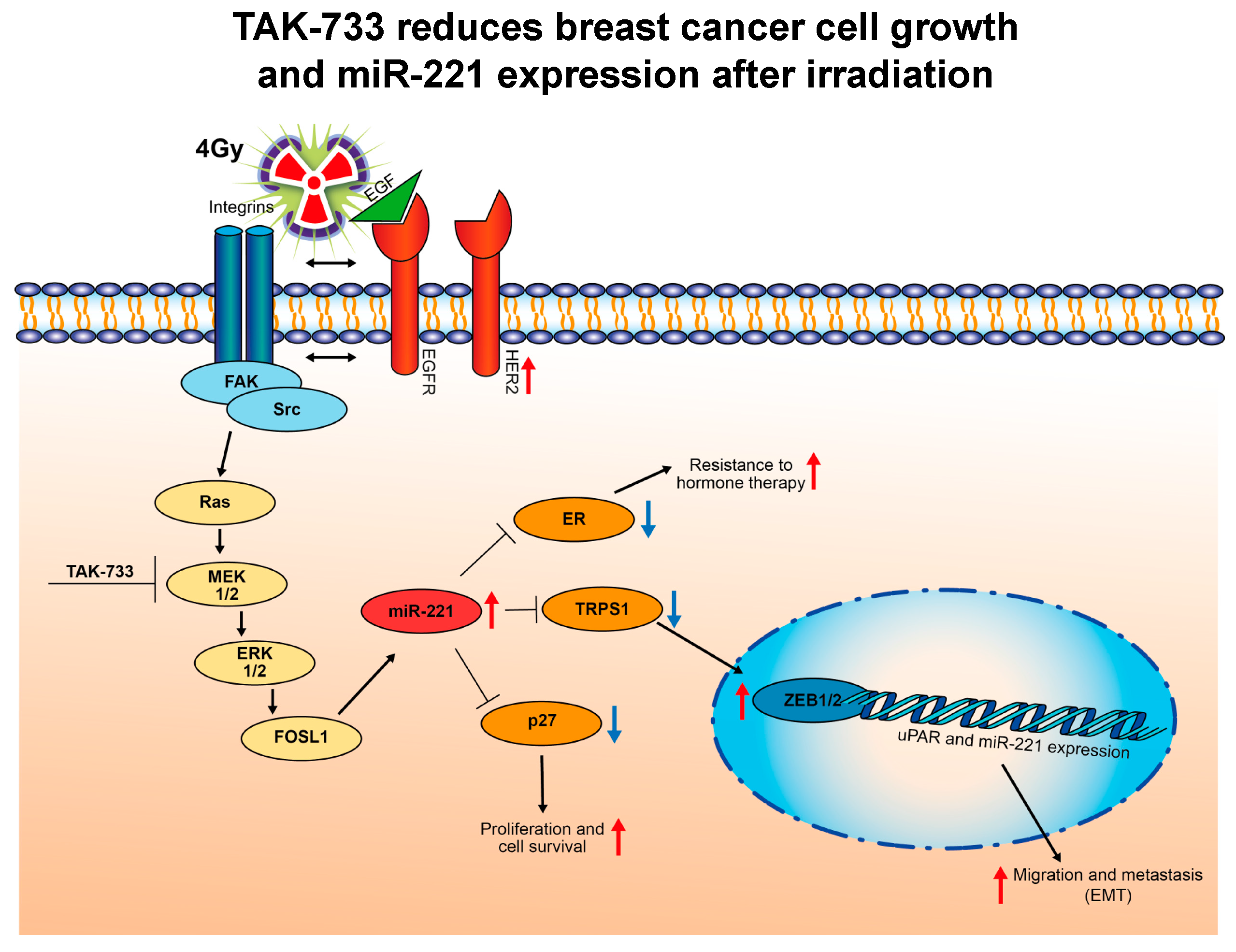
3. Discussion
4. Materials and Methods
4.1. Omics Data Analysis Using Web Databases with Clinical Follow-Up
4.2. Growth and Maintenance of Cell Lines and 3D-Microtissue Generation
4.3. Lentivirus Production and Infection of Breast Cancer Cell Lines
4.4. Generation of Tumour 3D-Microtissues and Treatment with Compounds and Irradiation
4.5. RNA Isolation for miRNA Expression Analysis
4.6. TaqMan-miRNA Assays and Data Analyses
4.7. Western Blot Analysis
4.8. Cell Viability and Caspase 3/7 Activity Assay
4.9. Migration Assay
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Early Breast Cancer Trialists’ Collaborative Group; Darby, S.; McGale, P.; Correa, C.; Taylor, C.; Arriagada, R.; Clarke, M.; Cutter, D.; Davies, C.; Ewertz, M.; et al. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: Meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet 2011, 378, 1707–1716. [Google Scholar] [CrossRef] [Green Version]
- Alabdulkareem, H.; Pinchinat, T.; Khan, S.; Landers, A.; Christos, P.; Simmons, R.; Moo, T.A. The impact of molecular subtype on breast cancer recurrence in young women treated with contemporary adjuvant therapy. Breast J. 2018, 24, 148–153. [Google Scholar] [CrossRef]
- Keklikoglou, I.; Cianciaruso, C.; Guc, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 2019, 21, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wu, X.; Hu, C.; Zhang, Z.; Zhang, L.; Liang, S.; Xu, Y.; Zhang, F. A meta-analysis of combination therapy versus single-agent therapy in anthracycline- and taxane-pretreated metastatic breast cancer: Results from nine randomized Phase III trials. Oncotargets Ther. 2016, 9, 4061–4074. [Google Scholar] [CrossRef] [Green Version]
- Miles, D.; von Minckwitz, G.; Seidman, A.D. Combination versus sequential single-agent therapy in metastatic breast cancer. Oncologist 2002, 7 (Suppl. 6), 13–19. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olea-Flores, M.; Zuniga-Eulogio, M.D.; Mendoza-Catalan, M.A.; Rodriguez-Ruiz, H.A.; Castaneda-Saucedo, E.; Ortuno-Pineda, C.; Padilla-Benavides, T.; Navarro-Tito, N. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial-Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 2885. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Ouyang, Y.; Che, J.; Li, X.; Zhao, Y.; Yang, K.; Zhao, X.; Chen, Y.; Fan, C.; Yuan, W. Potential Value of miR-221/222 as Diagnostic, Prognostic, and Therapeutic Biomarkers for Diseases. Front. Immunol. 2017, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, N.; Anastasov, N.; Rappl, K.; Braselmann, H.; Auer, G.; Walch, A.; Huber, M.; Hofig, I.; Schmitt, M.; Hofler, H.; et al. MiR-221/-222 differentiate prognostic groups in advanced breast cancers and influence cell invasion. Br. J. Cancer 2013, 109, 2714–2723. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.K.; Lin, H.Y.; Dou, X.W.; Chen, M.; Wei, X.L.; Zhang, Y.Q.; Wu, Y.; Chen, C.F.; Bai, J.W.; Xiao, Y.S.; et al. MiR-221/222 promote epithelial-mesenchymal transition by targeting Notch3 in breast cancer cell lines. NPJ Breast Cancer 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci. Signal. 2011, 4, ra41. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Calin, G.A. MicroRNAs miR-221 and miR-222: A new level of regulation in aggressive breast cancer. Genome Med. 2011, 3, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.J.; Lin, J.; Yang, H.; Kong, W.; He, L.; Ma, X.; Coppola, D.; Cheng, J.Q. MicroRNA-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J. Biol. Chem. 2008, 283, 31079–31086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Di Leva, G.; Li, M.; Fang, F.; Devlin, C.; Hartman-Frey, C.; Burow, M.E.; Ivan, M.; Croce, C.M.; Nephew, K.P. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene 2011, 30, 1082–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Guo, F.; Wang, P.; Hong, S.; Zhang, C. miR-221/222 confers radioresistance in glioblastoma cells through activating Akt independent of PTEN status. Curr. Mol. Med. 2014, 14, 185–195. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Anastasov, N.; Hofig, I.; Radulovic, V.; Strobel, S.; Salomon, M.; Lichtenberg, J.; Rothenaigner, I.; Hadian, K.; Kelm, J.M.; Thirion, C.; et al. A 3D-microtissue-based phenotypic screening of radiation resistant tumor cells with synchronized chemotherapeutic treatment. BMC Cancer 2015, 15, 466. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, N.; Hofig, I.; Rosemann, M.; Szumielewski, J.; Richter, S.; Schorpp, K.; Hadian, K.; Aubele, M.; Atkinson, M.J.; Anastasov, N. Three-dimensional microtissues essentially contribute to preclinical validations of therapeutic targets in breast cancer. Cancer Med. 2016, 5, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. MEK inhibitors in oncology: A patent review (2015-Present). Expert Opin. Ther. Pat. 2017, 27, 887–906. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.H.; Kundu, S.T.; Fradette, J.J.; Diao, L.; Tong, P.; Byers, L.A.; Wang, J.; Canales, J.R.; Villalobos, P.A.; Mino, B.; et al. ZEB1 suppression sensitizes KRAS mutant cancers to MEK inhibition by an IL17RD-dependent mechanism. Sci. Transl. Med. 2019, 11, eaaq1238. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Dougan, D.R.; Gong, X.; Halkowycz, P.; Jin, B.; Kanouni, T.; O’Connell, S.M.; Scorah, N.; Shi, L.; Wallace, M.B.; et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg. Med. Chem. Lett. 2011, 21, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet 2018, 14, e1007362. [Google Scholar] [CrossRef] [Green Version]
- Fardi, M.; Solali, S.; Farshdousti Hagh, M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis. 2018, 5, 304–311. [Google Scholar] [CrossRef]
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharm. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef]
- Li, S.; Li, Q.; Lu, J.; Zhao, Q.; Li, D.; Shen, L.; Wang, Z.; Liu, J.; Xie, D.; Cho, W.C.; et al. Targeted Inhibition of miR-221/222 Promotes Cell Sensitivity to Cisplatin in Triple-Negative Breast Cancer MDA-MB-231 Cells. Front. Genet. 2019, 10, 1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, C.; Gioffre, S.; Achilli, F.; Colombo, G.I.; D’Alessandra, Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: An overview of preclinical models and cancer patients. Heart Fail. Rev. 2018, 23, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, Y.; Tanaka, M.; Hayashi, H.; Yokoyama, K.; Osawa, M. Overexpression of miR-221 in sudden death with cardiac hypertrophy patients. Heliyon 2018, 4, e00639. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, M.; Pahl, R.; Huber, K.; Eilf, K.; Dunst, J. Cardiac Toxicity after Radiotherapy for Breast Cancer: Myths and Facts. Breast Care (Basel) 2015, 10, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Anastasov, N.; Klier, M.; Koch, I.; Angermeier, D.; Hofler, H.; Fend, F.; Quintanilla-Martinez, L. Efficient shRNA delivery into B and T lymphoma cells using lentiviral vector-mediated transfer. J. Hematop. 2009, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Anastasov, N.; Hofig, I.; Vasconcellos, I.G.; Rappl, K.; Braselmann, H.; Ludyga, N.; Auer, G.; Aubele, M.; Atkinson, M.J. Radiation resistance due to high expression of miR-21 and G2/M checkpoint arrest in breast cancer cells. Radiat. Oncol. 2012, 7, 206. [Google Scholar] [CrossRef] [Green Version]
- Anastasov, N.; Hofig, I.; Mall, S.; Krackhardt, A.M.; Thirion, C. Optimized Lentiviral Transduction Protocols by Use of a Poloxamer Enhancer, Spinoculation, and scFv-Antibody Fusions to VSV-G. Methods Mol. Biol. 2016, 1448, 49–61. [Google Scholar] [CrossRef]
- Anastasov, N.; Bonzheim, I.; Rudelius, M.; Klier, M.; Dau, T.; Angermeier, D.; Duyster, J.; Pittaluga, S.; Fend, F.; Raffeld, M.; et al. C/EBPbeta expression in ALK-positive anaplastic large cell lymphomas is required for cell proliferation and is induced by the STAT3 signaling pathway. Haematologica 2010, 95, 760–767. [Google Scholar] [CrossRef] [Green Version]
- Hofig, I.; Atkinson, M.J.; Mall, S.; Krackhardt, A.M.; Thirion, C.; Anastasov, N. Poloxamer synperonic F108 improves cellular transduction with lentiviral vectors. J. Gene Med. 2012, 14, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, N.; Anastasov, N.; Schaub, A.; Radulovic, V.; Schmitt, M.; Magdolen, V.; Aubele, M. Secreted uPAR isoform 2 (uPAR7b) is a novel direct target of miR-221. Oncotarget 2015, 6, 8103–8114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.C.; Mall, R.; Braselmann, H.; Feuchtinger, A.; Molatore, S.; Lindner, K.; Walch, A.; Gross, E.; Schmitt, M.; Falkenberg, N.; et al. uPAR enhances malignant potential of triple-negative breast cancer by directly interacting with uPA and IGF1R. BMC Cancer 2016, 16, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubele, M.; Auer, G.; Walch, A.K.; Munro, A.; Atkinson, M.J.; Braselmann, H.; Fornander, T.; Bartlett, J.M. PTK (protein tyrosine kinase)-6 and HER2 and 4, but not HER1 and 3 predict long-term survival in breast carcinomas. Br. J. Cancer 2007, 96, 801–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutschelknaus, L.; Azimzadeh, O.; Heider, T.; Winkler, K.; Vetter, M.; Kell, R.; Tapio, S.; Merl-Pham, J.; Huber, S.M.; Edalat, L.; et al. Radiation alters the cargo of exosomes released from squamous head and neck cancer cells to promote migration of recipient cells. Sci. Rep. 2017, 7, 12423. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anastasov, N.; Hirmer, E.; Klenner, M.; Ott, J.; Falkenberg, N.; Bao, X.; Mutschelknaus, L.; Moertl, S.; Combs, S.; Atkinson, M.J.; et al. MEK1 Inhibitor Combined with Irradiation Reduces Migration of Breast Cancer Cells Including miR-221 and ZEB1 EMT Marker Expression. Cancers 2020, 12, 3760. https://doi.org/10.3390/cancers12123760
Anastasov N, Hirmer E, Klenner M, Ott J, Falkenberg N, Bao X, Mutschelknaus L, Moertl S, Combs S, Atkinson MJ, et al. MEK1 Inhibitor Combined with Irradiation Reduces Migration of Breast Cancer Cells Including miR-221 and ZEB1 EMT Marker Expression. Cancers. 2020; 12(12):3760. https://doi.org/10.3390/cancers12123760
Chicago/Turabian StyleAnastasov, Nataša, Elisabeth Hirmer, Marbod Klenner, Jessica Ott, Natalie Falkenberg, Xuanwen Bao, Lisa Mutschelknaus, Simone Moertl, Stephanie Combs, Michael J. Atkinson, and et al. 2020. "MEK1 Inhibitor Combined with Irradiation Reduces Migration of Breast Cancer Cells Including miR-221 and ZEB1 EMT Marker Expression" Cancers 12, no. 12: 3760. https://doi.org/10.3390/cancers12123760
APA StyleAnastasov, N., Hirmer, E., Klenner, M., Ott, J., Falkenberg, N., Bao, X., Mutschelknaus, L., Moertl, S., Combs, S., Atkinson, M. J., & Schmid, T. (2020). MEK1 Inhibitor Combined with Irradiation Reduces Migration of Breast Cancer Cells Including miR-221 and ZEB1 EMT Marker Expression. Cancers, 12(12), 3760. https://doi.org/10.3390/cancers12123760