Functional Dependency Analysis Identifies Potential Druggable Targets in Acute Myeloid Leukemia

 ,
,  ,
,  ,
, 

Abstract
Simple Summary
Abstract
1. Introduction
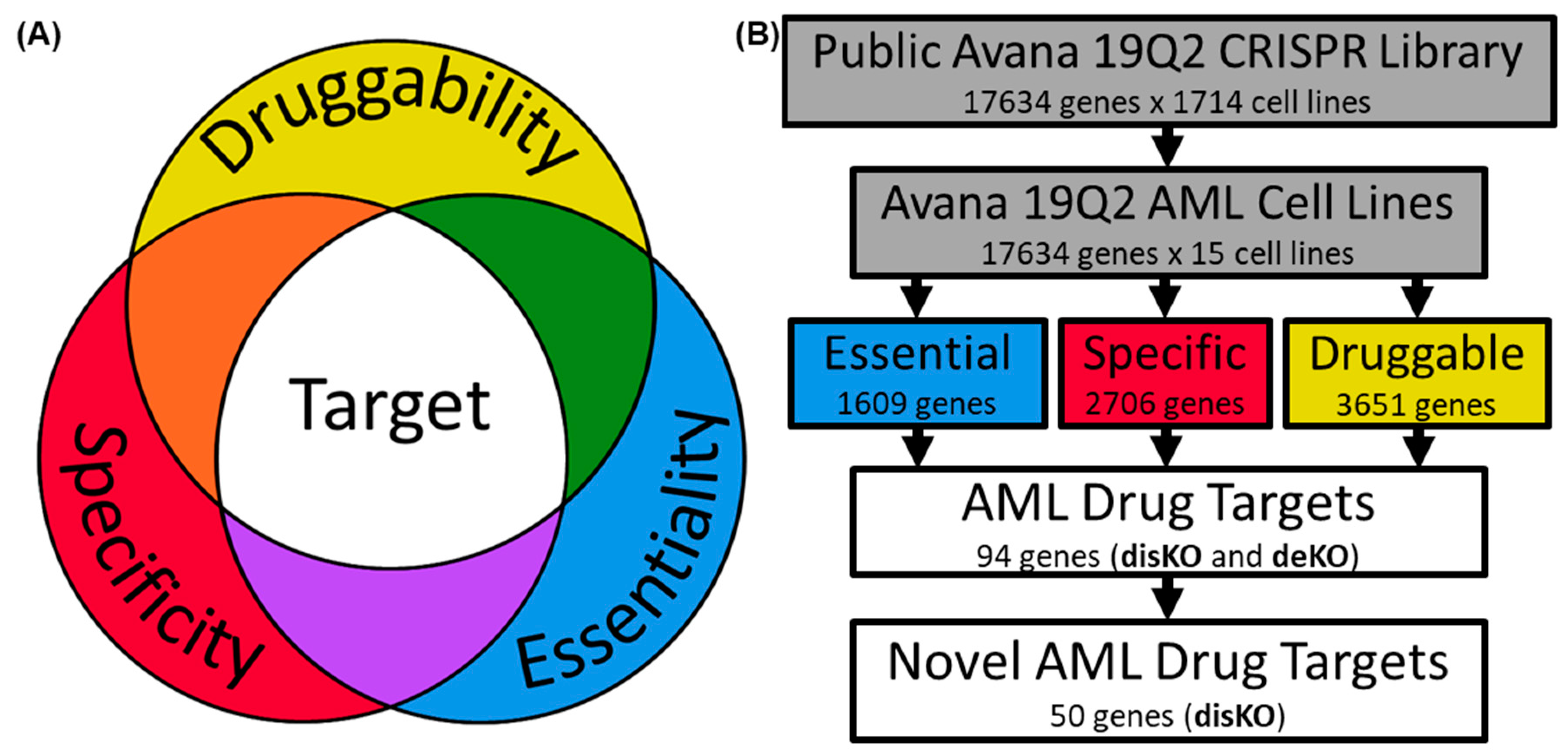
2. Methods
2.1. Examination of AML Dependencies
2.2. Assessing AML Gene Druggability
2.3. Determining AML Drug Target Screening Characteristics
2.4. Screening Genes for AML Drug Development
3. Results
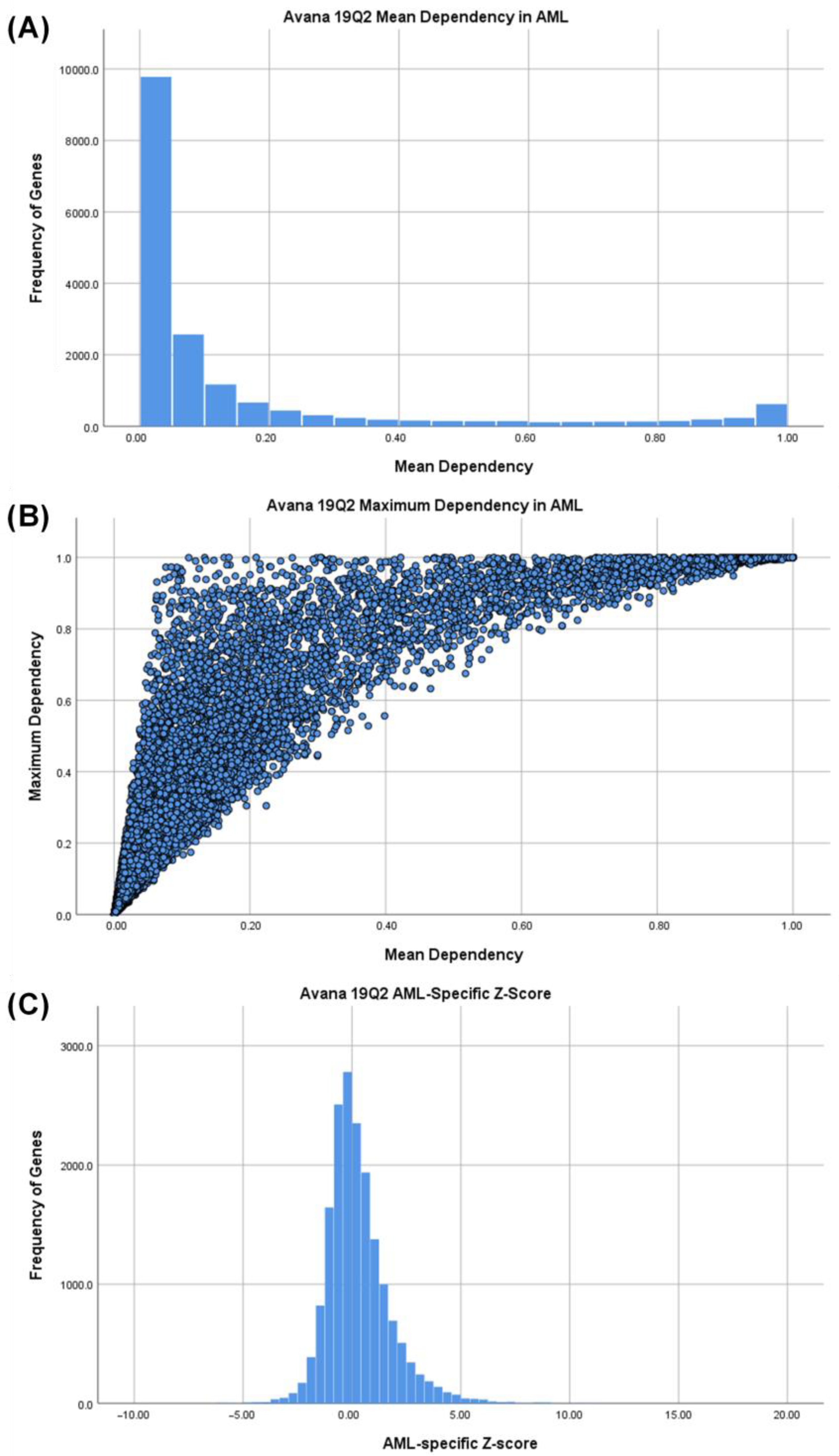
3.1. Identifying AML-Specific Essential Gene Characteristics
3.2. Identifying AML-Specific Essential Gene Characteristics
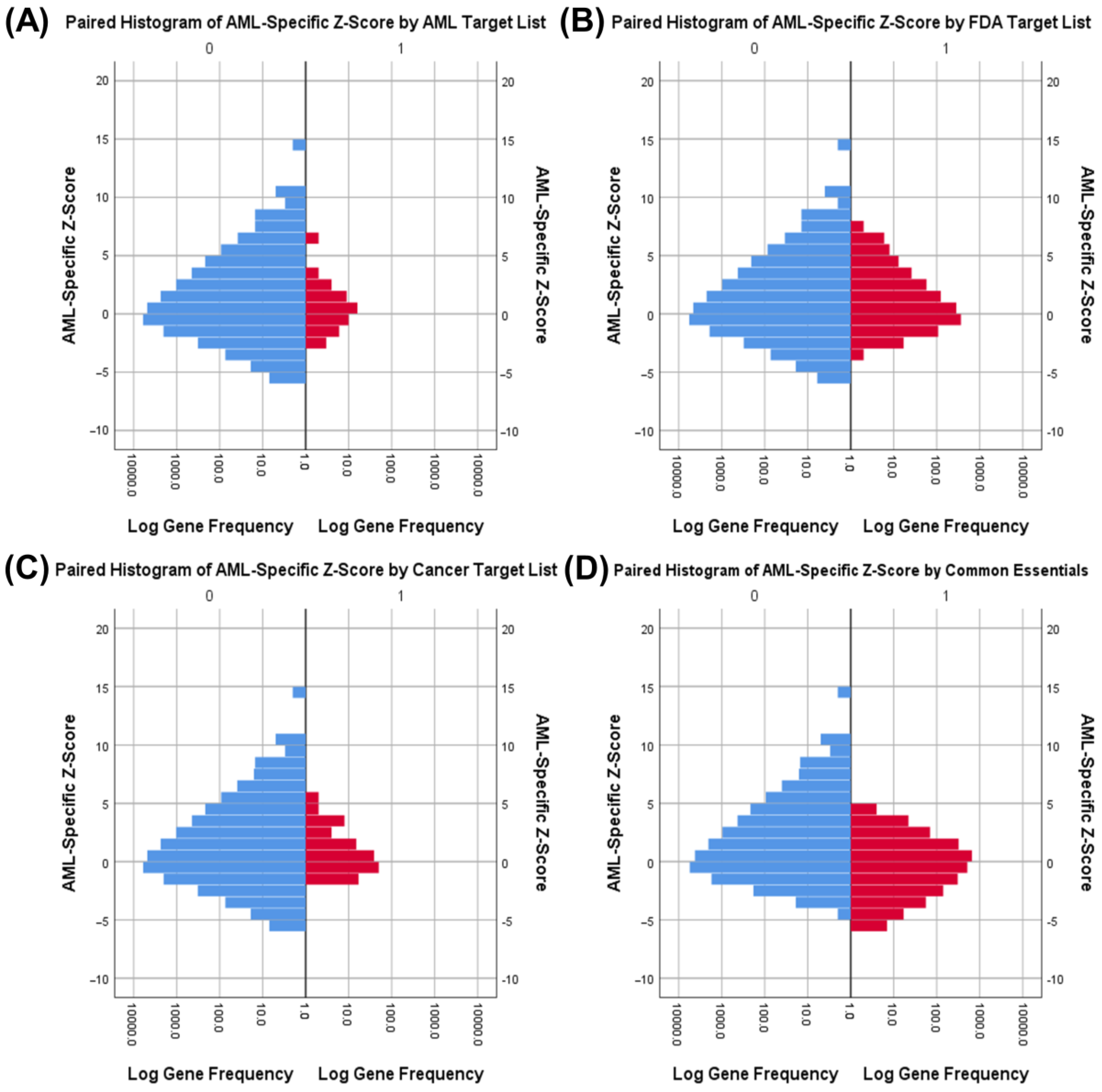
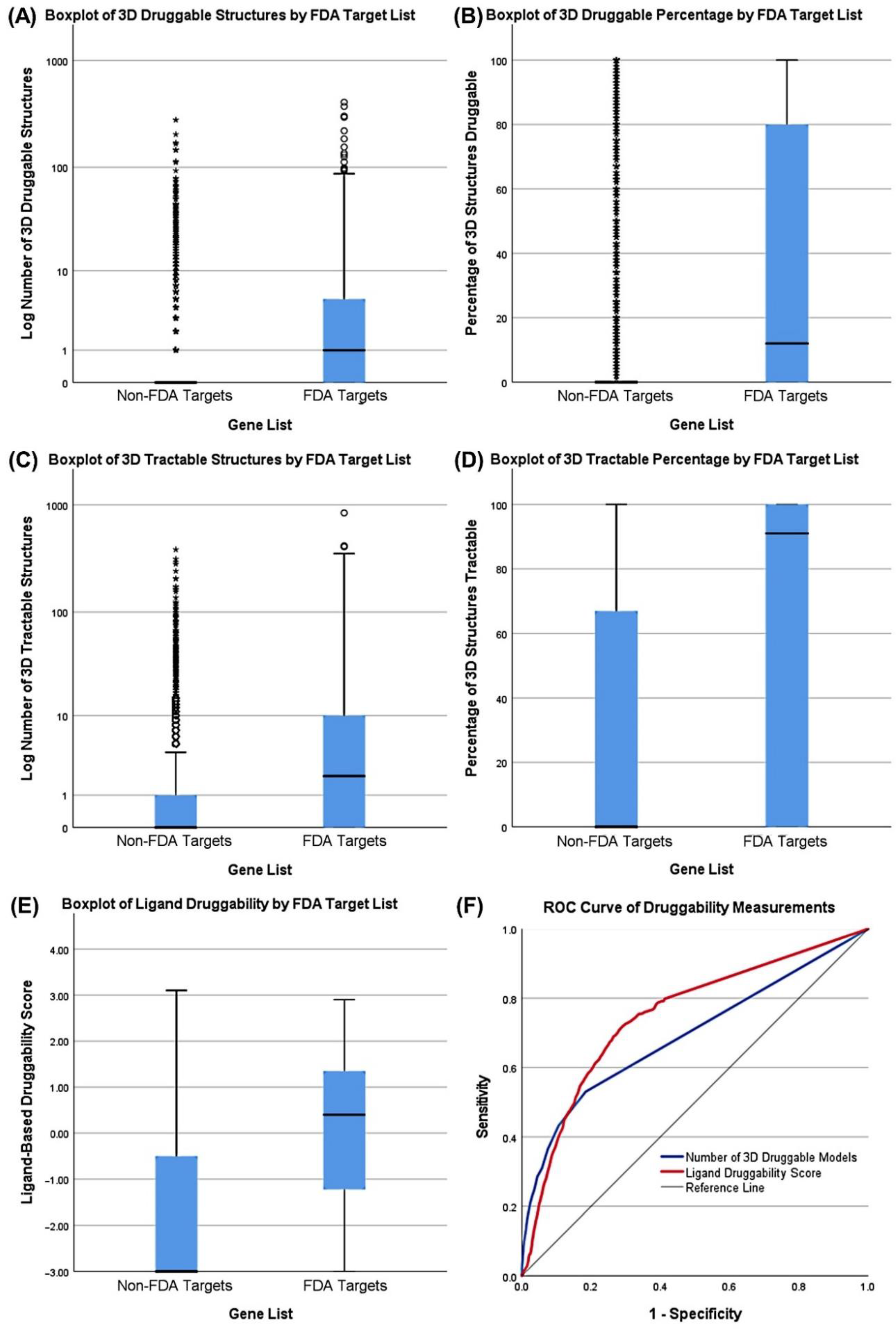
3.3. Druggability as a Discriminator
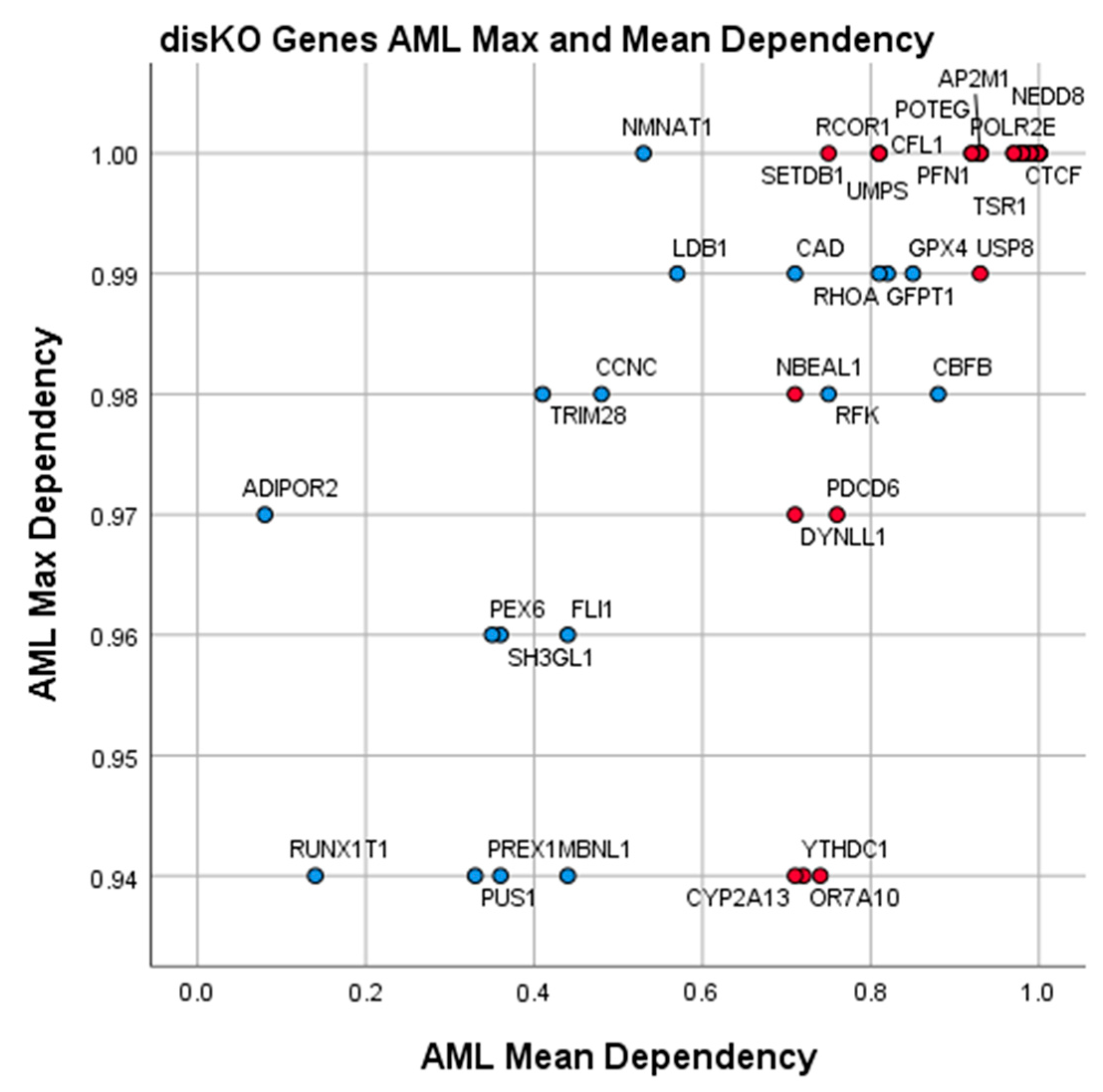
3.4. Essential Gene List for AML Drug Discovery
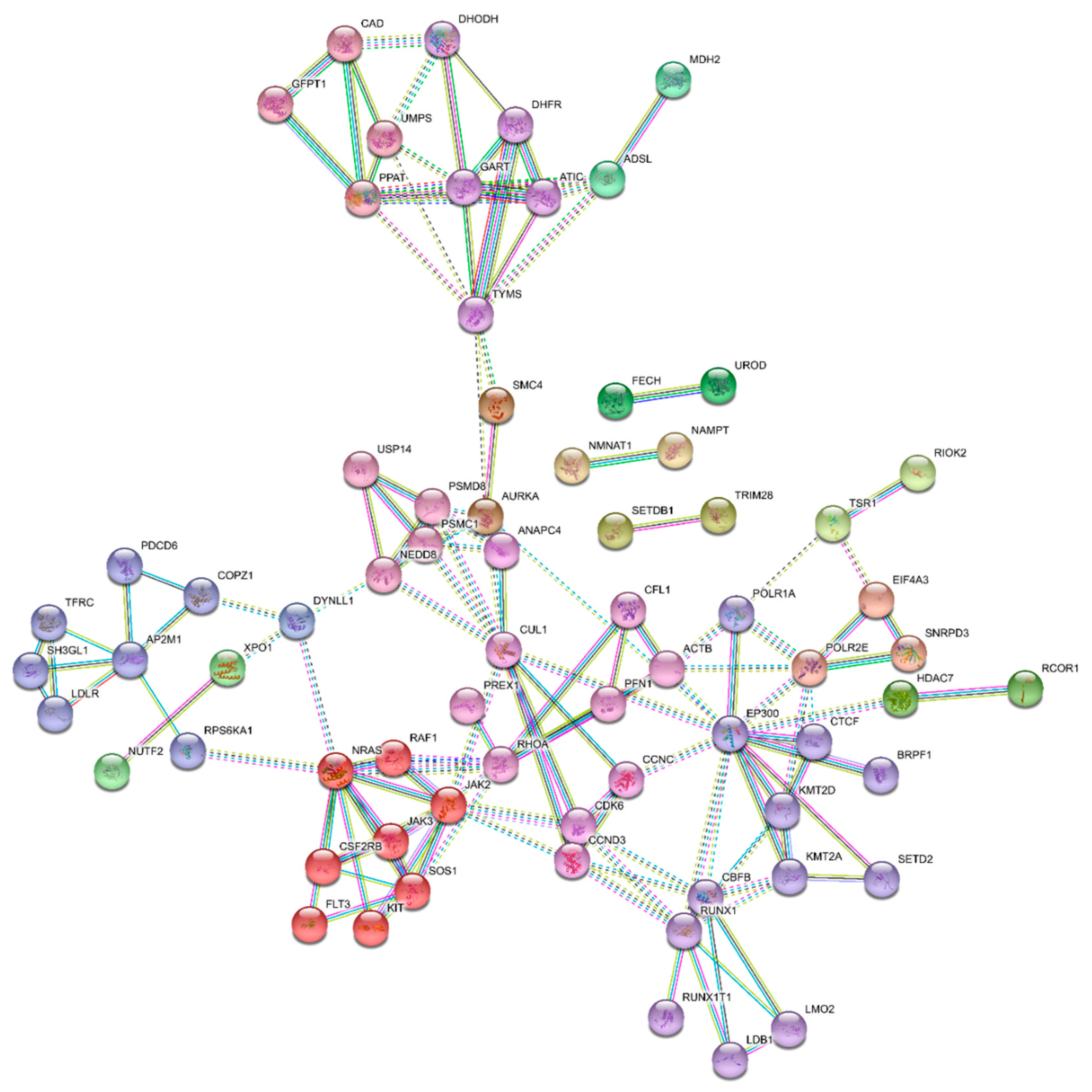
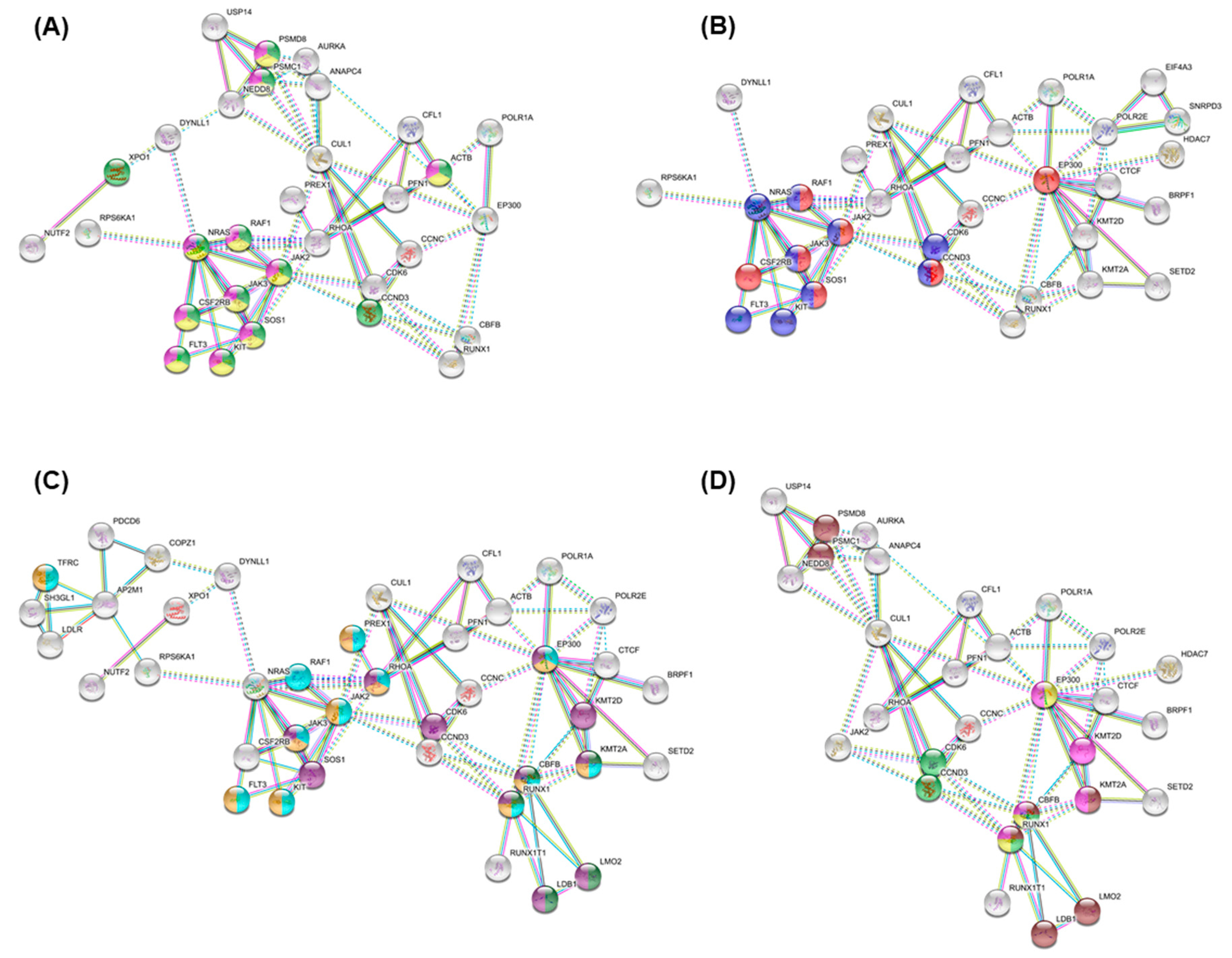
3.5. Essential Physiologic Processes in AML
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vasu, S.; Kohlschmidt, J.; Mrózek, K.; Eisfeld, A.K.; Nicolet, D.; Sterling, L.J.; Becker, H.; Metzeler, K.H.; Papaioannou, D.; Powell, B.L.; et al. Ten-year outcome of patients with acute myeloid leukemia not treated with allogeneic transplantation in first complete remission. Blood Adv. 2018, 2, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Perl, A.E. Advances in patient care through increasingly individualized therapy. Nat. Rev. Clin. Oncol. 2019, 16, 73–74. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e516. [Google Scholar] [CrossRef] [PubMed]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019, 720243. [Google Scholar] [CrossRef]
- Coker, E.A.; Mitsopoulos, C.; Tym, J.E.; Komianou, A.; Kannas, C.; Di Micco, P.; Villasclaras Fernandez, E.; Ozer, B.; Antolin, A.A.; Workman, P.; et al. canSAR: Update to the cancer translational research and drug discovery knowledgebase. Nucleic Acids Res. 2019, 47, D917–D922. [Google Scholar] [CrossRef] [PubMed]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.-Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018, 46, D1068–D1073. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown Kevin, R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2006, 35, D301–D303. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Hann, M.M.; Lakdawala, A.S.; Santos, R.; Thomas, P.J.; Todd, K. Approaches to target tractability assessment—A practical perspective. Medchemcomm 2018, 9, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef]
- Swaminathan, M.; Kantarjian, H.M.; Daver, N.; Borthakur, G.; Ohanian, M.; Kadia, T.; DiNardo, C.D.; Jain, N.; Estrov, Z.; Ferrajoli, A.; et al. The Combination of Quizartinib with Azacitidine or Low Dose Cytarabine Is Highly Active in Patients (Pts) with FLT3-ITD Mutated Myeloid Leukemias: Interim Report of a Phase I/II Trial. Blood 2017, 130, 723. [Google Scholar] [CrossRef]
- Azzi, J.; Cirrone, F.; Abdul Hay, M.; Kaminetzky, D.; Hymes, K.B.; Kreditor, M.; Al-Homsi, A.-S.; Grossbard, M.L.; Diefenbach, C.; Moskovits, T.; et al. Midostaurin in Combination with Idarubicin and Cytarabine (3+7) Induction for FLT3 Positive AML—Very High Complete Response Rates and Transition to Allogeneic Transplantation. Blood 2018, 132, 5216. [Google Scholar] [CrossRef]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef]
- Advani, A.S.; McDonough, S.; Copelan, E.; Willman, C.; Mulford, D.A.; List, A.F.; Sekeres, M.A.; Othus, M.; Appelbaum, F.R. SWOG0919: A Phase 2 study of idarubicin and cytarabine in combination with pravastatin for relapsed acute myeloid leukaemia. Br. J. Haematol. 2014, 167, 233–237. [Google Scholar] [CrossRef]
- Von Neuhoff, C.; Reinhardt, D.; Sander, A.; Zimmermann, M.; Bradtke, J.; Betts, D.R.; Zemanova, Z.; Stary, J.; Bourquin, J.P.; Haas, O.A.; et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J. Clin. Oncol. 2010, 28, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, F.R.; Kopecky, K.J.; Tallman, M.S.; Slovak, M.L.; Gundacker, H.M.; Kim, H.T.; Dewald, G.W.; Kantarjian, H.M.; Pierce, S.R.; Estey, E.H. The clinical spectrum of adult acute myeloid leukaemia associated with core binding factor translocations. Br. J. Haematol. 2006, 135, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Pirc-Danoewinata, H.; Dauwerse, H.G.; König, M.; Chudoba, I.; Mitterbauer, M.; Jäger, U.; Breuning, M.H.; Haas, O.A. CBFB/MYH11 fusion in a patient with AML-M4Eo and cytogenetically normal chromosomes 16. Genes Chromosomes Cancer 2000, 29, 186–191. [Google Scholar] [CrossRef]
- Morita, K.; Noura, M.; Tokushige, C.; Maeda, S.; Kiyose, H.; Kashiwazaki, G.; Taniguchi, J.; Bando, T.; Yoshida, K.; Ozaki, T.; et al. Autonomous feedback loop of RUNX1-p53-CBFB in acute myeloid leukemia cells. Sci. Rep. 2017, 7, 16604. [Google Scholar] [CrossRef]
- Illendula, A.; Pulikkan, J.A.; Zong, H.; Grembecka, J.; Xue, L.; Sen, S.; Zhou, Y.; Boulton, A.; Kuntimaddi, A.; Gao, Y.; et al. Chemical biology. A small-molecule inhibitor of the aberrant transcription factor CBFβ-SMMHC delays leukemia in mice. Science 2015, 347, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Dembitz, V.; Tomic, B.; Kodvanj, I.; Simon, J.A.; Bedalov, A.; Visnjic, D. The ribonucleoside AICAr induces differentiation of myeloid leukemia by activating the ATR/Chk1 via pyrimidine depletion. J. Biol. Chem. 2019, 294, 15257–15270. [Google Scholar] [CrossRef]
- Tricot, G.; Weber, G. Biochemically targeted therapy of refractory leukemia and myeloid blast crisis of chronic granulocytic leukemia with Tiazofurin, a selective blocker of inosine 5′-phosphate dehydrogenase activity. Anticancer Res. 1996, 16, 3341–3347. [Google Scholar]
- Amit, S.; Nidhi, J.; Courteney, K.L.; Anuhar, C.; Razif, G.; Kerstin, G.; Cecilia, M.; Jan, E.B.; Arnold, G.; Eric, B.; et al. Pyrimethamine as a Potent and Selective Inhibitor of Acute Myeloid Leukemia Identified by High-throughput Drug Screening. Curr. Cancer Drug Targets 2016, 16, 818–828. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e724. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fisher’s Exact Tests for Common Essential Genes by Gene List | ||||
|---|---|---|---|---|
| Target List | Common Essential Genes | Non-Common Essential Genes | Significance (2-Sided) | |
| AML Drug Targets | AML | Actual: 17 | Actual: 39 | p < 0.001 |
| Expected: 6.7 | Expected: 49.3 | |||
| Other | Actual: 2108 | Actual: 15,470 | ||
| Expected: 2118.3 | Expected: 15,459.7 | |||
| Cancer Drug Targets | Cancer | Actual: 18 | Actual: 122 | p = 0.806 |
| Expected: 16.9 | Expected: 123.1 | |||
| Other | Actual: 2107 | Actual: 15,387 | ||
| Expected: 2108.1 | Expected: 15,385.9 | |||
| All FDA Drug Targets | FDA | Actual: 104 | Actual: 914 | p = 0.069 |
| Expected: 122.7 | Expected: 895.3 | |||
| Other | Actual: 2021 | Actual: 14,595 | ||
| Expected: 2002.3 | Expected: 14,613.7 | |||
| Characteristic | Reference List | Measurement | Cutoff | Sensitivity |
|---|---|---|---|---|
| Essentiality | AML Drug Targets | AML Max. Dependency | 0.9361 | 0.309 |
| Specificity | Non-common essential genes | AML-Specific Z-Score | 1.4268 | 0.178 |
| Druggability (3D) | FDA Targets | Number of TracTable 3D Structures | 4.0652 | 0.400 |
| Druggability (Ligand) | FDA Targets | Ligand-Based Score | 0.8875 | 0.382 |
| Discovery Knockout Genes (AML disKO) | ||||||
|---|---|---|---|---|---|---|
| Gene | AML Mean Dependence | AML Max. Dependence | AML-Specific Z-Score | 3D Tractable Structures | Ligand Score | Gene Description |
| CBFB | 0.88 | 0.98 | 8.39 | 5 | −3.00 | Core-binding factor subunit beta |
| GPX4 | 0.85 | 0.99 | 2.85 | 6 | −3.00 | Glutathione peroxidase 4 |
| RHOA | 0.82 | 0.99 | 4.27 | 39 | −1.60 | Ras homolog family member A |
| UMPS | 0.81 | 1.00 | 3.49 | 33 | −1.30 | Uridine monophosphate synthetase |
| GFPT1 | 0.81 | 0.99 | 4.73 | 13 | −1.30 | Glutamine-F6P transaminase 1 |
| RFK | 0.75 | 0.98 | 3.92 | 4 | −3.00 | Riboflavin kinase |
| CAD | 0.71 | 0.99 | 4.30 | 36 | 0.90 | Carbamoyl-phosphate synthetase 2, aspar … |
| LDB1 | 0.57 | 0.99 | 6.37 | 6 | −0.90 | LIM domain binding 1 |
| NMNAT1 | 0.53 | 1.00 | 2.45 | 5 | −3.00 | Nicotinamide nucleotide acetyltransferase 1 |
| CCNC | 0.48 | 0.98 | 1.57 | 29 | 1.55 | Cyclin C |
| FLI1 | 0.44 | 0.96 | 8.83 | 5 | −3.00 | Fli-1 proto-oncogene, ETS transcription fa … |
| MBNL1 | 0.44 | 0.94 | 3.26 | 5 | −3.00 | Muscleblind like splicing regulator 1 |
| TRIM28 | 0.41 | 0.98 | 2.53 | 5 | −3.00 | Tripartite motif containing 28 |
| PREX1 | 0.36 | 0.94 | 8.55 | 9 | −0.95 | PIP3 dependent Rac exchange factor 1 |
| SH3GL1 | 0.36 | 0.96 | 3.07 | 0 | 1.35 | SH3 domain containing GRB2 like 1, endo … |
| PEX6 | 0.35 | 0.96 | 1.44 | 0 | 1.00 | Peroxisomal biogenesis factor 6 |
| PUS1 | 0.33 | 0.94 | 3.78 | 5 | 1.25 | Pseudouridine synthase 1 |
| RUNX1T1 | 0.14 | 0.94 | 3.89 | 4 | −3.00 | RUNX1 partner transcriptional corepressor 1 |
| ADIPOR2 | 0.08 | 0.97 | 3.85 | 4 | −3.00 | Adiponectin receptor 2 |
| POLR2E | 1.00 | 1.00 | 1.61 | 10 | −3.00 | RNA polymerase II, I and III subunit E |
| TSR1 | 1.00 | 1.00 | 2.09 | 5 | −3.00 | TSR1 ribosome maturation factor |
| CTCF | 1.00 | 1.00 | 1.43 | 6 | −3.00 | CCCTC-binding factor |
| NEDD8 | 1.00 | 1.00 | 1.55 | 11 | −3.00 | NEDD8 ubiquitin like modifier |
| MIS18A | 1.00 | 1.00 | 1.56 | 0 | 1.35 | MIS18 kinetochore protein A |
| ANAPC4 | 1.00 | 1.00 | 1.60 | 10 | −3.00 | Anaphase promoting complex subunit 4 |
| POLR1A | 1.00 | 1.00 | 1.51 | 0 | 1.40 | RNA polymerase I subunit A |
| EIF4A3 | 0.99 | 1.00 | 1.57 | 10 | −3.00 | Eukaryotic translation initiation factor 4A3 |
| SMC4 | 0.99 | 1.00 | 1.61 | 1 | 0.90 | Structural maintenance of chromosomes 4 |
| NUTF2 | 0.99 | 1.00 | 1.56 | 4 | −3.00 | Nuclear transport factor 2 |
| TFRC | 0.98 | 1.00 | 2.52 | 13 | −3.00 | Transferrin receptor |
| COPZ1 | 0.98 | 1.00 | 1.51 | 11 | −3.00 | COPI coat complex subunit zeta 1 |
| ADSL | 0.98 | 1.00 | 2.29 | 4 | −3.00 | Adenylosuccinate lyase |
| TERF2 | 0.98 | 1.00 | 1.70 | 11 | −3.00 | Telomeric repeat binding factor 2 |
| SNRPD3 | 0.98 | 1.00 | 1.52 | 9 | 0.30 | Small nuclear ribonucleoprotein D3 polype … |
| CUL1 | 0.97 | 1.00 | 1.57 | 8 | −0.50 | Cullin 1 |
| RIOK2 | 0.97 | 1.00 | 1.98 | 6 | 1.80 | RIO kinase 2 |
| AP2M1 | 0.93 | 1.00 | 2.43 | 13 | −0.95 | Adaptor related protein complex 2 subunit … |
| POTEG | 0.93 | 1.00 | 2.33 | 0 | 1.00 | POTE ankyrin domain family member G |
| USP8 | 0.93 | 0.99 | 1.64 | 5 | −1.85 | Ubiquitin specific peptidase 8 |
| PFN1 | 0.93 | 1.00 | 1.85 | 5 | −3.00 | Profilin 1 |
| CFL1 | 0.92 | 1.00 | 3.49 | 8 | −3.00 | Cofilin 1 |
| UROD | 0.92 | 1.00 | 3.12 | 19 | −3.00 | Uroporphyrinogen decarboxylase |
| SETDB1 | 0.81 | 1.00 | 2.35 | 15 | −3.00 | SET domain bifurcated histone lysine met … |
| PDCD6 | 0.76 | 0.97 | 1.62 | 10 | −3.00 | Programmed cell death 6 |
| RCOR1 | 0.75 | 1.00 | 2.66 | 9 | −0.05 | REST corepressor 1 |
| YTHDC1 | 0.74 | 0.94 | 2.36 | 36 | −3.00 | YTH domain containing 1 |
| OR7A10 | 0.72 | 0.94 | 1.96 | 0 | 2.75 | Olfactory receptor family 7 subfamily A me … |
| DYNLL1 | 0.71 | 0.97 | 1.87 | 7 | −3.00 | Dynein light chain LC8-type 1 |
| CYP2A13 | 0.71 | 0.94 | 2.65 | 8 | 1.15 | cytochrome P450 family 2 subfamily A me … |
| NBEAL1 | 0.71 | 0.98 | 2.65 | 0 | 0.95 | Neurobeachin like 1 |
| Developing Knockout Genes (AML deKO) | ||||||
|---|---|---|---|---|---|---|
| Gene | AML Mean Dependence | AML Max. Dependence | AML-Specific Z-Score | 3D Tractable Structures | Ligand Score | Gene Description |
| CDK6 | 0.86 | 1.00 | 3.30 | 16 | 0.20 | Cyclin dependent kinase 6 |
| NAMPT | 0.76 | 1.00 | 5.95 | 63 | 0.30 | Nicotinamide phosphoribosyltransferase |
| KMT2D | 0.76 | 0.99 | 3.40 | 1 | 1.35 | Lysine methyltransferase 2D |
| CCND3 | 0.73 | 0.99 | 6.64 | 1 | 1.65 | Cyclin D3 |
| EP300 | 0.72 | 1.00 | 3.19 | 32 | 1.05 | E1A binding protein p300 |
| PPAT | 0.71 | 1.00 | 2.29 | 0 | 1.65 | Phosphoribosyl pyrophosphate amidotrans... |
| PI4KB | 0.67 | 0.97 | 3.86 | 14 | 0.35 | Phosphatidylinositol 4-kinase beta |
| ATIC | 0.62 | 0.99 | 2.16 | 5 | −0.40 | 5-aminoimidazole-4-carboxamide ribonucl … |
| FECH | 0.62 | 0.97 | 3.60 | 23 | −3.00 | Ferrochelatase |
| TYMS | 0.59 | 1.00 | 1.43 | 60 | 1.10 | Thymidylate synthetase |
| GART | 0.56 | 0.98 | 3.72 | 29 | 0.05 | Phosphoribosylglycinamide formyltransfer … |
| BRPF1 | 0.53 | 0.94 | 2.71 | 56 | 1.10 | Bromodomain and PHD finger containing 1 |
| KMT2A | 0.50 | 0.96 | 4.01 | 19 | −0.25 | Lysine methyltransferase 2A |
| LMO2 | 0.49 | 0.99 | 14.21 | 5 | −3.00 | LIM domain only 2 |
| SOS1 | 0.49 | 0.99 | 3.23 | 59 | 0.05 | SOS Ras/Rac guanine nucleotide exchan … |
| RAF1 | 0.42 | 1.00 | 5.47 | 6 | 1.00 | Raf-1 proto-oncogene, serine/threonine ki … |
| RPS6KA1 | 0.41 | 0.94 | 7.38 | 6 | 0.80 | Ribosomal protein S6 kinase A1 |
| MDH2 | 0.36 | 0.99 | 3.41 | 7 | −0.55 | Malate dehydrogenase 2 |
| LDLR | 0.33 | 0.99 | 3.21 | 11 | 0.20 | Low density lipoprotein receptor |
| FLT3 | 0.31 | 0.99 | 3.05 | 7 | 1.65 | Fms related receptor tyrosine kinase 3 |
| NRAS | 0.30 | 1.00 | 2.89 | 4 | −0.10 | NRAS proto-oncogene, GTPase |
| RUNX1 | 0.30 | 1.00 | 6.77 | 5 | −1.45 | RUNX family transcription factor 1 |
| HDAC7 | 0.29 | 0.99 | 2.46 | 5 | 1.20 | Histone deacetylase 7 |
| USP14 | 0.25 | 0.95 | 2.52 | 8 | −1.90 | Ubiquitin specific peptidase 14 |
| JAK3 | 0.17 | 1.00 | 1.74 | 35 | 1.60 | Janus kinase 3 |
| ETV6 | 0.15 | 0.99 | 3.56 | 4 | 1.45 | ETS variant transcription factor 6 |
| JAK2 | 0.13 | 0.99 | 8.95 | 93 | 1.90 | Janus kinase 2 |
| KIT | 0.13 | 0.97 | 1.44 | 23 | 1.50 | KIT proto-oncogene, receptor tyrosine kina … |
| CSF2RB | 0.12 | 0.99 | 5.13 | 5 | −3.00 | Colony stimulating factor 2 receptor subun … |
| TYRP1 | 0.08 | 0.97 | 3.50 | 9 | −3.00 | Tyrosinase related protein 1 |
| XPO1 | 1.00 | 1.00 | 1.48 | 6 | −1.25 | Exportin 1 |
| HSPA5 | 0.99 | 1.00 | 1.56 | 26 | −0.95 | Heat shock protein family A (Hsp70) mem … |
| WNK1 | 0.99 | 1.00 | 2.08 | 6 | −1.90 | WNK lysine deficient protein kinase 1 |
| AURKA | 0.98 | 0.99 | 1.51 | 154 | 1.60 | Aurora kinase A |
| AHCY | 0.94 | 1.00 | 1.96 | 8 | 0.75 | Adenosylhomocysteinase |
| MCL1 | 0.88 | 1.00 | 3.61 | 95 | 0.15 | MCL1 apoptosis regulator, BCL2 family m … |
| PSMC1 | 0.88 | 0.97 | 1.75 | 25 | −3.00 | Proteasome 26S subunit, ATPase 1 |
| FDPS | 0.86 | 0.99 | 2.24 | 88 | 1.05 | Farnesyl diphosphate synthase |
| PSMD8 | 0.85 | 0.95 | 1.73 | 14 | −3.00 | Proteasome 26S subunit, non-ATPase 8 |
| ACTB | 0.82 | 0.99 | 1.92 | 18 | −3.00 | Actin beta |
| DHFR | 0.77 | 1.00 | 1.61 | 79 | 1.25 | Dihydrofolate reductase |
| HUS1 | 0.73 | 0.99 | 1.83 | 4 | −3.00 | HUS1 checkpoint clamp component |
| DHODH | 0.71 | 1.00 | 1.77 | 71 | 1.05 | Dihydroorotate dehydrogenase |
| SETD2 | 0.70 | 0.95 | 1.69 | 18 | 1.20 | SET domain containing 2, histone lysine m … |
| PANTHER Gene Overrepresentation Test | ||||||
|---|---|---|---|---|---|---|
| GO Biological Process Complete | Homo Sapiens REFLIST (20851) | Queried | Expected | Fold Enrichment | p-Value | FDR |
| ‘de novo’ UMP biosynthetic process (GO:0044205) | 3 | 3 | 0.01 | >100 | 2.03E−06 | 2.81E−04 |
| ‘de novo’ IMP biosynthetic process (GO:0006189) | 6 | 4 | 0.03 | >100 | 9.64E−08 | 2.85E−05 |
| dihydrofolate metabolic process (GO:0046452) | 3 | 2 | 0.01 | >100 | 2.19E−04 | 1.37E−02 |
| ‘de novo’ pyrimidine nucleobase biosynthetic process (GO:0006207) | 7 | 4 | 0.03 | >100 | 1.51E−07 | 3.71E−05 |
| vitellogenesis (GO:0007296) | 4 | 2 | 0.02 | >100 | 3.28E−04 | 1.87E−02 |
| positive regulation of CD8-positive, alpha-beta T cell differentiation (GO:0043378) | 4 | 2 | 0.02 | >100 | 3.28E−04 | 1.87E−02 |
| pyrimidine nucleobase biosynthetic process (GO:0019856) | 9 | 4 | 0.04 | 93.61 | 3.25E−07 | 6.56E−05 |
| tetrahydrofolate biosynthetic process (GO:0046654) | 7 | 3 | 0.03 | 90.26 | 1.20E−05 | 1.29E−03 |
| IMP biosynthetic process (GO:0006188) | 10 | 4 | 0.05 | 84.25 | 4.53E−07 | 8.41E−5 |
| myeloid progenitor cell differentiation (GO:0002318) | 6 | 2 | 0.03 | 70.21 | 6.08E−04 | 3.06E−02 |
| histone H3-K4 dimethylation (GO:0044648) | 6 | 2 | 0.03 | 70.21 | 6.08E−04 | 3.05E−02 |
| regulation of CD8-positive, alpha-beta T cell differentiation (GO:0043376) | 6 | 2 | 0.03 | 70.21 | 6.08E−04 | 3.04E−02 |
| folic acid-containing compound biosynthetic process (GO:0009396) | 9 | 3 | 0.04 | 70.21 | 2.18E−05 | 2.10E−03 |
| nucleobase biosynthetic process (GO:0046112) | 19 | 6 | 0.09 | 66.51 | 1.57E−09 | 9.30E−07 |
| IMP metabolic process (GO:0046040) | 13 | 4 | 0.06 | 64.8 | 1.07E−06 | 1.70E−04 |
| UMP metabolic process (GO:0046049) | 10 | 3 | 0.05 | 63.18 | 2.83E−05 | 2.58E−03 |
| UMP biosynthetic process (GO:0006222) | 10 | 3 | 0.05 | 63.18 | 2.83E−05 | 2.57E−03 |
| pyrimidine ribonucleoside monophosphate biosynthetic process (GO:0009174) | 10 | 3 | 0.05 | 63.18 | 2.83E−05 | 2.55E−03 |
| pyrimidine ribonucleoside monophosphate metabolic process (GO:0009173) | 10 | 3 | 0.05 | 63.18 | 2.83E−05 | 2.54E−03 |
| pyrimidine nucleobase metabolic process (GO:0006206) | 17 | 5 | 0.08 | 61.95 | 5.26E−08 | 1.65E−05 |
| regulation of ribonucleoprotein complex localization (GO:2000197) | 7 | 2 | 0.03 | 60.18 | 7.79E−04 | 3.73E−02 |
| brainstem development (GO:0003360) | 7 | 2 | 0.03 | 60.18 | 7.79E−04 | 3.72E−02 |
| pyrimidine nucleoside monophosphate biosynthetic process (GO:0009130) | 15 | 4 | 0.07 | 56.16 | 1.72E−06 | 2.55E−04 |
| positive regulation of CD8-positive, alpha-beta T cell activation (GO:2001187) | 8 | 2 | 0.04 | 52.65 | 9.71E−04 | 4.43E−02 |
| positive regulation of synaptic vesicle endocytosis (GO:1900244) | 8 | 2 | 0.04 | 52.65 | 9.71E−04 | 4.41E−02 |
| pyrimidine nucleoside monophosphate metabolic process (GO:0009129) | 16 | 4 | 0.08 | 52.65 | 2.14E−06 | 2.95E−04 |
| histone H3-K4 monomethylation (GO:0097692) | 8 | 2 | 0.04 | 52.65 | 9.71E−04 | 4.40E−02 |
| ribonucleoside monophosphate biosynthetic process (GO:0009156) | 33 | 8 | 0.16 | 51.06 | 1.57E−11 | 1.79E−08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Takacs, G.P.; Lamba, J.K.; Vulpe, C.; Cogle, C.R. Functional Dependency Analysis Identifies Potential Druggable Targets in Acute Myeloid Leukemia. Cancers 2020, 12, 3710. https://doi.org/10.3390/cancers12123710
Zhou Y, Takacs GP, Lamba JK, Vulpe C, Cogle CR. Functional Dependency Analysis Identifies Potential Druggable Targets in Acute Myeloid Leukemia. Cancers. 2020; 12(12):3710. https://doi.org/10.3390/cancers12123710
Chicago/Turabian StyleZhou, Yujia, Gregory P. Takacs, Jatinder K. Lamba, Christopher Vulpe, and Christopher R. Cogle. 2020. "Functional Dependency Analysis Identifies Potential Druggable Targets in Acute Myeloid Leukemia" Cancers 12, no. 12: 3710. https://doi.org/10.3390/cancers12123710
APA StyleZhou, Y., Takacs, G. P., Lamba, J. K., Vulpe, C., & Cogle, C. R. (2020). Functional Dependency Analysis Identifies Potential Druggable Targets in Acute Myeloid Leukemia. Cancers, 12(12), 3710. https://doi.org/10.3390/cancers12123710