The Tumor Microenvironment—A Metabolic Obstacle to NK Cells’ Activity

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Biological Aspects of NK Cell Cytotoxicity
2.1. NK Cells’ Metabolism
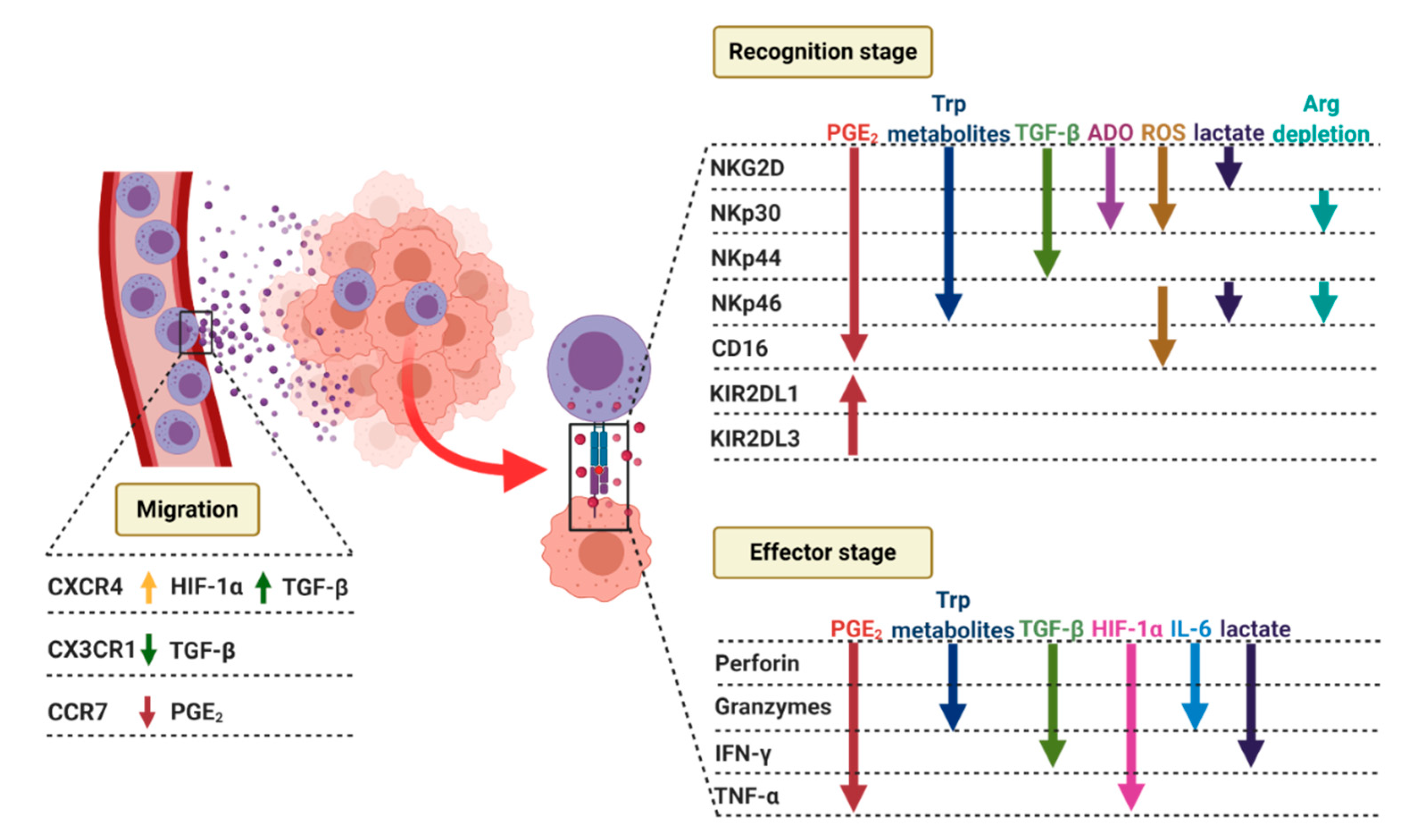
2.2. NK Cells Recruitment to the Tumor Site
2.3. Formation of the Lytic NK-Cell Immunological Synapse
2.3.1. Recognition Stage
2.3.2. Effector Stage
2.3.3. Termination Stage
2.4. NK Cells’ Cytokine Production
3. Characteristics of the Tumor Microenvironment
3.1. Tumor Hypoxia and Acidosis
3.2. Oxidative Stress
3.3. Cytokines
3.4. Amino Acid Deprivation
3.5. Alterations in the Key Enzymes of Lipid and Adenosine Metabolism
4. How Tumor Microenvironment Factors Inhibit NK Cells
4.1. NK Cells’ Metabolism
4.2. NK Cells Recruitment to the Tumor Site
4.3. NK Cells’ Lytic Synapse
4.3.1. Recognition Stage
4.3.2. Effector Stage
4.4. NK Cells’ Cytokines and Chemokines Production
5. Strategies to Overcome the Inhibitory Effects of TME on NK Cell Functions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sharma, P.; Kumar, P.; Sharma, R. Natural Killer Cells—Their Role in Tumour Immunosurveillance. J. Clin. Diagn. Res. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Scherer, D.C.; King, A.G.; Manz, M.G.; Weissman, I.L. Lymphocyte development from hematopoietic stem cells. Curr. Opin. Genet. Dev. 2001, 11, 520–526. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the CD56bright subset. Blood 2001, 97, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Zhang, X.R.; Iwatsuki, S.; Van Thiel, D.H.; Herberman, R.B.; Whiteside, T.L. Isolation, phenotyping, and functional analysis of lymphocytes from human liver. Clin. Immunol. Immunopathol. 1990, 56, 401–419. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Crinier, A.; Narni-Mancinelli, E.; Ugolini, S.; Vivier, E. SnapShot: Natural Killer Cells. Cell 2020, 180, 1280. [Google Scholar] [CrossRef]
- Blom, B.; Spits, H. Development of human lymphoid cells. Annu. Rev. Immunol. 2006, 24, 287–320. [Google Scholar] [CrossRef]
- Yu, J.; Freud, A.G.; Caligiuri, M.A. Location and cellular stages of natural killer cell development. Trends Immunol. 2013, 34, 573–582. [Google Scholar] [CrossRef]
- Ahn, Y.O.; Blazar, B.R.; Miller, J.S.; Verneris, M.R. Lineage relationships of human interleukin-22-producing CD56+ RORgammat+ innate lymphoid cells and conventional natural killer cells. Blood 2013, 121, 2234–2243. [Google Scholar] [CrossRef]
- Eckelhart, E.; Warsch, W.; Zebedin, E.; Simma, O.; Stoiber, D.; Kolbe, T.; Rülicke, T.; Mueller, M.; Casanova, E.; Sexl, V. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011, 117, 1565–1573. [Google Scholar] [CrossRef]
- Kennedy, M.K.; Glaccum, M.; Brown, S.N.; Butz, E.A.; Viney, J.L.; Embers, M.; Matsuki, N.; Charrier, K.; Sedger, L.; Willis, C.R.; et al. Reversible Defects in Natural Killer and Memory Cd8 T Cell Lineages in Interleukin 15–Deficient Mice. J. Exp. Med. 2000, 191, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Kündig, T.M.; Schorle, H.; Bachmann, M.F.; Hengartner, H.; Zinkernagel, R.M.; Horak, I. Immune responses in interleukin-2-deficient mice. Sciience 1993, 262, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Vosshenrich, C.A.J.; Di Santo, J.P. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat. Rev. Immunol. 2007, 7, 703–714. [Google Scholar] [CrossRef]
- Colucci, F.; Caligiuri, M.A.; Di Santo, J.P. What does it take to make a natural killer? Nat. Rev. Immunol. 2003, 3, 413–425. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Smyth, M.J. Close encounters of different kinds: Dendritic cells and NK cells take centre stage. Nat. Rev. Immunol. 2005, 5, 112–124. [Google Scholar] [CrossRef]
- Koka, R.; Burkett, P.; Chien, M.; Chai, S.; Boone, D.L.; Ma, A. Cutting edge: Murine dendritic cells require IL-15R alpha to prime NK cells. J. Immunol. 2004, 173, 3594–3598. [Google Scholar] [CrossRef]
- Crouse, J.; Xu, H.C.; Lang, P.A.; Oxenius, A. NK cells regulating T cell responses: Mechanisms and outcome. Trends Immunol. 2015, 36, 49–58. [Google Scholar] [CrossRef]
- Martin-Fontecha, A.; Thomsen, L.L.; Brett, S.; Gerard, C.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 2004, 5, 1260–1265. [Google Scholar] [CrossRef]
- Wu, Y.; Tian, Z.; Wei, H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front. Immunol. 2017, 8, 930. [Google Scholar] [CrossRef]
- Liang, S.; Wei, H.; Sun, R.; Tian, Z. IFNalpha regulates NK cell cytotoxicity through STAT1 pathway. Cytokine 2003, 23, 190–199. [Google Scholar] [CrossRef]
- Putz, E.M.; Majoros, A.; Gotthardt, D.; Prchal-Murphy, M.; Zebedin-Brandl, E.M.; Fux, D.A.; Schlattl, A.; Schreiber, R.D.; Carotta, S.; Müller, M.; et al. Novel non-canonical role of STAT1 in Natural Killer cell cytotoxicity. OncoImmunology 2016, 5, 1186314. [Google Scholar] [CrossRef]
- Gotthardt, D.; Putz, E.M.; Straka, E.; Kudweis, P.; Biaggio, M.; Poli, V.; Strobl, B.; Müller, M.; Sexl, V. Loss of STAT3 in murine NK cells enhances NK cell–dependent tumor surveillance. Blood 2014, 124, 2370–2379. [Google Scholar] [CrossRef]
- Gotthardt, D.; Putz, E.M.; Grundschober, E.; Prchal-Murphy, M.; Straka, E.; Kudweis, P.; Heller, G.; Bago-Horvath, Z.; Witalisz-Siepracka, A.; Cumaraswamy, A.A.; et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov. 2016, 6, 414–429. [Google Scholar] [CrossRef]
- Sim, G.C.; Radvanyi, L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef]
- Gotthardt, D.; Sexl, V. STATs in NK-Cells: The Good, the Bad, and the Ugly. Front. Immunol. 2017, 7, 694. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Cai, S.F.; Cao, X.; Bredemeyer, A.J.; Presti, R.M.; French, A.R.; Ley, T.J. Acquisition of Murine NK Cell Cytotoxicity Requires the Translation of a Pre-existing Pool of Granzyme B and Perforin mRNAs. Immunity 2007, 26, 798–811. [Google Scholar] [CrossRef]
- Enandagopal, N.; Ali, A.K.; Komal, A.K.; Lee, S.-H. The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front. Immunol. 2014, 5, 187. [Google Scholar] [CrossRef]
- Yamamoto, K.; Shibata, F.; Miyasaka, N.; Miura, O. The human perforin gene is a direct target of STAT4 activated by IL-12 in NK cells. Biochem. Biophys. Res. Commun. 2002, 297, 1245–1252. [Google Scholar] [CrossRef]
- Sabry, M.; Zubiak, A.; Hood, S.P.; Simmonds, P.; Arellano-Ballestero, H.; Cournoyer, E.; Mashar, M.; Pockley, A.G.; Lowdell, M.W. Tumor- and cytokine-primed human natural killer cells exhibit distinct phenotypic and transcriptional signatures. PLoS ONE 2019, 14, 0218674. [Google Scholar] [CrossRef]
- Salzberger, W.; Martrus, G.; Bachmann, K.; Goebels, H.; Heß, L.; Koch, M.; Langeneckert, A.; Lunemann, S.; Oldhafer, K.J.; Pfeifer, C.; et al. Tissue-resident NK cells differ in their expression profile of the nutrient transporters Glut1, CD98 and CD71. PLoS ONE 2018, 13, 0201170. [Google Scholar] [CrossRef]
- Keating, S.E.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Keane, C.; Brennan, K.; Finlay, D.K.; Gardiner, C.M. Metabolic Reprogramming Supports IFN-gamma Production by CD56bright NK Cells. J. Immunol. 2016, 196, 2552–2560. [Google Scholar] [CrossRef]
- Gardiner, C.M.; Finlay, D.K. What Fuels Natural Killers? Metabolism and NK Cell Responses. Front. Immunol. 2017, 8, 367. [Google Scholar] [CrossRef]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef]
- Loftus, R.M.; Assmann, N.; Kedia-Mehta, N.; O’Brien, K.L.; Garcia, A.; Gillespie, C.; Hukelmann, J.L.; Oefner, P.J.; Lamond, A.I.; Gardiner, C.M.; et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Messaoudene, M.; Fregni, G.; Fourmentraux-Neves, E.; Chanal, J.; Maubec, E.; Mazouz-Dorval, S.; Couturaud, B.; Girod, A.; Sastre-Garau, X.; Albert, S.; et al. Mature Cytotoxic CD56bright/CD16+ Natural Killer Cells Can Infiltrate Lymph Nodes Adjacent to Metastatic Melanoma. Cancer Res. 2014, 74, 81–92. [Google Scholar] [CrossRef]
- Ali, T.H.; Pisanti, S.; Ciaglia, E.; Mortarini, R.; Anichini, A.; Garofalo, C.; Tallerico, R.; Santinami, M.; Gulletta, E.; Ietto, C.; et al. Enrichment of CD56dimKIR+CD57+ highly cytotoxic NK cells in tumour-infiltrated lymph nodes of melanoma patients. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- Levi, I.; Amsalem, H.; Nissan, A.; Darash-Yahana, M.; Peretz, T.; Mandelboim, O.; Rachmilewitz, J. Characterization of tumor infiltrating Natural Killer cell subset. Oncotarget 2015, 6, 13835–13843. [Google Scholar] [CrossRef]
- Mamessier, E.; Sylvain, A.; Thibult, M.-L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Gonçalves, A.; André, P.; Romagné, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [PubMed]
- Berahovich, R.D.; Lai, N.L.; Wei, Z.; Lanier, L.L.; Schall, T.J. Evidence for NK Cell Subsets Based on Chemokine Receptor Expression. J. Immunol. 2006, 177, 7833–7840. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Qin, S.; Unutmaz, D.; Soler, D.; Murphy, K.E.; Hodge, M.R.; Wu, L.; Butcher, E.C. Unique Subpopulations of CD56+ NK and NK-T Peripheral Blood Lymphocytes Identified by Chemokine Receptor Expression Repertoire. J. Immunol. 2001, 166, 6477–6482. [Google Scholar] [CrossRef] [PubMed]
- Pachynski, R.K.; Zabel, B.A.; Kohrt, H.E.; Tejeda, N.M.; Monnier, J.; Swanson, C.D.; Holzer, A.K.; Gentles, A.J.; Sperinde, G.V.; Edalati, A.; et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J. Exp. Med. 2012, 209, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, C.; Ye, Y.-B.; Peng, F.; Chen, Q. Expression of Chemerin Correlates with a Favorable Prognosis in Patients with Non-Small Cell Lung Cancer. Lab. Med. 2011, 42, 553–557. [Google Scholar] [CrossRef]
- Sechler, J.M.; Barlic, J.; Grivel, J.-C.; Murphy, P.M. IL-15 alters expression and function of the chemokine receptor CX3CR1 in human NK cells. Cell. Immunol. 2004, 230, 99–108. [Google Scholar] [CrossRef]
- Inngjerdingen, M.; Damaj, B.; Maghazachi, A.M.A. Expression and regulation of chemokine receptors in human natural killer cells. Blood 2001, 97, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Wendel, M.; Galani, I.E.; Suri-Payer, E.; Cerwenka, A. Natural Killer Cell Accumulation in Tumors Is Dependent on IFN- and CXCR3 Ligands. Cancer Res. 2008, 68, 8437–8445. [Google Scholar] [CrossRef]
- Halama, N.; Braun, M.; Kahlert, C.; Spille, A.; Quack, C.; Rahbari, N.; Koch, M.; Weitz, J.; Kloor, M.; Zoernig, I.; et al. Natural Killer Cells are Scarce in Colorectal Carcinoma Tissue despite High Levels of Chemokines and Cytokines. Clin. Cancer Res. 2011, 17, 678–689. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Barber, D.F.; Ljunggren, H.G.; Long, E.O. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J. Exp. Med. 2005, 202, 1001–1012. [Google Scholar] [CrossRef]
- Barber, D.F.; Faure, M.; Long, E.O. LFA-1 Contributes an Early Signal for NK Cell Cytotoxicity. J. Immunol. 2004, 173, 3653–3659. [Google Scholar] [CrossRef]
- Riteau, B.; Barber, D.F.; Long, E.O. Vav1 phosphorylation is induced by beta2 integrin engagement on natural killer cells upstream of actin cytoskeleton and lipid raft reorganization. J. Exp. Med. 2003, 198, 469–474. [Google Scholar] [CrossRef]
- Orange, J.S.; Harris, K.E.; Andzelm, M.M.; Valter, M.M.; Geha, R.S.; Strominger, J.L. The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proc. Natl. Acad. Sci. USA 2003, 100, 14151–14156. [Google Scholar] [CrossRef]
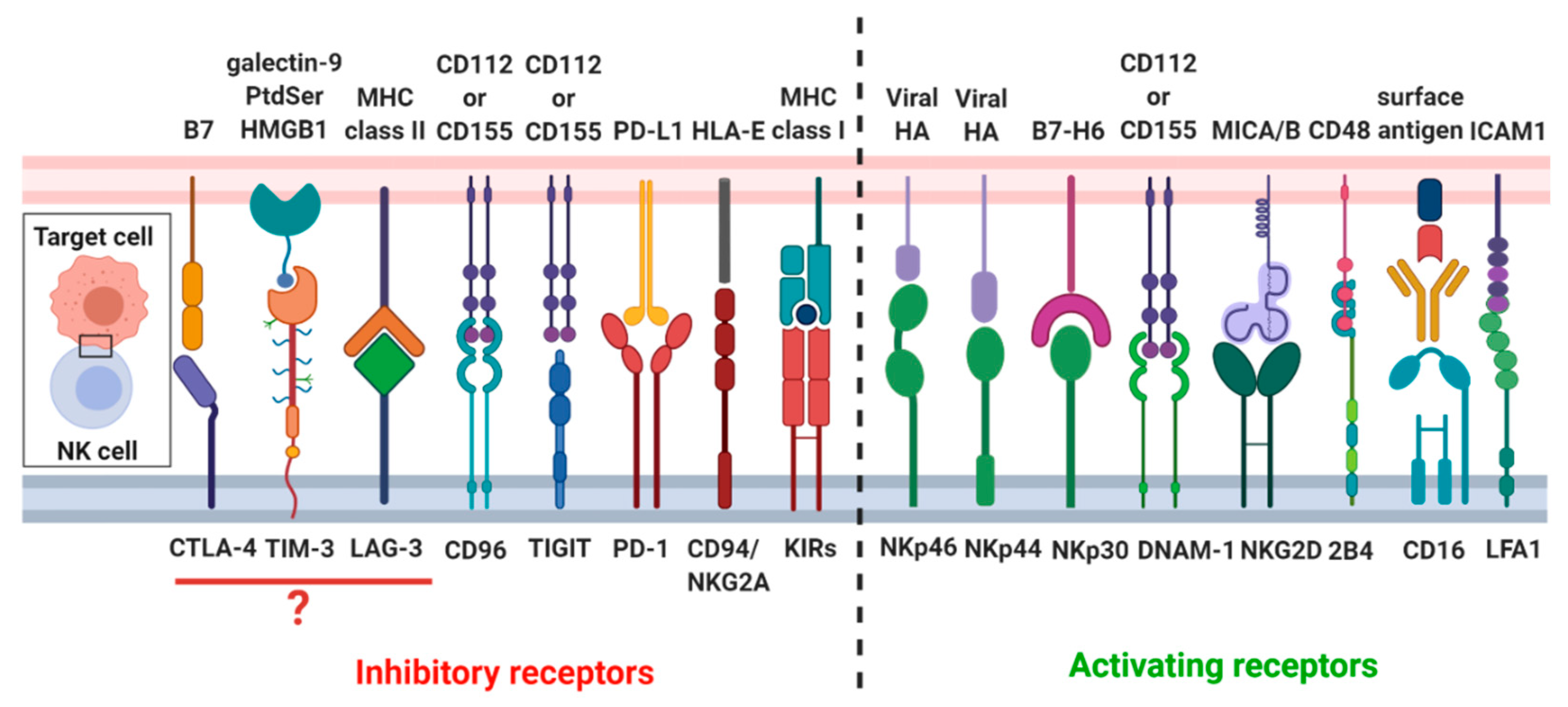
- Kumar, S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology 2018, 154, 383–393. [Google Scholar] [CrossRef]
- Echester, C.; Efritsch, K.; Kohrt, H.E. Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Front. Immunol. 2015, 6, 601. [Google Scholar] [CrossRef]
- Lanier, L.L. Nk Cell Receptors. Annu. Rev. Immunol. 1998, 16, 359–393. [Google Scholar] [CrossRef]
- Hudspeth, K.; Silva-Santos, B.; Mavilio, D. Natural Cytotoxicity Receptors: Broader Expression Patterns and Functions in Innate and Adaptive Immune Cells. Front. Immunol. 2013, 4, 69. [Google Scholar] [CrossRef]
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a Novel Triggering Surface Molecule Specifically Expressed by Activated Natural Killer Cells, Is Involved in Non–Major Histocompatibility Complex–restricted Tumor Cell Lysis. J. Exp. Med. 1998, 187, 2065–2072. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e3. [Google Scholar] [CrossRef]
- Stojanovic, A.; Fiegler, N.; Brunner-Weinzierl, M.; Cerwenka, A. CTLA-4 is expressed by activated mouse NK cells and inhibits NK Cell IFN-gamma production in response to mature dendritic cells. J. Immunol. 2014, 192, 4184–4191. [Google Scholar] [CrossRef]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Meng, F.; Li, L.J.; Lu, F.; Yue, J.; Liu, Z.; Zhang, W.; Fu, R. Overexpression of TIGIT in NK and T Cells Contributes to Tumor Immune Escape in Myelodysplastic Syndromes. Front. Oncol. 2020, 10, 1595. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef]
- Mariotti, F.R.; Petrini, S.; Ingegnere, T.; Tumino, N.; Besi, F.; Scordamaglia, F.; Munari, E.; Pesce, S.; Marcenaro, E.; Moretta, A.; et al. PD-1 in human NK cells: Evidence of cytoplasmic mRNA and protein expression. Oncoimmunology 2019, 8, 1557030. [Google Scholar] [CrossRef]
- Van Audenaerde, J.R.; De Waele, J.; Marcq, E.; Van Loenhout, J.; Lion, E.; Bergh, J.M.V.D.; Jesenofsky, R.; Masamune, A.; Roeyen, G.; Pauwels, P.; et al. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget 2017, 8, 56968–56979. [Google Scholar] [CrossRef]
- Da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-Cell Exhaustion in Advanced Melanoma by Tim-3 Blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Banas, H.; Casas-Aviles, I.; Duran, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Sriram, P.J.A.; Veonice, A.B.; Nivashini, K.; Seng, G.L.; Cheng, W.; Fairhurst, A.-M.; Connolly, J.E. LAG3 is a Central Regulator of NK Cell Cytokine Production. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dustin, M.L.; Long, E.O. Cytotoxic immunological synapses. Immunol. Rev. 2010, 235, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Vyas, Y.M.; Mehta, K.M.; Morgan, M.; Maniar, H.; Butros, L.; Jung, S.; Burkhardt, J.K.; Dupont, B. Spatial organization of signal transduction molecules in the NK cell immune synapses during MHC class I-regulated noncytolytic and cytolytic interactions. J. Immunol. 2001, 167, 4358–4367. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S.; Ramesh, N.; Remold-O’Donnell, E.; Sasahara, Y.; Koopman, L.; Byrne, M.; Bonilla, F.A.; Rosen, F.S.; Geha, R.S.; Strominger, J.L. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc. Natl. Acad. Sci. USA 2002, 99, 11351–11356. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.B.; Cella, M.; Giurisato, E.; Fujikawa, K.; Miletic, A.V.; Kloeppel, T.; Brim, K.; Takai, T.; Shaw, A.S.; Colonna, M.; et al. Vav1 Controls DAP10-Mediated Natural Cytotoxicity by Regulating Actin and Microtubule Dynamics. J. Immunol. 2006, 177, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- Mace, E.M.; Dongre, P.; Hsu, H.; Sinha, P.; James, A.M.; Mann, S.S.; Forbes, L.R.; Watkin, L.B.; Orange, J.S. Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol. Cell Biol. 2014, 92, 245–255. [Google Scholar] [CrossRef]
- Blott, E.J.; Griffiths, G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002, 3, 122–131. [Google Scholar] [CrossRef]
- Liu, D.; Xu, L.; Yang, F.; Li, D.; Gong, F.; Xu, T. Rapid biogenesis and sensitization of secretory lysosomes in NK cells mediated by target-cell recognition. Proc. Natl. Acad. Sci. USA 2005, 102, 123–127. [Google Scholar] [CrossRef]
- Metkar, S.S.; Wang, B.; Catalan, E.; Anderluh, G.; Gilbert, R.J.C.; Pardo, J.; Froelich, C. Perforin Rapidly Induces Plasma Membrane Phospholipid Flip-Flop. PLoS ONE 2011, 6, e24286. [Google Scholar] [CrossRef]
- Uellner, R.; Zvelebil, M.J.; Hopkins, J.; Jones, J.; MacDougall, L.K.; Morgan, B.; Podack, E.; Waterfield, M.D.; Griffiths, G.M. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J. 1997, 16, 7287–7296. [Google Scholar] [CrossRef] [PubMed]
- Kam, C.-M.; Hudig, D.; Powers, J.C. Granzymes (lymphocyte serine proteases): Characterization with natural and synthetic substrates and inhibitors. Biochim. Biophys. Acta 2000, 1477, 307–323. [Google Scholar] [CrossRef]
- McCann, F.E.; Vanherberghen, B.; Eleme, K.; Carlin, L.M.; Newsam, R.J.; Goulding, D.; Davis, D.M. The Size of the Synaptic Cleft and Distinct Distributions of Filamentous Actin, Ezrin, CD43, and CD45 at Activating and Inhibitory Human NK Cell Immune Synapses. J. Immunol. 2003, 170, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 2008, 8, 713–725. [Google Scholar] [CrossRef]
- Simhadri, V.R.; Andersen, J.F.; Calvo, E.; Choi, S.-C.; Coligan, J.E.; Borrego, F. Human CD300a binds to phosphatidylethanolamine and phosphatidylserine, and modulates the phagocytosis of dead cells. Blood 2012, 119, 2799–2809. [Google Scholar] [CrossRef]
- Srpan, K.; Ambrose, A.; Karampatzakis, A.; Saeed, M.; Cartwright, A.N.; Guldevall, K.; De Matos, G.D.S.C.; Önfelt, B.; Davis, D.M. Shedding of CD16 disassembles the NK cell immune synapse and boosts serial engagement of target cells. J. Cell Biol. 2018, 217, 3267–3283. [Google Scholar] [CrossRef]
- Paolini, R.; Bernardini, G.; Molfetta, R.; Santoni, A. NK cells and interferons. Cytokine Growth Factor Rev. 2015, 26, 113–120. [Google Scholar] [CrossRef]
- Reefman, E.; Kay, J.G.; Wood, S.M.; Offenhäuser, C.; Brown, D.L.; Roy, S.; Stanley, A.C.; Low, P.C.; Manderson, A.P.; Stow, J.L. Cytokine Secretion Is Distinct from Secretion of Cytotoxic Granules in NK Cells. J. Immunol. 2010, 184, 4852–4862. [Google Scholar] [CrossRef]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Maffey, A.; Storini, C.; Diceglie, C.; Martelli, C.; Sironi, L.; Calzarossa, C.; Tonna, N.; Lovchik, R.; Delamarche, E.; Ottobrini, L.; et al. Mesenchymal stem cells from tumor microenvironment favour breast cancer stem cell proliferation, cancerogenic and metastatic potential, via ionotropic purinergic signalling. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkolizadeh, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Dyck, L.; Lynch, L. Cancer, obesity and immunometabolism – Connecting the dots. Cancer Lett. 2018, 417, 11–20. [Google Scholar] [CrossRef]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell. Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Fischer, B.; Müller, B.; Fisch, P.; Kreutz, W. An Acidic Microenvironment Inhibits Antitumoral Non–Major Histocompatibility Complex-Restricted Cytotoxicity: Implications for Cancer Immunotherapy. J. Immunother. 2000, 23, 196–207. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander, H.M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative Metabolome Profiling of Colon and Stomach Cancer Microenvironment by Capillary Electrophoresis Time-of-Flight Mass Spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Mijatovic, S.A.; Savić-Radojević, A.; Plješa-Ercegovac, M.; Simić, T.; Nicoletti, F.; Maksimović-Ivanić, D. The Double-Faced Role of Nitric Oxide and Reactive Oxygen Species in Solid Tumors. Antioxidants 2020, 9, 374. [Google Scholar] [CrossRef]
- Rizi, B.S.; Achreja, A.; Nagrath, D. Nitric Oxide: The Forgotten Child of Tumor Metabolism. Trends Cancer 2017, 3, 659–672. [Google Scholar] [CrossRef]
- Ahmad, G.; Almasry, M.; Dhillon, A.S.; Abuayyash, M.M.; Kothandaraman, N.; Cakar, Z. Overview and Sources of Reactive Oxygen Species (ROS) in the Reproductive System. In Oxidative Stress in Human Reproduction; Springer: New York, NY, USA, 2017; Volume 4, pp. 1–16. [Google Scholar]
- Firczuk, M.; Bajor, M.; Graczyk-Jarzynka, A.; Fidyt, K.; Goral, A.; Zagozdzon, R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020, 471, 1–11. [Google Scholar] [CrossRef]
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef]
- Hardie, D.G. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin. Cancer Res. 2015, 21, 3836–3840. [Google Scholar] [CrossRef]
- Cai, Z.; Li, C.-F.; Han, F.; Liu, C.; Zhang, A.; Hsu, C.-C.; Peng, D.; Zhang, X.; Jin, G.; Rezaeian, A.-H.; et al. Phosphorylation of PDHA by AMPK Drives TCA Cycle to Promote Cancer Metastasis. Mol. Cell 2020, 80, 263–278.e7. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nat. Cell Biol. 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Kozlovsky, N.; Rudich, A.; Potashnik, R.; Ebina, Y.; Murakami, T.; Bashan, N. Transcriptional Activation of theGlut1Gene in Response to Oxidative Stress in L6 Myotubes. J. Biol. Chem. 1997, 272, 33367–33372. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef]
- Krstić, J.; Trivanović, D.; Mojsilović, S.; Santibañez, J.F. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxidative Med. Cell. Longev. 2015, 2015, 1–15. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.-H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Konjar, Š.; Sutton, V.R.; Hoves, S.; Repnik, U.; Yagita, H.; Reinheckel, T.; Peters, C.; Turk, V.; Turk, B.; Trapani, J.A.; et al. Human and mouse perforin are processed in part through cleavage by the lysosomal cysteine proteinase cathepsin L. Immunology 2010, 131, 257–267. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cifaldi, L.; Prencipe, G.; Caiello, I.; Bracaglia, C.; Locatelli, F.; De Benedetti, F.; Strippoli, R. Inhibition of Natural Killer Cell Cytotoxicity by Interleukin-6: Implications for the Pathogenesis of Macrophage Activation Syndrome. Arthritis Rheumatol. 2015, 67, 3037–3046. [Google Scholar] [CrossRef] [PubMed]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Chonov, D.C.; Ignatova, M.M.K.; Ananiev, J.R.; Gulubova, M.V. IL-6 Activities in the Tumour Microenvironment. Part 1. Open Access Maced. J. Med Sci. 2019, 7, 2391–2398. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, 301–368. [Google Scholar] [CrossRef]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
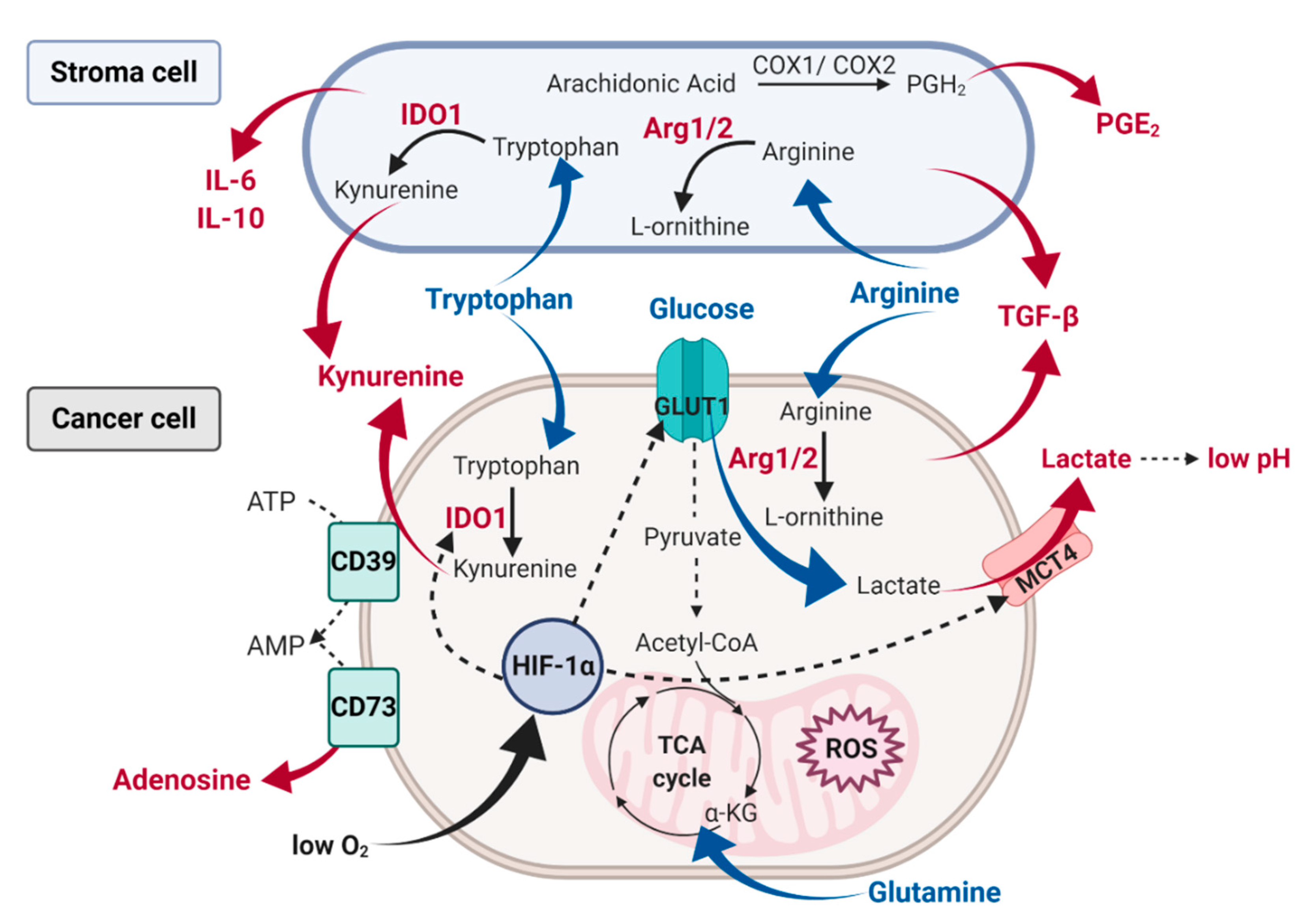
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Durán, R.V.; Hall, M.N. Glutaminolysis feeds mTORC1. Cell Cycle 2012, 11, 4107–4108. [Google Scholar] [CrossRef]
- Vaughn, A.E.; Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008, 10, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, H.; Jia, L.; Sun, H. Effects of Treg cells and IDO on human epithelial ovarian cancer cells under hypoxic conditions. Mol. Med. Rep. 2014, 11, 1708–1714. [Google Scholar] [CrossRef]
- Ohtaki, H.; Ito, H.; Ando, K.; Ishikawa, T.; Hoshi, M.; Tanaka, R.; Osawa, Y.; Yokochi, T.; Moriwaki, H.; Saito, K.; et al. Interaction between LPS-induced NO production and IDO activity in mouse peritoneal cells in the presence of activated Valpha14 NKT cells. Biochem. Biophys. Res. Commun. 2009, 389, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A.; Ng, H.; Jessup, W.; Kockx, M.; Cartland, S.; Thomas, S.; Hogg, P.J.; Wargon, O. Hypoxia regulates the production and activity of glucose transporter-1 and indoleamine 2,3-dioxygenase in monocyte-derived endothelial-like cells: Possible relevance to infantile haemangioma pathogenesis. Br. J. Dermatol. 2011, 164, 308–315. [Google Scholar] [CrossRef]
- Roman, J.; Rangasamy, T.; Guo, J.; Sugunan, S.; Meednu, N.; Packirisamy, G.; Shimoda, L.A.; Golding, A.; Semenza, G.; Georas, S.N. T-cell activation under hypoxic conditions enhances IFN-gamma secretion. Am. J. Respir. Cell Mol. Biol. 2010, 42, 123–128. [Google Scholar] [CrossRef]
- Von Bergwelt-Baildon, M.S.; Popov, A.; Saric, T.; Chemnitz, J.; Classen, S.; Stoffel, M.S.; Fiore, F.; Roth, U.; Beyer, M.; Debey, S.; et al. CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: Additional mechanisms of T-cell inhibition. Blood 2006, 108, 228–237. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Heiden, M.G.V.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I–Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II Expressed in Cancer-Associated Fibroblasts Indicates Tissue Hypoxia and Predicts Poor Outcome in Patients with Pancreatic Cancer. PLoS ONE 2013, 8, 55146. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Gannon, P.O.; Godin-Ethier, J.; Hassler, M.; Delvoye, N.; Aversa, M.; Poisson, A.O.; Péant, B.; Alam Fahmy, M.; Saad, F.; Lapointe, R.; et al. Androgen-Regulated Expression of Arginase 1, Arginase 2 and Interleukin-8 in Human Prostate Cancer. PLoS ONE 2010, 5, 12107. [Google Scholar] [CrossRef]
- Obiorah, I.E.; Chahine, J.; Ko, K.; Park, B.U.; DeGuzman, J.; Kallakury, B. Prognostic Implications of Arginase and Cytokeratin 19 Expression in Hepatocellular Carcinoma After Curative Hepatectomy: Correlation with Recurrence-Free Survival. Gastroenterol. Res. 2019, 12, 78–87. [Google Scholar] [CrossRef]
- Chang, C.I.; Liao, J.C.; Kuo, L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. 2001, 61, 1100–1106. [Google Scholar]
- Albaugh, V.L.; Pinzon-Guzman, C.; Barbul, A. Arginine-Dual roles as an onconutrient and immunonutrient. J. Surg. Oncol. 2017, 115, 273–280. [Google Scholar] [CrossRef]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Eligini, S.; Arenaz, I.; Barbieri, S.S.; Faleri, M.L.; Crisci, M.; Tremoli, E.; Colli, S. Cyclooxygenase-2 mediates hydrogen peroxide-induced wound repair in human endothelial cells. Free. Radic. Biol. Med. 2009, 46, 1428–1436. [Google Scholar] [CrossRef]
- Ma, X.; Holt, D.; Kundu, N.; Reader, J.; Goloubeva, O.; Take, Y.; Fulton, A.M. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. OncoImmunology 2013, 2, e22647. [Google Scholar] [CrossRef]
- Majumder, M.; Nandi, P.; Omar, A.; Ugwuagbo, K.C.; Lala, P.K. EP4 as a Therapeutic Target for Aggressive Human Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1019. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.L.; Obermajer, N.; Odunsi, K.; Edwards, R.P.; Kalinski, P. Synergistic COX2 Induction by IFNgamma and TNFalpha Self-Limits Type-1 Immunity in the Human Tumor Microenvironment. Cancer Immunol. Res. 2016, 4, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar] [PubMed]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin. Investig. 2011, 121, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, B.; Mandelboim, O. Sweet Killers: NK Cells Need Glycolysis to Kill Tumors. Cell Metab. 2018, 28, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef]
- Xie, D.; Zhu, S.; Bai, L. Lactic acid in tumor microenvironments causes dysfunction of NKT cells by interfering with mTOR signaling. Sci. China Life Sci. 2016, 59, 1290–1296. [Google Scholar] [CrossRef]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987. [Google Scholar] [CrossRef]
- Kedia-Mehta, N.; Finlay, D.K. Competition for nutrients and its role in controlling immune responses. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Harmon, C.; Robinson, M.W.; Hand, F.; Almuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol. Res. 2019, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, A.; Serganova, I.; Khanin, R.; Karabeber, H.; Ni, X.; Thakur, S.; Zakian, K.L.; Blasberg, R.; Koutcher, J.A. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res. 2013, 19, 5158–5169. [Google Scholar] [CrossRef]
- Xie, H.; Hanai, J.; Ren, J.G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting lactate dehydrogenase—A inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018, 28, 243–255. [Google Scholar] [CrossRef]
- Velásquez, S.Y.; Killian, D.; Schulte, J.; Sticht, C.; Thiel, M.; Lindner, H.A. Short Term Hypoxia Synergizes with Interleukin 15 Priming in Driving Glycolytic Gene Transcription and Supports Human Natural Killer Cell Activities. J. Biol. Chem. 2016, 291, 12960–12977. [Google Scholar] [CrossRef]
- Chambers, A.M.; Wang, J.; Lupo, K.B.; Yu, H.; Lanman, N.M.A.; Matosevic, S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front. Immunol. 2018, 9, 2533. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16−NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+T Cell Proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef]
- Almutairi, S.M.; Ali, A.K.; He, W.; Yang, D.-S.; Ghorbani, P.; Wang, L.; Fullerton, M.D.; Lee, S.-H. Interleukin-18 up-regulates amino acid transporters and facilitates amino acid–induced mTORC1 activation in natural killer cells. J. Biol. Chem. 2019, 294, 4644–4655. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Chow, M.T.; Chen, A.; Halse, H.M.; Wong, C.S.; Andrews, D.M.; Sloan, E.K.; Parker, B.S.; Bowtell, D.D.; Smyth, M.J.; et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012, 72, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. AJNR Am. J. Neuroradiol. 2018, 13, 612–632. [Google Scholar] [CrossRef] [PubMed]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef]
- Carrega, P.; Bonaccorsi, I.; Di Carlo, E.; Morandi, B.; Paul, P.; Rizzello, V.; Cipollone, G.; Navarra, G.; Mingari, M.C.; Moretta, L.; et al. CD56brightPerforinlow Noncytotoxic Human NK Cells Are Abundant in Both Healthy and Neoplastic Solid Tissues and Recirculate to Secondary Lymphoid Organs via Afferent Lymph. J. Immunol. 2014, 192, 3805–3815. [Google Scholar] [CrossRef]
- Bernardini, G.; Santoni, A. The Pathophysiological Role of Chemokines in the Regulation of NK Cell Tissue Homing. Crit. Rev. Oncog. 2014, 19, 77–90. [Google Scholar] [CrossRef]
- Regis, S.; Caliendo, F.; Dondero, A.; Casu, B.; Romano, F.; Loiacono, F.; Moretta, A.; Bottino, C.; Castriconi, R. TGF-beta1 Downregulates the Expression of CX3CR1 by Inducing miR-27a-5p in Primary Human NK. Cells Front. Immunol. 2017, 8, 868. [Google Scholar]
- Casu, B.; Dondero, A.; Regis, S.; Caliendo, F.; Petretto, A.; Bartolucci, M.; Bellora, F.; Bottino, C.; Castriconi, R. Novel Immunoregulatory Functions of IL-18, an Accomplice of TGF-beta1. Cancers 2019, 11, 75. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, R.; Schwarz, K.; Mangeat, M.; Schwarz, E.C.; Hamed, M.; Bogeski, I.; Helms, V.; Rieger, H.; Qu, B. Bystander cells enhance NK cytotoxic efficiency by reducing search time. Sci. Rep. 2017, 7, 44357. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Santoriello, C.; Mione, M.C.; Hurlstone, A.; Martin, P. Live Imaging of Innate Immune Cell Sensing of Transformed Cells in Zebrafish Larvae: Parallels between Tumor Initiation and Wound Inflammation. PLoS Biol. 2010, 8, e1000562. [Google Scholar] [CrossRef] [PubMed]
- Izawa, S.; Kono, K.; Mimura, K.; Kawaguchi, Y.; Watanabe, M.; Maruyama, T.; Fujii, H. H2O2 production within tumor microenvironment inversely correlated with infiltration of CD56dim NK cells in gastric and esophageal cancer: Possible mechanisms of NK cell dysfunction. Cancer Immunol. Immunother. 2011, 60, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Morandi, B.; Costa, R.; Frumento, G.; Forte, G.; Altavilla, G.; Ratto, G.B.; Mingari, M.C.; Moretta, L.; Ferlazzo, G. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56brightCD16− cells and display an impaired capability to kill tumor cells. Cancer 2008, 112, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, Q.; Jiang, Y.; Yu, J.; Hu, Y.; Mou, T.; Chen, G.; Li, G. Gastric cancer cells inhibit natural killer cell proliferation and induce apoptosis via prostaglandin E2. OncoImmunology 2015, 5, e1069936. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Sousa, C.R.E. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Holt, D.M.; Ma, X.; Kundu, N.; Collin, P.D.; Fulton, A.M. Modulation of Host Natural Killer Cell Functions in Breast Cancer via Prostaglandin E2 Receptors EP2 and EP4. J. Immunother. 2012, 35, 179–188. [Google Scholar] [CrossRef]
- Holt, D.; Ma, X.; Kundu, N.; Fulton, A. Prostaglandin E(2) (PGE (2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol. Immunother. 2011, 60, 1577–1586. [Google Scholar] [CrossRef]
- Walker, W.; Rotondo, D. Prostaglandin E2 is a potent regulator of interleukin-12- and interleukin-18-induced natural killer cell interferon-gamma synthesis. Immunology 2004, 111, 298–305. [Google Scholar] [CrossRef]
- Su, Y.; Huang, X.; Raskovalova, T.; Zacharia, L.; Lokshin, A.; Jackson, E.; Gorelik, E. Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP–PKA signaling and immunosuppression. Cancer Immunol. Immunother. 2008, 57, 1611–1623. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Cheng, L.; Langenbach, R.; Ju, C. Prostaglandin I2 and E2 mediate the protective effects of cyclooxygenase-2 in a mouse model of immune-mediated liver injury. Hepatology 2006, 45, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Hoshi, M.; Ohtaki, H.; Taguchi, A.; Ando, K.; Ishikawa, T.; Osawa, Y.; Hara, A.; Moriwaki, H.; Saito, K.; et al. Ability of IDO to Attenuate Liver Injury in α-Galactosylceramide–Induced Hepatitis Model. J. Immunol. 2010, 185, 4554–4560. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Saga, Y.; Mizukami, H.; Wang, D.; Takahashi, S.; Nonaka, H.; Fujiwara, H.; Takei, Y.; Machida, S.; Takikawa, O.; et al. Downregulation of indoleamine-2,3-dioxygenase in cervical cancer cells suppresses tumor growth by promoting natural killer cell accumulation. Oncol. Rep. 2012, 28, 1574–1578. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Saga, Y.; Mizukami, H.; Sato, N.; Nonaka, H.; Fujiwara, H.; Takei, Y.; Machida, S.; Takikawa, O.; Ozawa, K.; et al. Indoleamine-2,3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int. J. Oncol. 2012, 40, 929–934. [Google Scholar] [CrossRef]
- Ino, K.; Yamamoto, E.; Shibata, K.; Kajiyama, H.; Yoshida, N.; Terauchi, M.; Nawa, A.; Nagasaka, T.; Takikawa, O.; Kikkawa, F. Inverse Correlation between Tumoral Indoleamine 2,3-Dioxygenase Expression and Tumor-Infiltrating Lymphocytes in Endometrial Cancer: Its Association with Disease Progression and Survival. Clin. Cancer Res. 2008, 14, 2310–2317. [Google Scholar] [CrossRef]
- Crane, C.A.; Austgen, K.; Haberthur, K.; Hofmann, C.; Moyes, K.W.; Avanesyan, L.; Fong, L.; Campbell, M.J.; Cooper, S.; Oakes, S.A.; et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc. Natl. Acad. Sci. USA 2014, 111, 12823–12828. [Google Scholar] [CrossRef]
- Daneshmandi, S.; Seth, P.; Seth, P. Blockade of Lactate Dehydrogenase-A (LDH-A) Improves Efficacy of Anti-Programmed Cell Death-1 (PD-1) Therapy in Melanoma. Cancers 2019, 11, 450. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mule, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef]
- Beldi-Ferchiou, A.; Caillat-Zucman, S. Control of NK Cell Activation by Immune Checkpoint Molecules. Int. J. Mol. Sci. 2017, 18, 2129. [Google Scholar] [CrossRef]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef] [PubMed]
- Siemens, D.R.; Hu, N.; Sheikhi, A.K.; Chung, E.; Frederiksen, L.J.; Pross, H.; Graham, C. Hypoxia Increases Tumor Cell Shedding of MHC Class I Chain-Related Molecule: Role of Nitric Oxide. Cancer Res. 2008, 68, 4746–4753. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Yamanegi, K.; Ohyama, H.; Hata, M.; Nakasho, K.; Futani, H.; Okamura, H.; Terada, N. Hypoxia downregulates the expression of cell surface MICA without increasing soluble MICA in osteosarcoma cells in a HIF-1?-dependent manner. Int. J. Oncol. 2012, 41, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.I.; Thoren, F.B.; Brune, M.; Hellstrand, K. NKp46 and NKG2D receptor expression in NK cells with CD56dim and CD56bright phenotype: Regulation by histamine and reactive oxygen species. Br. J. Haematol. 2006, 132, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Hanson, M.G.V.; Johansson, C.C.; Sakurai, D.; Poschke, I.; Norell, H.; Malmberg, K.-J.; Kiessling, R. The CD16−CD56brightNK Cell Subset Is Resistant to Reactive Oxygen Species Produced by Activated Granulocytes and Has Higher Antioxidative Capacity Than the CD16+CD56dimSubset. J. Immunol. 2007, 179, 4513–4519. [Google Scholar] [CrossRef]
- Akhiani, A.A.; Werlenius, O.; Aurelius, J.; Movitz, C.; Martner, A.; Hellstrand, K.; Thorén, F.B. Role of the ERK Pathway for Oxidant-Induced Parthanatos in Human Lymphocytes. PLoS ONE 2014, 9, 89646. [Google Scholar] [CrossRef]
- Aurelius, J.; Hallner, A.; Werlenius, O.; Riise, R.E.; Möllgård, L.; Brune, M.; Hansson, M.; Martner, A.; Thorén, F.B.; Hellstrand, K. NOX2-dependent immunosuppression in chronic myelomonocytic leukemia. J. Leukoc. Biol. 2017, 102, 459–466. [Google Scholar] [CrossRef]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; de Lima, M.; Wald, D.N. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE 2018, 13, 0191358. [Google Scholar]
- Park, Y.P.; Choi, S.-C.; Kiesler, P.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Complex regulation of human NKG2D-DAP10 cell surface expression: Opposing roles of the γc cytokines and TGF-β1. Blood 2011, 118, 3019–3027. [Google Scholar] [CrossRef]
- Vacca, P.; Cantoni, C.; Vitale, M.; Prato, C.; Canegallo, F.; Fenoglio, D.; Ragni, N.; Moretta, L.; Mingari, M.C. Crosstalk between decidual NK and CD14+ myelomonocytic cells results in induction of Tregs and immunosuppression. Proc. Natl. Acad. Sci. USA 2010, 107, 11918–11923. [Google Scholar] [CrossRef]
- Ban, Y.; Zhao, Y.; Liu, F.; Dong, B.; Kong, B.; Qu, X. Effect of Indoleamine 2,3-Dioxygenase Expressed in HTR-8/SVneo Cells on Decidual NK Cell Cytotoxicity. Am. J. Reprod. Immunol. 2016, 75, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Park, H.; Kim, J.; Park, G.; Kim, Y.S.; Kim, S.M.; Jeong, J.-Y.; Kil Seo, S.; Lee, H.-K.; Cho, D.; et al. IDO metabolite produced by EBV-transformed B cells inhibits surface expression of NKG2D in NK cells via the c-Jun N-terminal kinase (JNK) pathway. Immunol. Lett. 2011, 136, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Vergnaud-Gauduchon, J.; Goncalves-Mendes, N.; Perche, O.; Vasson, M.P.; Farges, M.C. Altered functions of natural killer cells in response to L-Arginine availability. Cell Immunol. 2012, 280, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer–cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008, 111, 1327–1333. [Google Scholar] [CrossRef]
- Park, A.; Lee, Y.; Kim, M.S.; Kang, Y.J.; Park, Y.-J.; Jung, H.; Kim, T.-D.; Lee, H.G.; Choi, I.; Yoon, S.R. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front. Immunol. 2018, 9, 1859. [Google Scholar] [CrossRef]
- Meron, G.; Tishler, Y.; Shaashua, L.; Rosenne, E.; Levi, B.; Melamed, R.; Gotlieb, N.; Matzner, P.; Sorski, L.; Ben-Eliyahu, S. PGE2 suppresses NK activity in vivo directly and through adrenal hormones: Effects that cannot be reflected by ex vivo assessment of NK cytotoxicity. Brain, Behav. Immun. 2012, 28, 128–138. [Google Scholar] [CrossRef]
- Melamed, R.; Rosenne, E.; Shakhar, K.; Schwartz, Y.; Abudarham, N.; Ben-Eliyahu, S. Marginating pulmonary-NK activity and resistance to experimental tumor metastasis: Suppression by surgery and the prophylactic use of a β-adrenergic antagonist and a prostaglandin synthesis inhibitor. Brain Behav. Immun. 2005, 19, 114–126. [Google Scholar] [CrossRef]
- Mahaweni, N.M.; Bos, G.M.J.; Mitsiades, C.S.; Tilanus, M.G.J.; Wieten, L. Daratumumab augments alloreactive natural killer cell cytotoxicity towards CD38+ multiple myeloma cell lines in a biochemical context mimicking tumour microenvironment conditions. Cancer Immunol. Immunother. 2018, 67, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.C.; Zhou, X.; Cuchens, M.; Jones, Q. Prostaglandin E2Suppressed IL-15-Mediated Human NK Cell Function through Down-Regulation of Common γ-Chain. J. Immunol. 2001, 166, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Inbar, S.; Neeman, E.; Avraham, R.; Benish, M.; Rosenne, E.; Ben-Eliyahu, S. Do Stress Responses Promote Leukemia Progression? An Animal Study Suggesting a Role for Epinephrine and Prostaglandin-E2 through Reduced NK Activity. PLoS ONE 2011, 6, e19246. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, S.; Lambert, M.; Zucman, D.; Choukem, S.-P.; Tognarelli, S.; Pages, C.; Lebbé, C.; Caillatzucman, S. Human Herpesvirus 8 (HHV8) Sequentially Shapes the NK Cell Repertoire during the Course of Asymptomatic Infection and Kaposi Sarcoma. PLOS Pathog. 2012, 8, e1002486. [Google Scholar] [CrossRef] [PubMed]
- Croxatto, D.; Vacca, P.; Canegallo, F.; Conte, R.; Venturini, P.L.; Moretta, L.; Mingari, M.C. Stromal Cells from Human Decidua Exert a Strong Inhibitory Effect on NK Cell Function and Dendritic Cell Differentiation. PLoS ONE 2014, 9, 89006. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef] [PubMed]
- Yakar, I.; Melamed, R.; Shakhar, G.; Shakhar, K.; Rosenne, E.; Abudarham, N.; Page, G.G.; Ben-Eliyahu, S. Prostaglandin e (2) suppresses NK activity in vivo and promotes postoperative tumor metastasis in rats. Ann. Surg. Oncol. 2003, 10, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Ma, X.; Holt, D.; Goloubeva, O.; Ostrand-Rosenberg, S.; Fulton, A.M. Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res. Treat. 2008, 117, 235–242. [Google Scholar] [CrossRef]
- Ishida, N.; Ishiyama, K.; Saeki, Y.; Tanaka, Y.; Ohdan, H. Cotransplantation of preactivated mesenchymal stem cells improves intraportal engraftment of islets by inhibiting liver natural killer cells in mice. Arab. Archaeol. Epigr. 2019, 19, 2732–2745. [Google Scholar] [CrossRef]
- Wennerberg, E.; Pfefferle, A.; Ekblad, L.; Yoshimoto, Y.; Kremer, V.; Kaminskyy, V.O.; Juhlin, C.C.; Hoog, A.; Bodin, I.; Svjatoha, V.; et al. Human anaplastic thyroid carcinoma cells are sensitive to NK cell-mediated lysis via ULBP2/5/6 and chemoattract NK cells. Clin. Cancer Res. 2014, 20, 5733–5744. [Google Scholar] [CrossRef]
- Hodge, G.; Barnawi, J.; Jurisevic, C.; Moffat, D.; Holmes, M.; Reynolds, P.N.; Jersmann, H.; Hodge, S. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-gamma by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin. Exp. Immunol. 2014, 178, 79–85. [Google Scholar] [CrossRef]
- Gross, C.C.; Brzostowski, J.A.; Liu, D.; Long, E.O. Tethering of Intercellular Adhesion Molecule on Target Cells Is Required for LFA-1–Dependent NK Cell Adhesion and Granule Polarization. J. Immunol. 2010, 185, 2918–2926. [Google Scholar] [CrossRef]
- Al Absi, A.; Wurzer, H.; Guerin, C.L.; Hoffmann, C.; Moreau, F.; Mao, X.; Brown-Clay, J.; Petrolli, R.; Casellas, C.P.; Dieterle, M.; et al. Actin Cytoskeleton Remodeling Drives Breast Cancer Cell Escape from Natural Killer–Mediated Cytotoxicity. Cancer Res. 2018, 78, 5631–5643. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; Van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.K.; Spielman, J.; Zhao, D.Y.; Olsen, K.J.; Podack, E.R. Perforin, Fas ligand, and tumor necrosis factor are the major cytotoxic molecules used by lymphokine-activated killer cells. J. Immunol. 1996, 157, 1919–1925. [Google Scholar] [PubMed]
- Trotta, R.; Col, J.D.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J.; Wei, M.; Mao, H.; Byrd, J.C.; et al. TGF-β Utilizes SMAD3 to Inhibit CD16-Mediated IFN-γ Production and Antibody-Dependent Cellular Cytotoxicity in Human NK Cells1. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, Y.; Sun, R.; Wei, H.; Tian, Z. Impairment of hepatic NK cell development in IFN-gamma deficient mice. Cytokine 2012, 60, 616–625. [Google Scholar] [CrossRef]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef]
- Yu, J.; Wei, M.; Becknell, B.; Trotta, R.; Liu, S.; Boyd, Z.; Jaung, M.S.; Blaser, B.W.; Sun, J.; Benson, D.M.; et al. Pro- and Antiinflammatory Cytokine Signaling: Reciprocal Antagonism Regulates Interferon-gamma Production by Human Natural Killer Cells. Immunity 2006, 24, 575–590. [Google Scholar] [CrossRef]
- Tang, P.M.-K.; Zhou, S.; Meng, X.-M.; Wang, Q.-M.; Li, C.-J.; Lian, G.-Y.; Huang, X.-R.; Tang, Y.-J.; Yuan, G.X.; Yan, B.P.-Y.; et al. Smad3 promotes cancer progression by inhibiting E4BP4-mediated NK cell development. Nat. Commun. 2017, 8, 14677. [Google Scholar] [CrossRef]
- D’Andrea, A.; Aste-Amezaga, M.; Valiante, N.M.; Ma, X.; Kubin, M.; Trinchieri, G. Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med. 1993, 178, 1041–1048. [Google Scholar] [CrossRef]
- Mocellin, S.; Panelli, M.C.; Wang, E.; Rossi, C.R.; Pilati, P.; Nitti, D.; Lise, M.; Marincola, F.M. IL-10 stimulatory effects on human NK cells explored by gene profile analysis. Genes Immun. 2004, 5, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Yung, P.P.; Jing-Jing, Z.; Wen-Biao, L.; Min, T.; Zi-Peng, L.; Ji-Shu, W.; Kui-Rong, J.; Wen-Tao, G.; Jun-Li, W.; Ze-Kuan, X.; et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 2014, 14, 738. [Google Scholar]
- Harizi, H. Reciprocal crosstalk between dendritic cells and natural killer cells under the effects of PGE2 in immunity and immunopathology. Cell. Mol. Immunol. 2013, 10, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Sun, C. The Rise of NK Cell Checkpoints as Promising Therapeutic Targets in Cancer Immunotherapy. Front. Immunol. 2019, 10, 2354. [Google Scholar] [CrossRef] [PubMed]
- Huenecke, S.; Zimmermann, S.Y.; Kloess, S.; Esser, R.; Brinkmann, A.; Tramsen, L.; Koenig, M.; Erben, S.; Seidl, C.; Tonn, T.; et al. IL-2−driven Regulation of NK Cell Receptors With Regard to the Distribution of CD16+ and CD16− Subpopulations and In Vivo Influence After Haploidentical NK Cell Infusion. J. Immunother. 2010, 33, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Germeraad, W.T.V.; Rouschop, K.M.A.; Steeghs, E.M.P.; Van Gelder, M.; Bos, G.M.J.; Wieten, L. Hypoxia Induced Impairment of NK Cell Cytotoxicity against Multiple Myeloma Can Be Overcome by IL-2 Activation of the NK Cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef] [PubMed]
- Solocinski, K.; Padget, M.R.; Fabian, K.P.; Wolfson, B.; Cecchi, F.; Hembrough, T.; Benz, S.C.; Rabizadeh, S.; Soon-Shiong, P.; Schlom, J.; et al. Overcoming hypoxia-induced functional suppression of NK cells. J. Immunother. Cancer 2020, 8, e000246. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, X.; Stojanovic, A.; Zhang, Q.; Wincher, M.; Bühler, L.; Arnold, A.; Correia, M.P.; Winkler, M.; Koch, P.-S.; et al. Single-Cell RNA Sequencing of Tumor-Infiltrating NK Cells Reveals that Inhibition of Transcription Factor HIF-1α Unleashes NK Cell Activity. Immunity 2020, 52, 1075–1087.e8. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.-T. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- De Milito, A.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2010, 127, 207–219. [Google Scholar] [CrossRef]
- Bellone, M.; Calcinotto, A.; Filipazzi, P.; De Milito, A.; Fais, S.; Rivoltini, L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. OncoImmunology 2013, 2, e22058. [Google Scholar] [CrossRef]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P.; et al. GSK3 Inhibition Drives Maturation of NK Cells and Enhances Their Antitumor Activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef]
- Parameswaran, R.; Ramakrishnan, P.; Moreton, S.A.; Xia, Z.; Hou, Y.; Lee, D.A.; Gupta, K.; Lima, M.; Beck, R.C.; Wald, D.N. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 2016, 7, 11154. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Matsunaga, K.-I. Susceptibility of Natural Killer (NK) Cells to Reactive Oxygen Species (ROS) and Their Restoration by the Mimics of Superoxide Dismutase (SOD). Cancer Biother. Radiopharm. 1998, 13, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Mellqvist, U.H.; Hansson, M.; Brune, M.; Dahlgren, C.; Hermodsson, S.; Hellstrand, K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: Role of reactive oxygen species and regulation by histamine. Blood 2000, 96, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Betten, Å.; Dahlgren, C.; Mellqvist, U.-H.; Hermodsson, S.; Hellstrand, K. Oxygen radical-induced natural killer cell dysfunction: Role of myeloperoxidase and regulation by serotonin. J. Leukoc. Biol. 2004, 75, 1111–1115. [Google Scholar] [CrossRef]
- Werlenius, O.; Aurelius, J.; Hallner, A.; Akhiani, A.A.; Simpanen, M.; Martner, A.; Andersson, P.-O.; Hellstrand, K.; Thorén, F.B. Reactive oxygen species induced by therapeutic CD20 antibodies inhibit natural killer cell-mediated antibody-dependent cellular cytotoxicity against primary CLL cells. Oncotarget 2016, 7, 32046–32053. [Google Scholar] [CrossRef]
- Aydin, E.; Johansson, J.; Nazir, F.H.; Hellstrand, K.; Martner, A. Role of NOX2-Derived Reactive Oxygen Species in NK Cell–Mediated Control of Murine Melanoma Metastasis. Cancer Immunol. Res. 2017, 5, 804–811. [Google Scholar] [CrossRef]
- Rouce, R.H.; Shaim, H.; Sekine, T.; Weber, G.; Ballard, B.; Ku, S.; Barese, C.; Murali, V.; Wu, M.F.; Liu, H.; et al. The TGF-beta/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia 2016, 30, 800–811. [Google Scholar] [CrossRef]
- Yvon, E.S.; Burga, R.; Powell, A.; Cruz, C.R.; Fernandes, R.; Barese, C.; Nguyen, T.; Abdel-Baki, M.S.; Bollard, C.M. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 2017, 19, 408–418. [Google Scholar] [CrossRef]
- Zhang, J.; Han, X.; Hu, X.; Jin, F.; Gao, Z.; Yin, L.; Qin, J.; Yin, F.; Li, C.; Wang, Y. IDO1 impairs NK cell cytotoxicity by decreasing NKG2D/NKG2DLs via promoting miR-18a. Mol. Immunol. 2018, 103, 144–155. [Google Scholar] [CrossRef]
- Liu, X.; Shin, N.; Koblish, H.K.; Yang, G.; Wang, Q.; Wang, K.; Leffet, L.; Hansbury, M.J.; Thomas, B.; Rupar, M.; et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010, 115, 3520–3530. [Google Scholar] [CrossRef]
- Watanabe, T.; Gaedicke, S.; Guffart, E.; Firat, E.; Niedermann, G. Adding Indoximod to Hypofractionated Radiotherapy with Anti-PD-1 Checkpoint Blockade Enhances Early NK and CD8+ T-Cell–Dependent Tumor Activity. Clin. Cancer Res. 2019, 26, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, T.; Catton, E.C.; Rosko, A.E.; Efebera, Y.; Chaudhry, M.; Cottini, F.; Mundy-Bosse, B.L.; Hughes, T.; Benson, D.M., Jr. Aryl Hydrocarbon Receptor (AHR) Antagonism As a Transformative, Dual-Mechanism Novel Therapy for Multiple Myeloma. Blood 2018, 132, 1933. [Google Scholar] [CrossRef]
- Harish, A.; Hohana, G.; Fishman, P.; Arnon, O.; Bar-Yehuda, S. A3 adenosine receptor agonist potentiates natural killer cell activity. Int. J. Oncol. 2003, 23, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Morello, S.; Sorrentino, R.; Montinaro, A.; Luciano, A.; Maiolino, P.; Ngkelo, A.; Arra, C.; Adcock, I.M.; Pinto, A. NK1.1+ Cells and CD8+ T Cells Mediate the Antitumor Activity of Cl-IB-MECA in a Mouse Melanoma Model. Neoplasia 2011, 13, 365-IN20. [Google Scholar] [CrossRef] [PubMed]
- Ohana, G.; Baryehuda, S.; Arich, A.; Madi, L.; Dreznick, Z.; Rathwolfson, L.; Silberman, D.; Slosman, G.; Fishman, P.S. Inhibition of primary colon carcinoma growth and liver metastasis by the A3 adenosine receptor agonist CF101. Br. J. Cancer 2003, 89, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Lokshin, A.; Raskovalova, T.; Huang, X.; Zacharia, L.C.; Jackson, E.K.; Gorelik, E. Adenosine-Mediated Inhibition of the Cytotoxic Activity and Cytokine Production by Activated Natural Killer Cells. Cancer Res. 2006, 66, 7758–7765. [Google Scholar] [CrossRef]
- Raskovalova, T.; Huang, X.; Sitkovsky, M.; Zacharia, L.C.; Jackson, E.K.; Gorelik, E. GsProtein-Coupled Adenosine Receptor Signaling and Lytic Function of Activated NK Cells. J. Immunol. 2005, 175, 4383–4391. [Google Scholar] [CrossRef]
- Häusler, S.F.M.; Del Barrio, I.M.; Strohschein, J.; Chandran, P.A.; Engel, J.B.; Hönig, A.; Ossadnik, M.; Horn, E.; Fischer, B.; Krockenberger, M.; et al. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol. Immunother. 2011, 60, 1405–1418. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Madore, J.; Reinhardt, J.; Landsberg, J.; Chitsazan, A.; Rautela, J.; Bald, T.; Barkauskas, D.S.; Ahern, E.; et al. Targeting Adenosine in BRAF-Mutant Melanoma Reduces Tumor Growth and Metastasis. Cancer Res. 2017, 77, 4684–4696. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Barkauskas, D.S.; Sult, E.; Hay, C.; Blake, S.J.; Huang, Q.; Liu, J.; Takeda, K.; Teng, M.W.; et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell 2016, 30, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2Areceptors potently suppresses the metastasis of CD73+tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [PubMed]
- Häusler, S.F.M.; Del Barrio, I.M.; Diessner, J.; Stein, R.G.; Strohschein, J.; Hönig, A.; Dietl, J.; Wischhusen, J. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am. J. Transl. Res. 2014, 6, 129–139. [Google Scholar] [PubMed]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Muller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Vijayan, D.; Li, X.-Y.; Robson, S.C.; Geetha, N.; Teng, M.W.L.; Smyth, M.J. The role of NK cells and CD39 in the immunological control of tumor metastases. OncoImmunology 2019, 8, e1593809. [Google Scholar] [CrossRef]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.L.; McMichael, E.L.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric Oxide Production by Myeloid-Derived Suppressor Cells Plays a Role in Impairing Fc Receptor–Mediated Natural Killer Cell Function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Silinda, N.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Blaszczyk, R.; Brzezinska, J.; Dymek, B.; Stanczak, P.S.; Mazurkiewicz, M.; Olczak, J.; Nowicka, J.; Dzwonek, K.; Zagozdzon, A.; Golab, J.; et al. Discovery and Pharmacokinetics of Sulfamides and Guanidines as Potent Human Arginase 1 Inhibitors. ACS Med. Chem. Lett. 2020, 11, 433–438. [Google Scholar] [CrossRef]
- Stanczak, P.S.G.; Wolska, P.; Zdziarska, A.M.; Mazurkiewicz, M.; Blaszczyk, R.; Nowicka, J.; Sosnowska, A.; Ramji, K.; Nowis, D.; Golab, J.; et al. Development of OAT-1746, a novel arginase 1 and 2 inhibitor for cancer immunotherapy. Ann. Oncol. 2017, 28, 418. [Google Scholar] [CrossRef]
- Kim, S.-J.; Ha, G.-H.; Bae, J.-H.; Kim, G.R.; Son, C.-H.; Park, Y.-S.; Yang, K.; Oh, S.-O.; Kim, S.H.; Kang, C.-D. COX-2- and endoplasmic reticulum stress-independent induction of ULBP-1 and enhancement of sensitivity to NK cell-mediated cytotoxicity by celecoxib in colon cancer cells. Exp. Cell Res. 2015, 330, 451–459. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Chemokines | Chemokines | Chemokine Receptor | Chemokine Receptor Expression on the NK Cell Population | |
|---|---|---|---|---|
| NKbright | NKdim | |||
| Tumor | CCL3, CCL5, CCL7, CCL9, CCL14-16, CCL23 | CCR1 | + | − |
| CXCL1-3, CXCL5-8 | CXCR2 | − | ++ | |
| CXCL9-11 | CXCR3 | ++ | − | |
| CXCL12 | CXCR4 | + | ++ | |
| CXCL8 | CXCR1 | − | ++ | |
| CX3CL1 | CX3CR1 | − | ++ | |
| CCL3, CCL4 | CCR5 | ++ | + | |
| Lymph nodes | CCL19, CCL21 | CCR7 | ++ | − |
| TME Factor | Strategies | Examples of Clinical Trials (NCT: ClinicalTrials.gov Identifier) |
|---|---|---|
| Hypoxia | Priming of NK cells with IL-2 increases the expression of activating receptors and thus overcomes the inhibitory effects of hypoxia [244,245]. | Natural Killer Cells Plus IL-2 Following Chemotherapy to Treat Advanced Melanoma or Kidney Cancer NCT00328861 Intraperitoneal Delivery of Adaptive Natural Killer (NK) Cells (FATE-NK100) With Intraperitoneal Interleukin-2 in Women with Recurrent Ovarian, Fallopian Tube, and Primary Peritoneal Cancer NCT03213964 |
| Modification of NK cells to increase ADCC potential and activity—high-affinity NK cells (haNK) expressing CD16 and IL-2 are resistant to acute hypoxia [246]. | Phase 1 Study of haNK™ for Infusion in Subjects with Metastatic or Locally Advanced Solid Tumors NCT03027128 | |
| Inhibition of HIF-1α (either by genetic modifications or small molecular HIF-1α inhibitor) enhances effector functions of activated NK cells (degranulation, production of IFN-γ and TNF-α [247]. | None | |
| Lactic acid/Low pH | Genetic blockade of LDHA (mice with LDHA deficiency) heightens infiltration of NK cells in the melanoma tumors. Infiltrated NK cells have an elevated production of IFN-γ and granzyme B [199]. Novel LDHA inhibitor reduces lactate production, thus decrease TME acidity [248]. | None |
| Bicarbonate monotherapy neutralises tumor acidity and increases effector cells infiltration [249,250]. | Extended Use of Sodium Bicarbonate in Patients with Cancer NCT02531919 | |
| Blocking the mitochondrial ROS accumulation to prevent NK cells mitochondria dysfunction and apoptosis [162]. | None | |
| Glucose depletion | FBP1 inhibition during tumor promotion, but not tumor progression, can restore NK cell function [168]. | None |
| GSK-3 inhibitors, CHIR99021, blocks proteasomal degradation of cMYC and thus promotes glucose consumption in NK cells [251]. CHIR99021 was shown to improve NK-cells function in ovarian cancer [251]. Moreover, other GSK-3 inhibitors, including LY-2090314 were shown to augment NK cells cytotoxicity in AML patients [252]. | Phase 1 trials evaluating the application of NK-cells expanded ex-vivo and pre-treated with CHIR99021 in patients with AML (NCT03081780), ovarian cancer (NCT03213964) and other solid tumors (NCT03319459) Phase 2 trial of LY2090314 and Chemotherapy in Participants With Metastatic Pancreatic Cancer (NCT01632306) | |
| ROS | Superoxide dismutase and other SOD-mimicking substances partially restore the NK-cell mediated killing of YAC-1 cells inhibited by superoxide [253]. | None |
| Histamine reverses granulocyte-induced inhibition of human NK-cell mediated killing of K562 cells [254]. Serotonin restores NK cell-mediated killing of K562 cells inhibited by mononuclear phagocytes [255] | Maintenance Therapy With Ceplene® (Histamine) and IL-2 on Immune Response and MRD in Acute Myeloid Leukemia NCT01347996 -A Study of HDC/IL-2 Treatment in Chronic Myelomonocytic Leukemia (CMML) NCT03040401 | |
| Catalase protects human NK cells from H2O2 induced apoptosis [206,253,256]. | None | |
| Genetic inhibition of NOX2 (Nox2−/− mice that lack the myeloid gp91phox subunit of NOX2) or NOX2 inhibitor HDC reduces melanoma metastasis in a murine NK cell-dependent model of melanoma metastasis [257]. NOX2 inhibitors HDC and diphenylene iodonium chloride (DPI) play a protective role from monocyte-derived ROS-dependent NK cell apoptosis and mostly restore NK cell-mediated ADCC of primary CLL cells. NOX2 inhibitor HDC promotes degranulation of NK cells toward CMML cells in ADCC process and reverses CMML-induced NK cell apoptosis [208,256,257]. | None | |
| ERK1/2 inhibitor PD98059 protects NK cell from H2O2-induced or monocyte-dependent apoptosis [207]. | None | |
| TGF-β | Chemical inhibitors- TGF-β receptor kinase inhibitor, galunisertib (LY2157299) improves the activity of NK cells in metastatic colon cancer mouse model [209]. | A Study of Galunisertib on the Immune System in Participants with Cancer NCT02304419 ExIST Study of LY2157299 (Galunisertib) in Rectal Cancer NCT02688712 |
| anti-TGF-β antibodies- are shown to restore NK cells degranulation and cytokine release [258]. | -Anti-TGF Monoclonal Antibody (GC1008) in Relapsed Malignant Pleural Mesothelioma NCT01112293 -Safety and Efficacy Study of GC1008 to Treat Renal Cell Carcinoma or Malignant Melanoma NCT00356460 | |
| Genetic modification strategies- TGF-β dominant-negative receptor knockout receptor coupled to NK-activating domains (DAP12 or synNotch-RELA) enhance the cytotoxic activity of NK cells (particularly with DAP12 domain) [259] | None | |
| Glutamine depletion | CB-839 It has been reported that glutaminolysis can be inhibited without reducing NK cell functional responses [34]. | Study of the Glutaminase Inhibitor CB-839 in Solid Tumors NCT02071862 |
| Tryptophan metabolites | IDO1 inhibition restores NKG2D expression on NK cells and promotes their proliferation [260,261]. IDO pathway inhibition enhances NK cell tumor infiltration and antitumor activity [262]. | -Intraperitoneal Natural Killer Cells and INCB024360 for Recurrent Ovarian, Fallopian Tube, and Primary Peritoneal Cancer NCT02118285 -NLG802 Indoleamine 2,3-Dioxygenase (IDO) Inhibitor in Advanced Solid Tumors NCT03164603 |
| AHR antagonism increases cancer cell susceptibility to NK cell-mediated cytotoxicity and enhances NK cell-mediated ADCC [263]. | -A First-in-Humans Dose Finding Study for an Aryl Hydrocarbon Receptor Inhibitor (AhRi) in Patients with Advanced Cancer NCT04069026 -IK-175 in Patients with Advanced or Metastatic Solid Tumors and Urothelial Carcinoma NCT04200963 | |
| IL-18 treatment reversed IDO-mediated NK cell inhibition by upregulating NKG2D receptor [213]. | None | |
| Adenosine | A3R agonists: C1-IB-MECA increases activation and NK cells infiltration of B16-F10 melanoma. CF101 potentiation of NK cells’ activity [264,265,266]. | None |
| A2aR antagonists: SCH58261- enhances NK cells maturation, cytokine production, cytotoxic function against tumor cell lines, increases expression of granzyme B and reduces metastasis in a perforin-dependent manner. Increases NK cells infiltration of BRAFV600E-mutant melanoma. Promotes mouse NK cells proliferation and differentiation of human CD56bright into CD56dim mature NK cells. ZM241385- restores the cytotoxic function of IL-2 activated NK cells and cytokines production [267,268,269,270,271,272,273]. | -A Study to Evaluate Immunotherapy Combinations in Participants with Gastrointestinal Malignancies NCT03720678 -A Study to Evaluate the Safety and Tolerability of Immunotherapy Combinations in Participants with Advanced Malignancies NCT03629756 - A Study to Evaluate Safety/Tolerability of Immunotherapy Combinations in Participants with Triple-Negative Breast Cancer or Gynecologic Malignancies NCT03719326 | |
| CD73 inhibitor: APCP- reduces metastasis trough decreased A2aR-mediated suppression of NK cell-mediated cytotoxicity. Improves lytic activity of NK cells [270,273]. | A Study of the CD73 Inhibitor LY3475070 Alone or in Combination with Pembrolizumab in Participants with Advanced Cancer NCT04148937 | |
| anti-mouse CD73 antibody: TY/23- enhances anti-metastatic activity derived by NK cells [272]. Anti-human CD73 antibody increases the cytotoxicity of NK cells against ovarian cancer cell lines overexpressing CD73 [274]. | A Study of AK119 (Anti-CD73) in Combination with AK104 (PD-1/CTLA-4) in Subjects with Advanced Solid Tumors -Study of GS-1423 (Anti-CD73-TGFβ-Trap Bifunctional Antibody) in Participants with Advanced Solid Tumors NCT03954704 | |
| Anti-human CD39 antibody increases the cytotoxicity of NK cells against ovarian cancer cell lines overexpressing CD39 [274]. | - Study of SRF617 (anti-CD39 antibody) in Patients with Advanced Solid Tumors NCT04336098 - TTX-030 (anti-CD39 antibody) Single Agent and in Combination With Immunotherapy or Chemotherapy for Patients With Advanced Cancers NCT03884556 | |
| CD39 inhibitors: Polyoxometalate-1 (POM-1)- reverses Treg-mediated suppression of NK cells cytotoxicity and enhances their anti-metastatic activity. ARL67156- enhances the lytic activity of polyclonal NK cells [270,275,276]. | None | |
| Arginine | MDSCs upregulate arginase and catabolise arginine to NO. It has been found that NO impairs NK cell antibody-dependent cellular cytotoxicity and that the inhibition of iNOS can rescue this function [277]. | |
| Inhibition of the arginase activity by CB-1158 reduces tumor growth and increases tumor-infiltrating NK cells in vitro and in vivo [278]. OATD-02, another arginase inhibitor, has been shown to delay cancer progression [279,280]. | Arginase Inhibitor INCB001158 as a Single Agent and in Combination with Immune Checkpoint Therapy in Patients with Advanced/Metastatic Solid Tumors NCT02903914 | |
| Arachidonic acid metabolites | Selective COX-2 inhibitors increase cancer cell sensitivity to NK cell-mediated lysis [281]. | -Perioperative Administration of COX 2 Inhibitors and Beta Blockers to Women Undergoing Breast Cancer Surgery NCT00502684 -Perioperative Intervention to Reduce Metastatic Processes in Pancreatic Cancer Patients Undergoing Curative Surgery (BC-PC) NCT03838029 |
| EP2 antagonists restore tumor NK cell-mediated lysis [216]. EP4 antagonists restore NK cell antitumor activity cytokine production and migratory potential. Also, they decrease MHC I expression on cancer cells rendering them more sensitive to NK cell-mediated cytotoxicity [151,226]. | Phase 1a/1b Study of TPST-1495 (EP2/EP4 antagonist) Alone and With Pembrolizumab in Subjects with Solid Tumors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domagala, J.; Lachota, M.; Klopotowska, M.; Graczyk-Jarzynka, A.; Domagala, A.; Zhylko, A.; Soroczynska, K.; Winiarska, M. The Tumor Microenvironment—A Metabolic Obstacle to NK Cells’ Activity. Cancers 2020, 12, 3542. https://doi.org/10.3390/cancers12123542
Domagala J, Lachota M, Klopotowska M, Graczyk-Jarzynka A, Domagala A, Zhylko A, Soroczynska K, Winiarska M. The Tumor Microenvironment—A Metabolic Obstacle to NK Cells’ Activity. Cancers. 2020; 12(12):3542. https://doi.org/10.3390/cancers12123542
Chicago/Turabian StyleDomagala, Joanna, Mieszko Lachota, Marta Klopotowska, Agnieszka Graczyk-Jarzynka, Antoni Domagala, Andriy Zhylko, Karolina Soroczynska, and Magdalena Winiarska. 2020. "The Tumor Microenvironment—A Metabolic Obstacle to NK Cells’ Activity" Cancers 12, no. 12: 3542. https://doi.org/10.3390/cancers12123542
APA StyleDomagala, J., Lachota, M., Klopotowska, M., Graczyk-Jarzynka, A., Domagala, A., Zhylko, A., Soroczynska, K., & Winiarska, M. (2020). The Tumor Microenvironment—A Metabolic Obstacle to NK Cells’ Activity. Cancers, 12(12), 3542. https://doi.org/10.3390/cancers12123542