Disulfiram as a Therapeutic Agent for Metastatic Malignant Melanoma—Old Myth or New Logos?


 , and
, and
Abstract
:Simple Summary
Abstract
1. The History of the Discovery and Development of Disulfiram
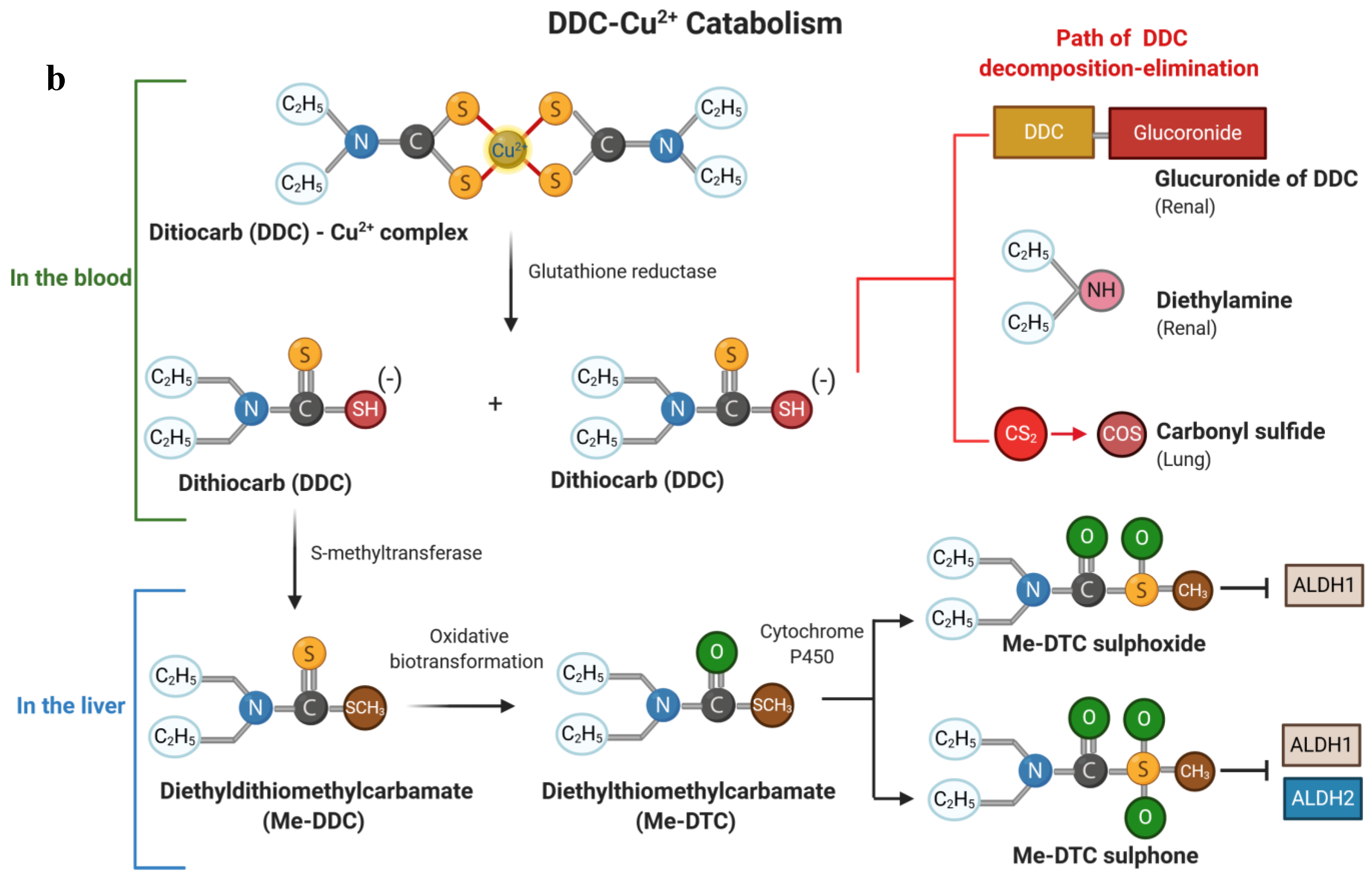
2. Chemical Structure of Disulfiram and Its Metabolites
3. Pharmacology and Metabolism of Disulfiram
4. In Vitro Activity of Disulfiram against Cancer Cells
4.1. Activity against Cancer Cells
4.2. Activity against Melanoma Cells
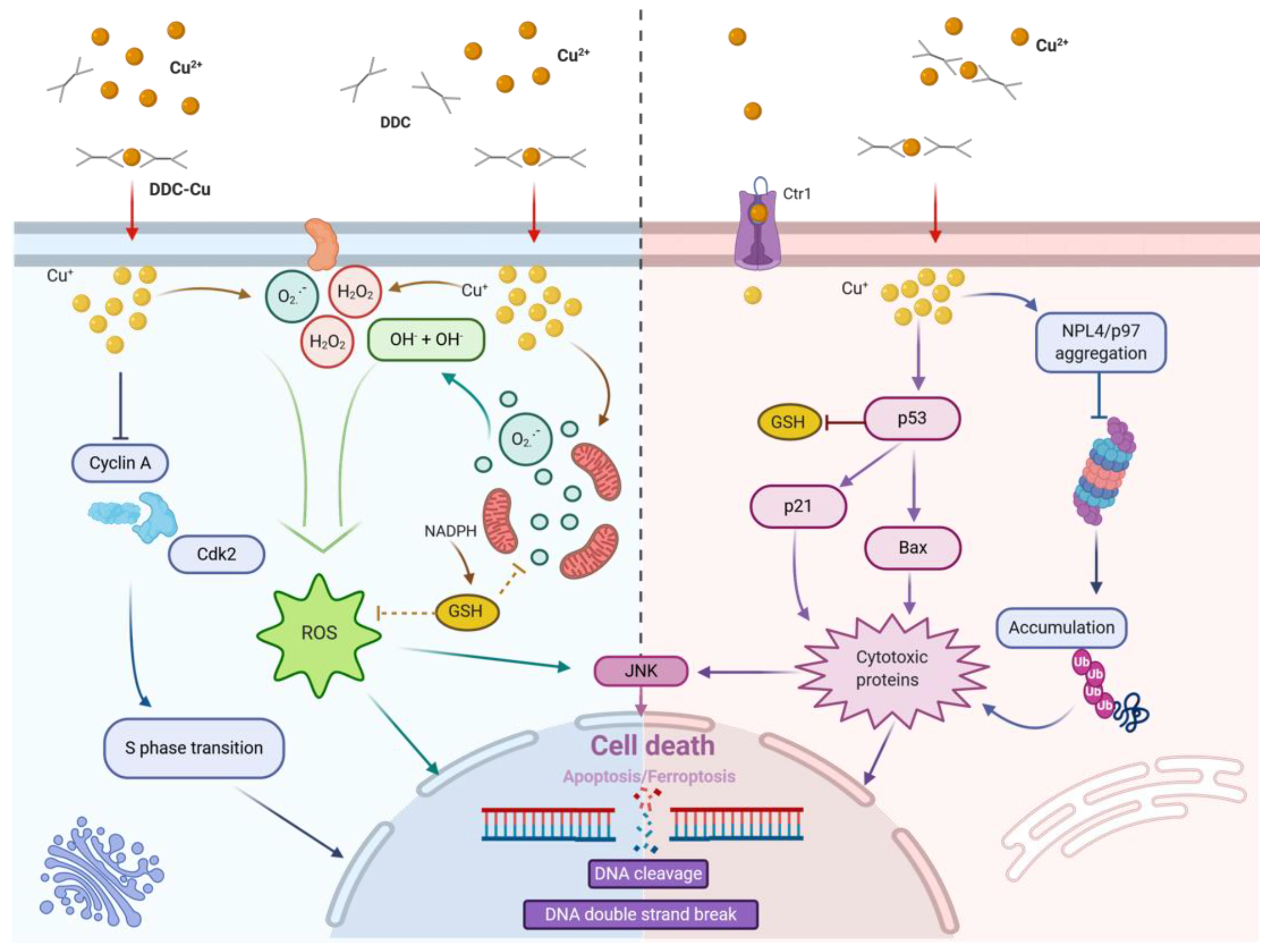
5. Mode of Action of Disulfiram on Cancer Cells
5.1. Disulfiram and ROS Formation
5.2. Disulfiram and Cell Death
5.3. Disulfiram and Proteasome Inhibition
5.4. Disulfiram and Transcription Factor Modifications
5.5. Disulfiram and Cancer Cell Invasion
6. The Use of Disulfiram in Cancer Patients
6.1. Clinical Trials
6.2. Epidemiological Data
7. Drug Combinations with Disulfiram
7.1. Combinations of Disulfiram with Chemotherapy
7.2. Combination of Disulfiram with Radiotherapy
7.3. Combination of Disulfiram with ROS Inducers
7.4. Combination of Disulfiram with Targeted Therapies
7.5. Combination of Disulfiram with Immunotherapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grodzki, M. Ueber äthylirte Sulfoharnstoffe. Eur. J. Inorg. Chem. 1881, 14, 2754–2758. [Google Scholar] [CrossRef] [Green Version]
- Twiss, D.; Bazier, S.A.; Thomas, F. The Dithiocarbamate Accelerators of Vulcanization. J. Soc. Chem. Ind. 1922, 41, 81–88. [Google Scholar]
- Kragh, H. From Disulfiram to Antabuse: The Invention of a Drug. Bull. Hist. Chem. 2008, 33, 82–88. [Google Scholar]
- Treatment Improvement Protocol (TIP) Series, No. 49. Incorporating Alcohol Pharmacotherapies into Medical Practice; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2009.
- Hald, J.; Jacobsen, E. The Formation of Acetaldehyde in the Organism after Ingestion of Antabuse (Tetraethylthiuram disulphide) and Alcohol. Acta Pharmacol. Toxicol. 2009, 4, 305–310. [Google Scholar] [CrossRef]
- De Sousa, A. Disulfiram, Its Use in Alcohol Dependence and Other Disorders; Springer: Gateway East, Singapore, 2019. [Google Scholar]
- The Mark Collection. Disulfiram [Mak Value Documentation, 1993]. In The Mak—Collection for Occupational Health and Safety: Annual Thresholds and Classifications for the Workplace; Wiley-VCH Verlag GmbH & Co. KGaA: Bonn, Germany, 2012; pp. 165–180. [Google Scholar]
- Mattei, E.; Delpino, A.; Mileo, A.M.; Ferrini, U. Induction of Stress Proteins in Murine and Human Melanoma Cell Cultures. Tumori J. 1986, 72, 129–134. [Google Scholar] [CrossRef]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nat. Cell Biol. 2017, 552, 194–199. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Wu, C.; Wang, L.; Chen, Z.-S.; Cui, W. The combination of disulfiram and copper for cancer treatment. Drug Discov. Today 2020, 25, 1099–1108. [Google Scholar] [CrossRef]
- Information, National Center for Biotechnology. Pubchem Compound Summary for Cid 3117, Disulfiram. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Disulfiram (accessed on 1 October 2020).
- Johansson, B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr. Scand. 1992, 86, 15–26. [Google Scholar] [CrossRef]
- Hajdú, I.; Kardos, J.; Major, B.; Fabó, G.; Lőrincz, Z.; Cseh, S.; Dormán, G. Inhibition of the LOX enzyme family members with old and new ligands. Selectivity analysis revisited. Bioorg. Med. Chem. Lett. 2018, 28, 3113–3118. [Google Scholar] [CrossRef]
- Petersen, E.N. The pharmacology and toxicology of disulfiram and its metabolites. Acta Psychiatr. Scand. 1992, 86, 7–13. [Google Scholar] [CrossRef]
- Askgaard, G.; Friis, S.; Hallas, J.; Thygesen, L.C.; Pottegård, A. Use of disulfiram and risk of cancer. Eur. J. Cancer Prev. 2014, 23, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, F.; Chen, J.; Chan, S.; He, Y.; Liu, W.; Zhang, G. Disulfiram/Copper Induces Antitumor Activity against Both Nasopharyngeal Cancer Cells and Cancer-Associated Fibroblasts through ROS/MAPK and Ferroptosis Pathways. Cancers 2020, 12, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvek, B.; Dvorak, Z. The Value of Proteasome Inhibition in Cancer. Can the Old Drug, Disulfiram, Have a Bright New Future as a Novel Proteasome Inhibitor? Drug Discov. Today 2008, 13, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.W.; Doudican, N.A.; Patel, K.R.; Orlow, S.J. Disulfiram induces copper-dependent stimulation of reactive oxygen species and activation of the extrinsic apoptotic pathway in melanoma. Melanoma Res. 2010, 20, 11–20. [Google Scholar] [CrossRef]
- Calderon-Aparicio, A.; Strasberg-Rieber, M.; Rieber, M. Disulfiram anti-cancer efficacy without copper overload is enhanced by extracellular H2O2 generation: Antagonism by tetrathiomolybdate. Oncotarget 2015, 6, 29771–29781. [Google Scholar] [CrossRef] [Green Version]
- Nobel, S.; Burgess, D.H.; Zhivotovsky, B.; Burkitt, M.J.; Orrenius, S.; Slater, A.F.G. Mechanism of Dithiocarbamate Inhibition of Apoptosis: Thiol Oxidation by Dithiocarbamate Disulfides Directly Inhibits Processing of the Caspase-3 Proenzyme. Chem. Res. Toxicol. 1997, 10, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, J.P.; Trapp, V. Reactive oxygen species: An Achilles’ heel of melanoma? Expert Rev. Anticancer. Ther. 2008, 8, 1751–1757. [Google Scholar] [CrossRef]
- Brar, S.S.; Grigg, C.; Wilson, K.S.; Holder, W.D.; Dréau, D.; Austin, C.; Foster, M.; Ghio, A.J.; Whorton, A.R.; Stowell, G.W.; et al. Disulfiram inhibits activating transcription factor/cyclic AMP-responsive element binding protein and human melanoma growth in a metal-dependent manner in vitro, in mice and in a patient with metastatic disease. Mol. Cancer Ther. 2004, 3, 1049–1060. [Google Scholar]
- Cen, D.; Brayton, D.; Shahandeh, B.; Meyskens, F.L.; Farmer, P.J. Disulfiram Facilitates Intracellular Cu Uptake and Induces Apoptosis in Human Melanoma Cells. J. Med. Chem. 2004, 47, 6914–6920. [Google Scholar] [CrossRef] [Green Version]
- Trapp, V.; Lee, K.; Doñate, F.; Mazar, A.P.; Fruehauf, J.P. Redox-related antimelanoma activity of ATN-224. Melanoma Res. 2009, 19, 350–360. [Google Scholar] [CrossRef]
- Cen, D.; Gonzalez, I.; Buckmeier, J.A.; Kahlon, R.S.; Tohidian, N.B.; Meyskens, F.L. Disulfiram induces apoptosis in human melanoma cells: A redox-related process. Mol. Cancer Ther. 2002, 1, 197–204. [Google Scholar] [PubMed]
- Balakirev, M.; Zimmer, G. Mitochondrial injury by disulfiram: Two different mechanisms of the mitochondrial permeability transition. Chem. Interact. 2001, 138, 299–311. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, Q.; Feng, X.; Dai, Y.; Jiang, Y.; Jiang, W.; Liu, X.; Xing, X.; Wang, Y.; Ni, Y.; et al. Disulfiram/copper markedly induced myeloma cell apoptosis through activation of JNK and intrinsic and extrinsic apoptosis pathways. Biomed. Pharmacother. 2020, 126, 110048. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Shi, P.; Fombon, I.S.; Xu, B.; Huang, F.; Wang, W.; Zhou, S. Disulfiram/copper complex activated JNK/c-jun pathway and sensitized cytotoxicity of doxorubicin in doxorubicin resistant leukemia HL60 cells. Blood Cells Mol. Dis. 2011, 47, 264–269. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive Oxygen Species Promote Tnfalpha-Induced Death and Sustained Jnk Activation by Inhibiting Map Kinase Phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Henkel, T.; Machleidt, T.; Alkalay, I.; Kronke, M.; Ben-Neriah, Y.; Baeuerle, P.A. Rapid Proteolysis of I Kappa B-Alpha Is Necessary for Activation of Transcription Factor Nf-κB. Nature 1993, 365, 182–185. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent Engagement of the Classical and Alternative Nf-Kappab Pathways by Diverse Genetic Abnormalities in Multiple Myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Knabb, J.R.; Papa, S.; Kuntzen, C.; Franzoso, G. Upregulation of Twist-1 by NF-κB Blocks Cytotoxicity Induced by Chemotherapeutic Drugs. Mol. Cell. Biol. 2007, 27, 3920–3935. [Google Scholar] [CrossRef] [Green Version]
- Di, C. Inhibition of prostate cancer cellular proteasome activity by a pyrrolidine dithiocarbamate-copper complex is associated with suppression of proliferation and induction of apoptosis. Front. Biosci. 2005, 10, 2932–2939. [Google Scholar] [CrossRef] [Green Version]
- Daniel, K.G.; Chen, D.; Orlu, S.; Cui, Q.C.; Miller, F.R.; Dou, Q.P. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005, 7, R897–R908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Kim, C.H.; Kim, J.H.; Lee, J.; Choi, J.J.; Chen, Z.A.; Lee, M.G.; Chung, K.C.; Hsu, C.Y.; Ahn, Y.S. Pyrrolidine dithiocarbamate and zinc inhibit proteasome-dependent proteolysis. Exp. Cell Res. 2004, 298, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.J.; Gidanian, S.; Shahandeh, B.; Di Bilio, A.J.; Tohidian, N.; Jr, F.L.M. Melanin as a Target for Melanoma Chemotherapy: Pro-oxidant Effect of Oxygen and Metals on Melanoma Viability. Pigment. Cell Res. 2003, 16, 273–279. [Google Scholar] [CrossRef]
- Majera, D.; Skrott, Z.; Chroma, K.; Merchut-Maya, J.M.; Mistrik, M.; Bartek, J. Targeting the Npl4 Adaptor of P97/Vcp Segregase by Disulfiram as an Emerging Cancer Vulnerability Evokes Replication Stress and DNA Damage While Silencing the Atr Pathway. Cells 2020, 9, 469. [Google Scholar] [CrossRef] [Green Version]
- Brar, S.S.; Kennedy, T.P.; Whorton, A.R.; Sturrock, A.B.; Huecksteadt, T.P.; Ghio, A.J.; Hoidal, J.R. Reactive oxygen species from NAD(P)H:quinone oxidoreductase constitutively activate NF-κb in malignant melanoma cells. Am. J. Physiol. Physiol. 2001, 280, 659–676. [Google Scholar] [CrossRef]
- Meyskens, F.L.; Buckmeier, J.A.; McNulty, S.E.; Tohidian, N.B. Activation of nuclear factor-kappa B in human metastatic melanomacells and the effect of oxidative stress. Clin. Cancer Res. 1999, 5, 1197–1202. [Google Scholar]
- Yakisich, J.; Sidén, A.; Eneroth, P.; Cruz, M. Disulfiram Is a Potent In Vitro Inhibitor of DNA Topoisomerases. Biochem. Biophys. Res. Commun. 2001, 289, 586–590. [Google Scholar] [CrossRef]
- Shian, S.-G.; Kao, Y.-R.; Wu, F.Y.-H.; Wu, C.-W. Inhibition of Invasion and Angiogenesis by Zinc-Chelating Agent Disulfiram. Mol. Pharmacol. 2003, 64, 1076–1084. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, K.F.; Blankenship, M.B.; Akerley, W.; Terrazas, M.C.; Kosak, K.M.; Boucher, K.M.; Buys, S.S.; Jones, K.; Werner, T.L.; Agarwal, N.; et al. Abstract 1308: A phase I clinical study investigating disulfiram and copper gluconate in patients with advanced treatment-refractory solid tumors involving the liver. Clin. Trials 2011, 71, 1308. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Lin, J.; Blackford, A.; Bardia, A.; King, S.; Armstrong, A.J.; Rudek, M.A.; Yegnasubramanian, S.; Carducci, M.A. Pharmacodynamic Study of Disulfiram in Men with Non-Metastatic Recurrent Prostate Cancer. Prostate Cancer Prostatic Dis. 2013, 16, 357–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechushtan, H.; Hamamreh, Y.; Nidal, S.; Gotfried, M.; Baron, A.; Shalev, Y.I.; Nisman, B.; Peretz, T.; Peylan-Ramu, N. A Phase IIb Trial Assessing the Addition of Disulfiram to Chemotherapy for the Treatment of Metastatic Non-Small Cell Lung Cancer. Oncology 2015, 20, 366–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Kephart, J.; Bronson, E.; Anand, M.; Daly, C.; Spasojevic, I.; Berg, H.; James, O.G.; Healy, P.; Halabi, S.; et al. Disulfiram (DSF) pharmacokinetics (PK) and copper PET imaging in a phase Ib study of intravenous (IV) copper loading with oral DSF for patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38 (Suppl. S6), 96. [Google Scholar] [CrossRef]
- Grossmann, K.F.; Terrazas, M.; Khong, H.T.; Akerley, W.; Kosak, K.; Boucher, K.; Kennedy, T.P.; Shami, P.J. Abstract LB-175: Phase II study of disulfiram and chelated Zn for the treatment of disseminated metastatic melanoma. Clin. Trials 2013, 73 (Suppl. S8). [Google Scholar] [CrossRef]
- Huang, J.; Campian, J.L.; Gujar, A.D.; Tsien, C.; Ansstas, G.; Tran, D.D.; DeWees, T.A.; Lockhart, A.C.; Kim, A.H. Final results of a phase I dose-escalation, dose-expansion study of adding disulfiram with or without copper to adjuvant temozolomide for newly diagnosed glioblastoma. J. Neuro-Oncol. 2018, 138, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; DeWees, T.; Campian, J.L.; Chheda, M.G.; Ansstas, G.; Tsien, C.; Zipfel, G.J.; Dunn, G.P.; Ippolito, J.E.; Cairncross, J.G. A TITE-CRM Phase I/II Study of Disulfiram and Copper with Concurrent Radiation Therapy and Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2019, 37 (Suppl. 15), 2033. [Google Scholar] [CrossRef]
- Huang, J.; Chaudhary, R.; Cohen, A.L.; Fink, K.; Goldlust, S.; Boockvar, J.; Chinnaiyan, P.; Wan, L.; Marcus, S.; Campian, J.L. A multicenter phase II study of temozolomide plus disulfiram and copper for recurrent temozolomide-resistant glioblastoma. J. Neuro-Oncol. 2019, 142, 537–544. [Google Scholar] [CrossRef]
- Koh, H.K.; Seo, S.Y.; Kim, J.H.; Kim, H.J.; Chie, E.K.; Kim, S.-K.; Kim, I.H. Disulfiram, a Re-positioned Aldehyde Dehydrogenase Inhibitor, Enhances Radiosensitivity of Human Glioblastoma Cells In Vitro. Cancer Res. Treat. 2018, 51, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Rae, C.; Tesson, M.; Babich, J.W.; Boyd, M.; Sorensen, A.; Mairs, R.J. The Role of Copper in Disulfiram-Induced Toxicity and Radiosensitization of Cancer Cells. J. Nucl. Med. 2013, 54, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; McLeod, H.L.; Cassidy, J. Disulfiram-Mediated Inhibition of Nf-Kappab Activity Enhances Cytotoxicity of 5-Fluorouracil in Human Colorectal Cancer Cell Lines. Int. J. Cancer 2003, 104, 504–511. [Google Scholar] [CrossRef]
- Huang, H.; Liao, Y.; Liu, N.; Hua, X.; Cai, J.; Yang, C.; Long, H.; Zhao, C.; Chen, X.; Lan, X.; et al. Two clinical drugs deubiquitinase inhibitor auranofin and aldehyde dehydrogenase inhibitor disulfiram trigger synergistic anti-tumor effects in vitro and in vivo. Oncotarget 2016, 7, 2796–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Wang, Y.; Kang, X.; Wu, A.; Yin, W.; Tang, Y.; Wang, J.; Zhang, M.; Duan, Y.; Huang, Y. Dual-Targeting Biomimetic Delivery for Anti-Glioma Activity Via Remodeling the Tumor Microenvironment and Directing Macrophage-Mediated Immunotherapy. Chem. Sci. 2018, 9, 2674–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triscott, J.; Lee, C.; Hu, K.; Fotovati, A.; Berns, R.; Pambid, M.; Luk, M.; Kast, R.E.; Kong, E.; Toyota, E. Disulfiram, a Drug Widely Used to Control Alcoholism, Suppresses the Self-Renewal of Glioblastoma and over-Rides Resistance to Temozolomide. Oncotarget 2012, 3, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon-Aparicio, A.; Cornejo, A.; Orue, A.; Rieber, M. Anticancer response to disulfiram may be enhanced by co-treatment with MEK inhibitor or oxaliplatin: Modulation by tetrathiomolybdate, KRAS/BRAF mutations and c-MYC/p53 status. Ecancermedicalscience 2019, 13, 890. [Google Scholar] [CrossRef] [Green Version]
- Paranjpe, A.; Zhang, R.; Ali-Osman, F.; Bobustuc, G.C.; Srivenugopal, K.S. Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis 2013, 35, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Bodenner, D.L.; Dedon, P.C.; Keng, P.C.; Katz, J.C.; Borch, R.F. Selective protection against cis-diamminedichloroplatinum(II)-induced toxicity in kidney, gut, and bone marrow by diethyldithiocarbamate. Cancer Res. 1986, 46, 2751–2755. [Google Scholar]
- Loo, T.W.; Clarke, D.M. Blockage of drug resistance in vitro by disulfiram, a drug used to treat alcoholism. J. Natl. Cancer Inst. 2000, 92, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nat. Cell Biol. 2014, 509, 492–496. [Google Scholar] [CrossRef] [Green Version]
- Sammons, S.; Brady, D.; Vahdat, L.; Salama, A.K.S. Copper suppression as cancer therapy: The rationale for copper chelating agents inBRAFV600mutated melanoma. Melanoma Manag. 2016, 3, 207–216. [Google Scholar] [CrossRef]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.-E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A Novel Role for Copper in Ras/Mitogen-Activated Protein Kinase Signaling. Mol. Cell. Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef] [Green Version]
- Del Ama, L.F.; Jones, M.; Walker, P.; Chapman, A.; Braun, J.A.; Mohr, J.; Hurlstone, A. Reprofiling using a zebrafish melanoma model reveals drugs cooperating with targeted therapeutics. Oncotarget 2016, 7, 40348–40361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terashima, Y.; Toda, E.; Itakura, M.; Otsuji, M.; Yoshinaga, S.; Okumura, K.; Shand, F.H.W.; Komohara, Y.; Takeda, M.; Kokubo, K.; et al. Targeting FROUNT with disulfiram suppresses macrophage accumulation and its tumor-promoting properties. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Phase | Title | Status | Study Results | Cancer | Drug (Monotherapy) | Trial ID & Ref. |
|---|---|---|---|---|---|---|
| I | Phase I Study of Disulfiram and Copper Gluconate for the Treatment of Refractory Solid Tumors Involving the Liver | Completed | Well tolerated; no dose limiting toxicity; one stable disease | Cancer | Disulfiram Copper Gluconate | NCT00742911 [43] |
| I | Bioavailability of Disulfiram and Metformin in Glioblastomas | Recruiting | N/A | Glioblastoma | Disulfiram | NCT03151772 |
| Ib | A Phase Ib Study of Intravenous Copper Loading with Oral Disulfiram in Metastatic, Castration Resistant Prostate Cancer | Terminated | No grade > 3 toxicities; no effect on PSA; 64Cu-PET shows Cu-uptake in some metastases | Prostate Cancer | Disulfiram Copper gluconate | NCT02963051 [45] |
| I/II | Disulfiram in Patients with Metastatic Melanoma | Completed | N/A | Stage IV Melanoma | Disulfiram | NCT00256230 |
| II | Study of Recurrent Prostate Cancer with Rising Prostate Specific Antigen (PSA) | Completed | Moderate tolerability (6/19 with grade 3); 5/19 (26%) patients with change in 5-methyl-cytosine; no effect on PSA levels | Prostate Cancer | Disulfiram | NCT01118741 [46] |
| II | Phase II Trial of Disulfiram with Copper in Metastatic Breast Cancer | Recruiting | N/A | Metastatic Breast Cancer | Disulfiram Copper | NCT03323346 |
| II | Disulfiram and Cisplatin in Refractory TGCTs. | Recruiting | N/A | Germ Cell Tumor | Disulfiram | NCT03950830 |
| Phase | Title | Status | Study Results | Cancer | Drug (Combination Therapy) | Trial ID & Ref. |
|---|---|---|---|---|---|---|
| I | Disulfiram Plus Arsenic Trioxide in Patients with Metastatic Melanoma and at Least One Prior Systemic Therapy | Terminated | N/A | Metastatic Melanoma | Disulfiram + Arsenic trioxide | NCT00571116 |
| I | Disulfiram in Treating Patients with Glioblastoma Multiforme After Radiation Therapy with Temozolomide | Completed | Max. tolerated dose 500 mg/day | Glioblastoma | Temozolomide Disulfiram Copper gluconate | NCT01907165 [44] |
| I | Disulfiram and Gemcitabine Hydrochloride in Treating Patients with Unresectable Solid Tumors or Metastatic Pancreatic Cancer | Recruiting | N/A | Stage IV Pancreatic Cancer | Disulfiram + Gemcitabine Hydrochloride | NCT02671890 |
| I | Phase 1 study of Disulfiram and Nivolumab for gastric cancer | Recruiting | N/A | Gastric cancer | Disulfiram + Nivolumab | jRCTs0311883 |
| I/II | Disulfiram/Copper with Concurrent Radiation Therapy and Temozolomide in Patients with Newly Diagnosed Glioblastoma | Recruiting | Low toxicity at 250 mg/day. Median follow-up of 12.3 months, 1-year PFS: 57%; 1-year OS: 69%. | Glioblastoma Multiforme | Disulfiram Copper Gluconate Surgery/ Radiation Temozolomide | NCT02715609 [47] |
| I/II | A Proof-of-concept Clinical Trial Assessing 9 Repurposed Drugs Combined with Metronomic Temozolomide (CUSP9v3 Treatment Protocol) for Recurrent Glioblastoma | Active, not recruiting | N/A | Glioblastoma | Temozolomide + Disulfiram | NCT02770378 |
| II | Disulfiram/Copper Combination in The Treatment of Newly Diagnosed Glioblastoma Multiforme | Unknown status | N/A | Glioblastoma Multiforme | Temozolomide Disulfiram Copper | NCT01777919 |
| II | Disulfiram and Chelated Zinc for the Rx of Disseminated Mets Mel That Has Failed First Line Therapy | Completed | 1 grade 3+ toxicity (confusional episode); ORR 0/12; 1 target lesion –27% | Melanoma | Disulfiram and chelated zinc | NCT02101008 [48] |
| II | Safety, Tolerability and Efficacy of Disulfiram and Copper Gluconate in Recurrent Glioblastoma | Completed | Low toxicity (1/23 with grade 3 elevated ALT); ORR 0/23; 14% with clinical benefit | Recurrent Glio-blastoma | Disulfiram/Copper Temozolomide (TMZ) | NCT03034135 [49] |
| II | Disulfiram and Copper Gluconate with Temozolomide in Unmethylated Glioblastoma Multiforme | Recruiting | N/A | Glio-blastoma Multiforme | Disulfiram Copper gluconate Temozolomide | NCT03363659 |
| II | Disulfiram-Copper Gluconate in Met Pancreas Cancer w Rising CA19-9 on Abraxane-Gemzar, FOLFIRINOX or Gemcitabine | Recruiting | N/A | Metastatic Pancreatic Cancer | Disulfiram Copper + Nab-Paclitaxel/Gemcitabine, Gemcitabine or FOLFIRINOX | NCT03714555 |
| II | Vinorelbine, Cisplatin, Disulfiram and Copper in CTC_EMT Positive Refractory Metastatic Breast Cancer. | Recruiting | N/A | Metastatic Breast Cancer | Vinorelbine, Cisplatin, Disulfiram and Copper | NCT04265274 |
| IIb | Initial Assessment of the Effect of the Addition of Disulfiram (Antabuse) to Standard Chemotherapy in Lung Cancer | Completed | Benefit in PFS (5.9 vs. 4.9 mo) and OS (10.0 vs. 7.1 mo) | Non-small Cell Lung Cancer | Chemotherapy ± disulfiram | NCT00312819 [50] |
| II/III | Disulfiram in Recurrent Glioblastoma | Active, not recruiting | N/A | Glio-blastoma | Disulfiram Copper Alkylating Agents | NCT02678975 |
| Therapy | Description | Cancer Type and Model | Effect of Combination |
|---|---|---|---|
| Temozolomide (TMZ) | Imidazo-tetrazine derived alkylating chemotherapy | Glioblastoma in vitro: BT74, GBM4 and short-term cultures | Inhibtion of chemoresistance development |
| Oxaliplatin | Platinum-based chemotherapy | Colorectal cancer in vitro: SW-620KRAS G12V p53 mutation | Additive cytotoxic effect to the Oxaliplatin |
| BCNU (1,3-bis-2- chloroethyl- nitrosourea) | MGMT (O6-methylguanine methyltransferase)- alkylating agent | Human glioblastoma in vitro: U87 and T98G | The preincubation with 50 µM DSF for 12 h enhance 3-fold the cytotoxicity effect of BCNU compared with BCNU alone. |
| 5-fluorouracil (5-FU) | Antineoplastic agent | Colorectal cancer in vitro: DLD-1, RKOWT | Enhancement of the 5-FU cytotoxicity |
| Cisplatin (Cl2H6N2Pt) | Platinum-based chemotherapy | Melanoma Lung carcinoma Colon carcinoma in vivo: L1210 and P388 leukemia B16 melanoma Lewis lung Colon 26 in B6D2F1 (C57BL/6xDBA/2F1) mice | Provision of a chemoprotective effect |
| Radio therapy | High energy doses of radiation | Glioblastoma in vitro: U138MG, T98G, U251MG, U87M and U373MG | Enhancement of radio-sensitivity |
| Radio therapy | High energy doses of radiation | Neuroblastoma Glioblastoma in vitro: SK-N-BE(2c), UVW-NAT in vivo: SK-N-BE(2c), UVW-NAT in CD-1 nu/nu mice | Induction of radio-sensitization |
| Radio therapy | High energy doses of radiation | Breast cancer in vitro: MDA-MB-231, MDA-MB-231-luc-D3H1, SUM149, UACC-812 in vivo: MDA-MB-231-luc in NOD/SCID mice 4T1 cells in BALB/c mice | Reduction of tumor growth and lung metastases formation |
| Auranofin | Gold salt | Hepatocellular carcinoma in vitro: HepG2 and SMMC-7721 in vivo: HepG2 and SMMC-7721 in BALB/c nude mice | Enhancement of Auranofin-induced apoptosis |
| Tetra-thio molybdate (ATN-224) | Superoxide dismutase inhibitor | Melanoma in vitro: M14 and YUZAZ6 | Antagonistic effect |
| Arsenic trioxide (As2O3) | A pentavalent semimetal | Melanoma In vitro: A375 | Increase of ROS production |
| UO126 | MEK1/2 inhibitor | Melanoma in vitro: C8161KRASmut, A375BRAFmut | Synergistic improvement of the tumor suppression effect of MEK inhibition. |
| GSK 1120212 (Trametinib), PD184352 | MEK1/2 inhibitor | Melanoma in vitro: WM852NRASmut, D04NRASmut, A375BRAFmut, 501MelBRAFmut, WM266-4BRAFmut in vivo: V12RAS zebrafish | Additive cytotoxic effect in combination with MEK inhibitors. |
| Regorafenib | Macrophage modulator | Glioblastoma in vitro: U87 and GL261 in vivo: U87 in BALB/c mice GL261 in C57BL/6 mice | Synergistic effect in the conversion and polarization of the TUMs, leading to an increased antitumoral effect. |
| Clone J43, BioXcell | Antibody against the immune-checkpoint PD-1 | Melanoma Lung carcinoma in vitro: Lewis lung carcinoma (LLC), B16F10 (B16), THP-1 cells, and CHO cells. 16-DsRed and LLC-DsRed (For visualization) in vivo: B16F10 and LLC in C57BL/6 mice | FROUNT inhibition and thereby decrease of tumor progression and TAMs activity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meraz-Torres, F.; Plöger, S.; Garbe, C.; Niessner, H.; Sinnberg, T. Disulfiram as a Therapeutic Agent for Metastatic Malignant Melanoma—Old Myth or New Logos? Cancers 2020, 12, 3538. https://doi.org/10.3390/cancers12123538
Meraz-Torres F, Plöger S, Garbe C, Niessner H, Sinnberg T. Disulfiram as a Therapeutic Agent for Metastatic Malignant Melanoma—Old Myth or New Logos? Cancers. 2020; 12(12):3538. https://doi.org/10.3390/cancers12123538
Chicago/Turabian StyleMeraz-Torres, Francisco, Sarah Plöger, Claus Garbe, Heike Niessner, and Tobias Sinnberg. 2020. "Disulfiram as a Therapeutic Agent for Metastatic Malignant Melanoma—Old Myth or New Logos?" Cancers 12, no. 12: 3538. https://doi.org/10.3390/cancers12123538
APA StyleMeraz-Torres, F., Plöger, S., Garbe, C., Niessner, H., & Sinnberg, T. (2020). Disulfiram as a Therapeutic Agent for Metastatic Malignant Melanoma—Old Myth or New Logos? Cancers, 12(12), 3538. https://doi.org/10.3390/cancers12123538