Molecular and Immune Biomarkers for Cutaneous Melanoma: Current Status and Future Prospects


 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Biomarkers in Target Therapy
2.1. BRAF
2.2. NRAS
2.3. KIT
2.4. Other Single Gene Candidate Predictive Biomarkers
2.5. Resistance to Target Therapy
3. Biomarkers in Immunotherapy
3.1. PD-1/PD-L1 Signaling
3.2. Baseline Immune Factors
3.3. Tumor Mutational Burden
3.4. Tumor Microenvironment
3.5. Microbiome
4. Circulating Total DNA
4.1. Micro RNAs
4.2. Circulating Tumor Cells
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| AEC | Absolute eosinophil count |
| AMC | Absolute monocyte count |
| AJCC | American Joint Committee for Cancer |
| AKT | Protein Kinase B |
| BEAMing | Beads, emulsion, amplification and magnetics |
| BIM | Bcl-2-like protein 11 |
| BTLA | B-T lymphocyte attenuator |
| BRAF | v-Raf murine sarcoma viral oncogene homolog B1 |
| CCR | Cytokeratin receptor |
| CDKN | Cyclin-Dependent Kinase Inhibitor |
| CDK4 | Cyclin-Dependent Kinase 4 |
| CpG | Cytosine linked to a guanine by a phosphate bond |
| CSD | Chronic sun damage |
| CSF-1 | Colony-Stimulating Factor-1 |
| CTC | Circulating tumor cells |
| ctDNA | Cellular tumor DNA |
| CTLA-4 | Cyotoxic T-Lymphocyte associated protein 4 |
| CXCL | Chemokine (C-X-C motif) ligand |
| DC | Dendritic cells |
| DFI | Disease-free survival |
| DNA | Desosiiribonucleic acid |
| ddPCR | Digital droplets polymerase chain reaction |
| EpCAM | Epithelial cell adhesion |
| Fn | Fusobacterium nucleatum |
| FOXP3 | Forkhead box P3 |
| GNAQ | G protein subunit alpha q |
| GNA11 | Guanine nucleotide-binding protein subunit alpha-11 |
| HLA | Human leukocyte antigen |
| ICIs | Immune checkpoint inhibitors |
| IFN | Interferon |
| IHC | Immunohistochemistry |
| IL-12 | Interleukin |
| KIT | Proto-oncogene receptor tyrosine kinase |
| LAG-3 | Lymphocyte-activation gene 3 |
| LDH | Lactate dehydrogenase |
| MAPK | Mitogen-activated protein kinases |
| MDSC | Myeloid-derived suppressor cell |
| MET | MET proto-oncogene |
| MIA | Melanoma inhibitory activity |
| miRNA | Microrna |
| MITF | Melanocyte inducing transcription factor |
| MSS | Melanoma-specific survival |
| mTOR | Mammalian target of rapamycin |
| NF1 | Neurofibromin 1 |
| NGS | Next-generation sequencing |
| NK | Natural killer |
| NRAS | Neuroblastoma RAS Viral Oncogene Homolog |
| NSCLC | Non-small cell lung cancer |
| ORR | Overall response rate |
| OS | Overall survival |
| PD-L1 | Programmed Death—Ligand 1 |
| PCR | Polymerase chain reaction |
| PFS | Progression Free Survival |
| PI3K | Phosphatidyl inositol 3 kinases |
| PKC | Protein kinase c |
| PTEN | Phosphatase and TENsin homolog |
| RLC | Relative lymphocyte count |
| RNA | Ribonucleic acid |
| RTK | Receptor tyrosine kinase |
| TAM | Tumor-Associated Macrophages |
| TGFβ | Transforming Growth factor β |
| TIGIT | T-cell immunoreceptor with immunoglobulin and ITIM domains |
| TIL | Tumor-infiltrating lymphocytes |
| TLR | Toll-like receptor |
| TMB | Tumor mutational burden |
| TME | Tumor Microenvironment |
| Treg | Regulatory T-cell |
| WES | Whole exome sequencing |
| Bi | Bliography |
References
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Flaherty, K.; Goff, S. Emerging strategies in systemic therapy for the treatment of melanoma. Am. Soc. Clin. Oncol. Educ. B 2018, 38, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in stage IIIBRAF-mutated melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Mandalà, M.; Del Vecchio, M.; Gogas, H.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab vs. Ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandalà, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.-A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring C-kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef]
- Wei, X.; Mao, L.; Chi, Z.; Sheng, X.; Cui, C.; Kong, Y.; Dai, J.; Wang, X.; Li, S.; Tang, B.; et al. Efficacy evaluation of Imatinib for the treatment of melanoma: Evidence from a retrospective study. Oncol. Res. 2019, 27, 495–501. [Google Scholar] [CrossRef]
- Guo, J.; Carvajal, R.D.; Dummer, R.; Hauschild, A.; Daud, A.; Bastian, B.C.; Markovic, S.N.; Queirolo, P.; Arance, A.; Berking, C.; et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 2017, 28, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Davies, M.A.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and therapeutic targets in melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef] [PubMed]
- Furney, S.J.; Turajlic, S.; Stamp, G.; Nohadani, M.; Carlisle, A.; Thomas, J.M.; Hayes, A.; Strauss, D.; Gore, M.; Oord, J.V.D.; et al. Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J. Pathol. 2013, 230, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.M.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Davies, H.N.; Bignell, G.R.; Cox, C.E.; Stephens, P.J.; Edkins, S.; Clegg, S.; Teague, J.W.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nat. Cell Biol. 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Kim, S.; Hahn, H.J.; Lee, Y.W.; Choe, Y.B.; Ahn, K.J.; Kim, S.-N. Metaanalysis of BRAF mutations and clinicopathologic characteristics in primary melanoma. J. Am. Acad. Derm. 2015, 72, 1036–1046. [Google Scholar] [CrossRef]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nat. Cell Biol. 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes Dabrafenib Plus Trametinib Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanical acts RAF-Erk signal pathway by oncological mutations B-Raf. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Hall, A.; Marshall, C.J.; Spurr, N.K.; Weiss, R.A. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nat. Cell Biol. 1983, 303, 396–400. [Google Scholar] [CrossRef]
- Jakob, J.A.; Bassett, R.L.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Apn, M.L.R.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef]
- Chiappetta, C.; Proietti, I.; Soccodato, V.; Puggioni, C.; Zaralli, R.; Pacini, L.; Porta, N.; Skroza, N.; Petrozza, V.; Potenza, C.; et al. BRAF and NRAS mutations are heterogeneous and not mutually exclusive in nodular melanoma. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 172–177. [Google Scholar] [CrossRef]
- Lee, J.-H.; Choi, J.-W.; Kim, Y.-S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta-analysis. Br. J. Dermatol. 2011, 164, 776–784. [Google Scholar] [CrossRef]
- Lattanzi, M.; Lee, Y.; Simpson, D.; Moran, U.; Darvishian, F.; Kim, R.H.; Hernando, E.; Polsky, D.; Hanniford, D.; Shapiro, R.; et al. Primary melanoma histologic subtype: Impact on survival and response to therapy. J. Natl. Cancer Inst. 2018, 111, 180–188. [Google Scholar] [CrossRef]
- Ugurel, S.; Thirumaran, R.K.; Bloethner, S.; Gast, A.; Sucker, A.; Mueller-Berghaus, J.; Rittgen, W.; Hemminki, K.; Becker, J.C.; Kumar, R.; et al. B-RAF and N-RAS mutations are preserved during short time in vitro propagation and differentially impact prognosis. PLoS ONE 2007, 2, e236. [Google Scholar] [CrossRef]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Groben, P.A.; Parrish, E.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncol. 2015, 1, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.L.; Aplin, A.E. Targeting mutant NRAS signaling pathways in melanoma. Pharm. Res. 2016, 107, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.; Daniel, M.; Guhan, S.M.; Tsao, H. KIT and melanoma: Biological insights and clinical implications. Yonsei Med. J. 2020, 61, 562–571. [Google Scholar] [CrossRef]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F.; Azar, S.; et al. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, T.M.; Kim, Y.J.; Jang, K.; Lee, H.J.; Lee, S.N.; Ahn, M.S.; Hwang, I.G.; Lee, S.; Lee, M.; et al. Phase II trial of Nilotinib in patients with metastatic malignant melanoma harboring KIT gene aberration: A multicenter trial of Korean cancer study group (UN10-06). Oncology 2015, 20, 1312–1319. [Google Scholar] [CrossRef]
- Delyon, J.; Chevret, S.; Jouary, T.; Dalac, S.; Dalle, S.; Guillot, B.; Arnault, J.-P.; Avril, M.-F.; Bedane, C.; Bens, G.; et al. STAT3 mediates Nilotinib response in KIT-altered melanoma: A phase II multicenter trial of the French skin cancer network. J. Investig. Dermatol. 2018, 138, 58–67. [Google Scholar] [CrossRef]
- Kluger, H.M.; Dudek, A.Z.; McCann, C.; Ritacco, J.; Southard, N.; Jilaveanu, L.B.; Molinaro, A.; Sznol, M.; Dudek, A.Z.; Jilaveanu, L.B. A phase 2 trial of dasatinib in advanced melanoma. Cancer 2010, 117, 2202–2208. [Google Scholar] [CrossRef]
- DeCoster, L.; Broek, I.V.; Neyns, B.; Majois, F.; Baurain, J.F.; Rottey, S.; Rorive, A.; Anckaert, E.; De Mey, J.; De Brakeleer, S.; et al. Biomarker analysis in a phase II Study of Sunitinib in patients with advanced melanoma. Anticancer Res. 2015, 35, 6893–6899. [Google Scholar]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef]
- Whittaker, S.R.; Theurillat, J.-P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Kivela, T.; Simpson, E.R.; Grossniklaus, H.E.; Jager, M.J.; Singh, A.D.; Caminal, J.M.; Pavlick, A.C.; Kujala, E.; Coupland, S.E.; Finger, P. Uveal melanoma. In AJCC Cancer Staging Manual; Springer: New York, NY, USA, 2017; pp. 805–817. [Google Scholar]
- Bol, K.F.; Donia, M.; Heegaard, S.; Kiilgaard, J.F.; Svane, I.M. Genetic biomarkers in melanoma of the ocular region: What the medical oncologist should know. Int. J. Mol. Sci. 2020, 21, 5231. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: A phase III, multicenter, randomized trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Hussussian, C.J.; Struewing, J.P.; Goldstein, A.M.; Higgins, P.A.T.; Ally, D.S.; Sheahan, M.D.; Clark, W.H.; Tucker, M.A.; Dracopoli, N.C. Germline p16 mutations in familial melanoma. Nat. Genet. 1994, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.T. Geographical Variation in the Penetrance of CDKN2A Mutations for Melanoma. J. Natl. Cancer Inst. 2002, 94, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Conway, C.; Beswick, S.; Elliott, F.; Chang, Y.-M.; Randerson-Moor, J.; Harland, M.; Affleck, P.; Marsden, J.; Sanders, D.S.; Boon, A.; et al. Deletion at chromosome arm 9p in relation to BRAF/NRAS mutations and prognostic significance for primary melanoma. Genes Chromosom. Cancer 2010, 49, 425–438. [Google Scholar] [CrossRef]
- Cachia, A.R.; Indsto, J.O.; McLaren, K.M.; Mann, G.J.; Arends, M.J. CDKN2A mutation and deletion status in thin and thick primary melanoma. Clin. Cancer Res. 2000, 6, 3511–3515. [Google Scholar]
- Guo, L.; Qi, J.; Wang, H.; Chen, Y.; Liu, Y. Getting under the skin: The role of CDK4/6 in melanomas. Eur. J. Med. Chem. 2020, 204, 112531. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Flaherty, K.T. Biomarkers in melanoma: Lessons from translational medicine. Trends Cancer 2016, 2, 305–312. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nat. Cell Biol. 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nat. Cell Biol. 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Martin, A.-M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.-T.; et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Austin-Breneman, J.L.; Jiang, H.; Haymaker, C.; Gopalakrishnan, V.; Tetzlaff, M.T.; Frederick, D.T.; et al. Distinct clinical patterns and immune infiltrates are observed at time of progression on targeted therapy versus immune checkpoint blockade for melanoma. OncoImmunology 2016, 5, e1136044. [Google Scholar] [CrossRef]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; Van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Investig. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Madore, J.; Vilain, R.E.; Menzies, A.M.; Kakavand, H.; Wilmott, J.S.; Hyman, J.; Yearley, J.H.; Kefford, R.F.; Thompson, J.F.; Long, G.V.; et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: Implications for anti-PD-1/PD-L1 clinical trials. Pigment. Cell Melanoma Res. 2014, 28, 245–253. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Daud, A.I.; Loo, K.; Pauli, M.L.; Sanchez-Rodriguez, R.; Sandoval, P.M.; Taravati, K.; Tsai, K.; Nosrati, A.; Nardo, L.; Alvarado, M.D.; et al. Tumor immune profiling predicts response to anti–PD-1 therapy in human melanoma. J. Clin. Investig. 2016, 126, 3447–3452. [Google Scholar] [CrossRef]
- Maccalli, C.; Giannarelli, D.; Capocefalo, F.; Pilla, L.; Fonsatti, E.; Di Giacomo, A.M.; Parmiani, G.; Maio, M. Immunological markers and clinical outcome of advanced melanoma patients receiving ipilimumab plus fotemustine in the NIBIT-M1 study. OncoImmunology 2015, 5, e1071007. [Google Scholar] [CrossRef]
- Martens, A.; Wistuba-Hamprecht, K.; Foppen, M.G.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Peripheral CD8 effector-memory type 1 T-cells correlate with outcome in ipilimumab-treated stage IV melanoma patients. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Martens, A.; Hassel, J.C.; Berking, C.; Postow, M.A.; Bisschop, K.; Simeone, E.; Mangana, J.; Schilling, B.; Di Giacomo, A.M.; et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin. Cancer Res. 2016, 22, 5487–5496. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.A.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Dubin, K.; Callahan, M.K.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, B.R.C.; et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 2016, 7, 10391. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Nishino, M.; Ramaiya, N.H.; Hatabu, M.N.N.H.R.H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Bence, C.; Hofman, V.; Chamorey, E.; Long-Mira, E.; Lassalle, S.; Albertini, A.; Liolios, I.; Zahaf, K.; Picard, A.; Montaudié, H.; et al. Association of combined PD -L1 expression and tumour-infiltrating lymphocyte features with survival and treatment outcomes in patients with metastatic melanoma. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 984–994. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, F.; Liu, L. Prognostic significance of PD-L1 in solid tumor. Medicine 2017, 96, e6369. [Google Scholar] [CrossRef]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef]
- Kluger, H.M.; Zito, C.R.; Barr, M.L.; Baine, M.K.; Chiang, V.L.; Sznol, M.; Rimm, D.L.; Chen, L.; Jilaveanu, L.B. Characterization of PD-L1 expression and associated T-cell infiltrates in metastatic melanoma samples from variable anatomic sites. Clin. Cancer Res. 2015, 21, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Massi, D.; Brusa, D.; Merelli, B.; Ciano, M.; Audrito, V.; Serra, S.; Buonincontri, R.; Baroni, G.; Nassini, R.; Minocci, D.; et al. PD-L1 marks a subset of melanomas with a shorter overall survival and distinct genetic and morphological characteristics. Ann. Oncol. 2014, 25, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Blank, C.U.; Mandalà, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nat. Cell Biol. 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasić, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nat. Cell Biol. 2017, 545, 60–65. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer—Dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nat. Cell Biol. 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Xu, H.; Xu, X.; Guo, T.; Ge, W. High tumor mutation burden predicts better efficacy of immunotherapy: A pooled analysis of 103078 cancer patients. OncoImmunology 2019, 8, e1629258. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [PubMed]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Hajjami, H.M.-E.; et al. T cell–induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Iii, E.E.K.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Jóźwicki, W.; Roszkowski, K.; Filipiak, J.; Slominski, A.T. Melanin content in melanoma metastases affects the outcome of radiotherapy. Oncotarget 2016, 7, 17844–17853. [Google Scholar] [CrossRef]
- Galván, I.; Inácio, Â.; Dañino, M.; Corbí-Llopis, R.; Monserrat, M.T.; Bernabeu-Wittel, J. High SLC7A11 expression in normal skin of melanoma patients. Cancer Epidemiol. 2019, 62, 101582. [Google Scholar] [CrossRef]
- Osrodek, M.; Hartman, M.L.; Czyz, M. Physiologically relevant oxygen concentration (6% O2) as an important component of the microenvironment impacting melanoma phenotype and melanoma response to targeted therapeutics in vitro. Int. J. Mol. Sci. 2019, 20, 4203. [Google Scholar] [CrossRef]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 1–16. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Domingue, J.C.; Sears, C.L. Microbiota dysbiosis in select human cancers: Evidence of association and causality. Semin. Immunol. 2017, 32, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Hemann, M.T. Drugs, bugs, and cancer: Fusobacterium nucleatum promotes chemoresistance in colorectal cancer. Cell 2017, 170, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; Kaiser, A.; Wrzesinski, C.; Hinrichs, C.S.; Cassard, L.; Boni, A.; Muranski, P.; Sanchez-Perez, L.; Palmer, D.C.; Yu, Z.; et al. Toll-like receptors in tumor immunotherapy. Clin. Cancer Res. 2007, 13, 5280–5289. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.M.; Pacey, S.; Baird, C.C.S.P.R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.-F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef]
- Chang, G.A.; Tadepalli, J.S.; Shao, Y.; Zhang, Y.; Weiss, S.A.; Robinson, E.; Spittle, C.; Furtado, M.; Shelton, D.N.; Karlin-Neumann, G.; et al. Sensitivity of plasma BRAFmutant and NRASmutant cell-free DNA assays to detect metastatic melanoma in patients with low RECIST scores and non-RECIST disease progression. Mol. Oncol. 2016, 10, 157–165. [Google Scholar] [CrossRef]
- Tan, L.; Sandhu, S.; Lee, R.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. 2019, 30, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Gremel, G.; Marshall, A.; Myers, K.; Fisher, N.; Dunn, J.; Dhomen, N.; Corrie, P.; Middleton, M.; Lorigan, P.; et al. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbé, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.-J.; et al. Phase II trial (BREAK-2) of the BRAF inhibitor Dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Circulating tumor DNA analysis and functional imaging provide complementary approaches for comprehensive disease monitoring in metastatic melanoma. JCO Precis. Oncol. 2017, 1–14. [Google Scholar] [CrossRef]
- Cortez, M.A.; Buesoramos, C.E.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids—The mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef]
- Friedman, E.B.; Shang, S.; De Miera, E.V.-S.; Fog, J.U.; Teilum, M.W.; Ma, M.W.; Berman, R.; Shapiro, R.; Pavlick, A.C.; Hernando, E.; et al. Serum microRNAs as biomarkers for recurrence in melanoma. J. Transl. Med. 2012, 10, 155. [Google Scholar] [CrossRef]
- Gray, E.; Reid, A.L.; Bowyer, S.; Calapre, L.; Siew, K.; Pearce, R.; Cowell, L.; Frank, M.H.; Millward, M.; Ziman, M. Circulating melanoma cell subpopulations: Their heterogeneity and differential responses to treatment. J. Investig. Dermatol. 2015, 135, 2040–2048. [Google Scholar] [CrossRef]
- Reid, A.; Millward, M.; Pearce, R.; Lee, M.; Frank, M.; Ireland, A.; Monshizadeh, L.; Rai, T.; Heenan, P.; Medic, S.; et al. Markers of circulating tumour cells in the peripheral blood of patients with melanoma correlate with disease recurrence and progression. Br. J. Dermatol. 2013, 168, 85–92. [Google Scholar] [CrossRef]
- Thress, K.S.; Brant, R.; Carr, T.H.; Dearden, S.; Jenkins, S.; Brown, H.; Hammett, T.; Cantarini, M.; Barrett, J.C. EGFR mutation detection in ctDNA from NSCLC patient plasma: A cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer 2015, 90, 509–515. [Google Scholar] [CrossRef]
- Hayashi, M.; Chu, D.; Meyer, C.F.; Llosa, N.J.; Bs, G.M.; Morris, C.D.; Levin, A.S.; Wolinsky, J.-P.; Albert, C.M.; Steppan, D.A.; et al. Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer 2016, 122, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.L.; Freeman, J.B.; Millward, M.; Ziman, M.; Gray, E.S. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin. Biochem. 2015, 48, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, Á. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.-C.; Madic, J.; Mariani, P.; Piperno-Neumann, S.; Rampanou, A.; Servois, V.; Cassoux, N.; Desjardins, L.; Milder, M.; Vaucher, I.; et al. Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int. J. Cancer 2014, 134, 1207–1213. [Google Scholar] [CrossRef]
- Lee, J.; Saw, R.; Thompson, J.; Lo, S.; Spillane, A.; Shannon, K.; Stretch, J.; Howle, J.; Menzies, A.; Carlino, M.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Schreuer, M.; Meersseman, G.; Herrewegen, S.V.D.; Jansen, Y.; Chevolet, I.; Bott, A.; Wilgenhof, S.; Seremet, T.; Jacobs, B.; Buyl, R.; et al. Quantitative assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J. Transl. Med. 2016, 14, 95. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.J.; Saturno, G.; Furney, S.J.; et al. Application of sequencing, liquid biopsies, and patient-derived xenografts for personalized medicine in melanoma. Cancer Discov. 2015, 6, 286–299. [Google Scholar] [CrossRef]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.T.; Kefford, R.F.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Cancer Res. 2015, 22, 567–574. [Google Scholar] [CrossRef]
- Redzic, Z.B. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Gao, G.; Cao, Y. Long noncoding RNAs as novel biomarkers have a promising future in cancer diagnostics. Dis. Markers 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Deng, H.; Wang, J.M.; Li, M.; Tang, R.; Tang, K.; Su, Y.; Hou, Y.; Zhang, J. Long non-coding RNAs: New biomarkers for prognosis and diagnosis of colon cancer. Tumor Biol. 2017, 39, 1010428317706332. [Google Scholar] [CrossRef]
- Kanemaru, H.; Fukushima, S.; Yamashita, J.; Honda, N.; Oyama, R.; Kakimoto, A.; Masuguchi, S.; Ishihara, T.; Inoue, Y.; Jinnin, M.; et al. The circulating microRNA-221 level in patients with malignant melanoma as a new tumor marker. J. Dermatol. Sci. 2011, 61, 187–193. [Google Scholar] [CrossRef]
- Dupin, E.; Le Douarin, N.M. Development of melanocyte precursors from the vertebrate neural crest. Oncogene 2003, 22, 3016–3023. [Google Scholar] [CrossRef]
- Rosenberg, R.; Gertler, R.; Friederichs, J.; Fuehrer, K.; Dahm, M.; Phelps, R.; Thorban, S.; Nekarda, H.; Siewert, J.R. Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytol. J. 2002, 49, 150–158. [Google Scholar] [CrossRef]
- Freeman, J.B.; Gray, E.; Millward, M.; Pearce, R.; Ziman, M. Evaluation of a multi-marker immunomagnetic enrichment assay for the quantification of circulating melanoma cells. J. Transl. Med. 2012, 10, 192. [Google Scholar] [CrossRef]
- Khoja, L.; Lorigan, P.C.; Zhou, C.; Lancashire, M.; Booth, J.; Cummings, J.; Califano, R.; Clack, G.; Hughes, A.; Dive, C. Biomarker utility of circulating tumor cells in metastatic cutaneous melanoma. J. Investig. Dermatol. 2013, 133, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- De Souza, L.M.; Robertson, B.M.; Robertson, G.P. Future of circulating tumor cells in the melanoma clinical and research laboratory settings. Cancer Lett. 2017, 392, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Lee, J.H.; Diefenbach, R.J.; Kefford, R.F.; Rizos, H. Liquid biomarkers in melanoma: Detection and discovery. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Klinac, D.; Gray, E.; Freeman, J.B.; Reid, A.L.; Bowyer, S.; Millward, M.; Ziman, M. Monitoring changes in circulating tumour cells as a prognostic indicator of overall survival and treatment response in patients with metastatic melanoma. BMC Cancer 2014, 14, 423. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, K.; Kuo, C.; Nakagawa, T.; Mori, T.; Ueno, H.; Lorico, A.R.; Wang, H.-J.; Hseuh, E.; O’Day, S.J.; Hoon, D.S. Multimarker quantitative real-time PCR detection of circulating melanoma cells in peripheral blood: Relation to disease stage in melanoma patients. Clin. Chem. 2005, 51, 981–988. [Google Scholar] [CrossRef]
- Terstappen, L.W.M.M.; Rao, C.; Bui, T.; Connelly, M.; Doyle, G.; Karydis, I.; Middleton, M.R.; Clack, G.; Malone, M.; Coumans, F.A.W. Circulating melanoma cells and survival in metastatic melanoma. Int. J. Oncol. 2011, 38, 755–760. [Google Scholar] [CrossRef]
- Anagnostou, V.; Yarchoan, M.; Hansen, A.R.; Wang, H.; Verde, F.; Sharon, E.; Collyar, D.; Chow, L.Q.; Forde, P.M. Immuno-oncology trial endpoints: Capturing clinically meaningful activity. Clin. Cancer Res. 2017, 23, 4959–4969. [Google Scholar] [CrossRef]


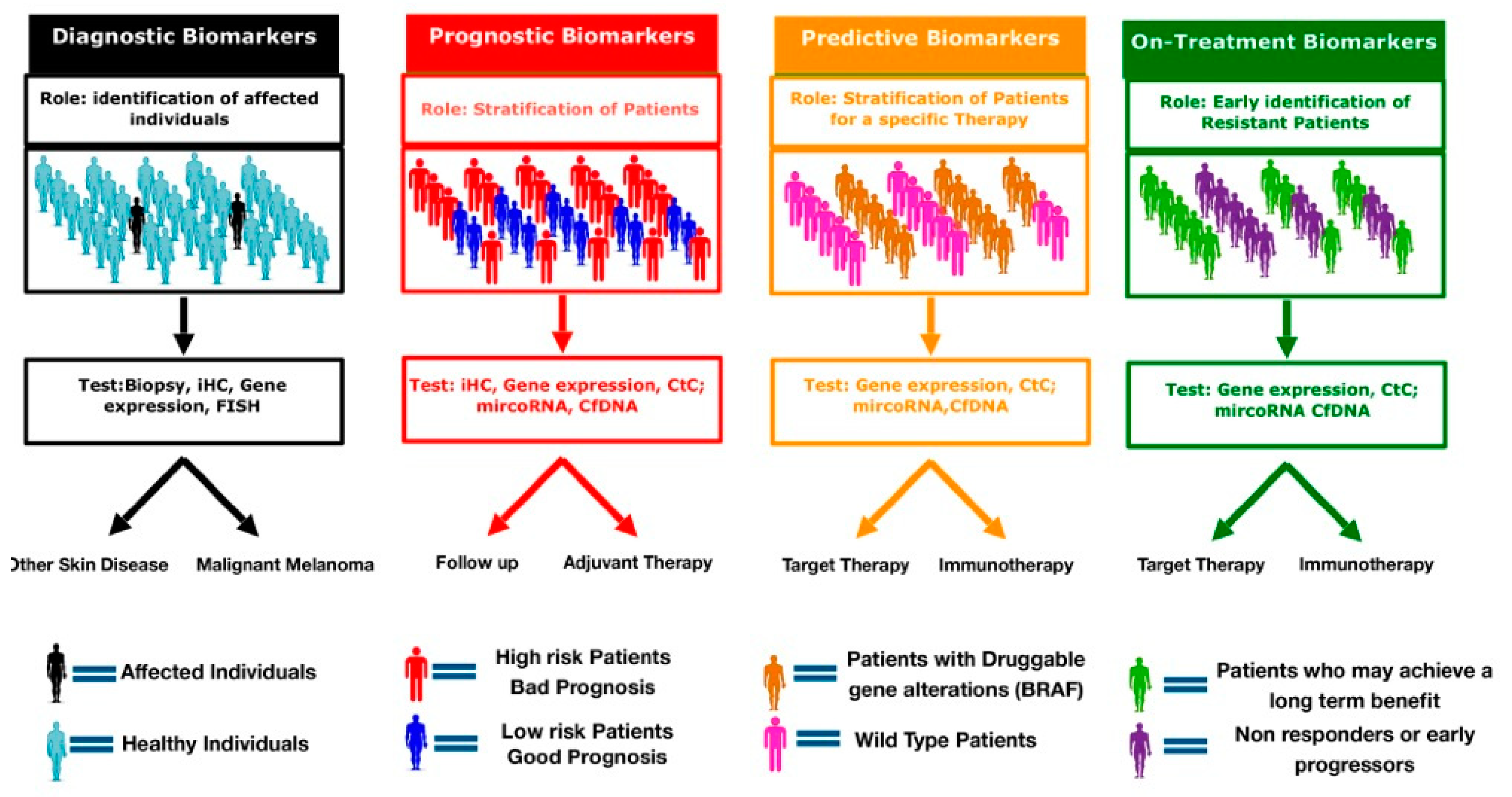{kind=link}
| Gene | Incidence | Comments | References |
|---|---|---|---|
| BRAF | 40–60% | Correlated with response to BRAF-targeted therapies. has led to FDA approval of amplification and sequencing technologies, and multiple laboratory tests to assess BRAF mutation status | [3] |
| NRAS | 20% | Correlated with response to MEK inhibitors | [6] |
| C-KIT | 3% of melanoma; 20–30% melanomas arising from (CSD) skin, acral and mucosal sites | KIT-inhibitors have shown activity in patients with specific mutations | [7,8,9,10,11] |
| GNAQ | 80% of uveal melanoma | MEK inhibitors failed to show efficacy in Phase III trials | [12] |
| NF1 | 46% of cases with BRAF and NRAS wild type | Early clinical trials | NCT02645149 |
| PTEN | 25–30% | Implicated in mechanism of resistance to MAPK inhibition | [13] |
| CDK2 | 11% | Early clinical trials | NCT02645149 |
| Biomarker | Clinical Validation | Tissue for Assessment | Assay | Comments | References |
|---|---|---|---|---|---|
| PD-L1 | Yes; Phase III Trial | Tumor; TME | IHC | Clinical responses in PD-L1 negative tumors. Variability of the assays | [64,67] |
| TMB | Yes; Phase III Trial | Blood; TME | NGS; WES | Lack of standardized TMB thresholds. Variability in quantification methods. | [68] |
| GEP | No; early clinical development | Tumor | IMPRES (RNA-seq) | Costs | [69] |
| TIL | No; early clinical development | Tumor | IF, IHC | Tumor tissue availability | [70] |
| Peripheral lymphocytes | No; early clinical development | Blood | IF | Role of T-cell subpopulations in predicticting clinical benefit | [71,72,73] |
| Gut Microbiota | No; early clinical development | Oral, gut | PCR; NGS | Inter-patients variability. Role in predicting toxicity | [74,75] |
| Biomarker | Predictive/Prognostic | Clinical Validation | Assay | Comments | References |
|---|---|---|---|---|---|
| ctDNA | Prognostic | Advanced clinical investigation | PCR based BEAMing | ctDNA has the potential to anticipate clinical progression. Need of a specific gene target | [121,122,123] |
| ctDNA | Predictive | Advanced clinical investigation | PCR based | Prognostic and predictive to dabrafenib and Trametinib. Limited sensitivity for brain metastasis | [124] |
| ctDNA | Prognostic | Advanced clinical investigation | PCR based | Prognostic and predictive to target therapy and Immunotherapy | [125,126] |
| MicroRNAs | Prognostic/Predictive | Pre-clinical | Luciferase assay | miRNAs are more stable compared to ctDNA. Low Tumor specificity | [127,128] |
| CTC | Prognostic/Predictive | Pre-clinical | PCR based | Lack of standardized technology | [129,130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilla, L.; Alberti, A.; Di Mauro, P.; Gemelli, M.; Cogliati, V.; Cazzaniga, M.E.; Bidoli, P.; Maccalli, C. Molecular and Immune Biomarkers for Cutaneous Melanoma: Current Status and Future Prospects. Cancers 2020, 12, 3456. https://doi.org/10.3390/cancers12113456
Pilla L, Alberti A, Di Mauro P, Gemelli M, Cogliati V, Cazzaniga ME, Bidoli P, Maccalli C. Molecular and Immune Biomarkers for Cutaneous Melanoma: Current Status and Future Prospects. Cancers. 2020; 12(11):3456. https://doi.org/10.3390/cancers12113456
Chicago/Turabian StylePilla, Lorenzo, Andrea Alberti, Pierluigi Di Mauro, Maria Gemelli, Viola Cogliati, Marina Elena Cazzaniga, Paolo Bidoli, and Cristina Maccalli. 2020. "Molecular and Immune Biomarkers for Cutaneous Melanoma: Current Status and Future Prospects" Cancers 12, no. 11: 3456. https://doi.org/10.3390/cancers12113456
APA StylePilla, L., Alberti, A., Di Mauro, P., Gemelli, M., Cogliati, V., Cazzaniga, M. E., Bidoli, P., & Maccalli, C. (2020). Molecular and Immune Biomarkers for Cutaneous Melanoma: Current Status and Future Prospects. Cancers, 12(11), 3456. https://doi.org/10.3390/cancers12113456