Extracellular MicroRNAs as Intercellular Mediators and Noninvasive Biomarkers of Cancer


Abstract
Simple Summary
Abstract
1. Introduction
2. MicroRNAs
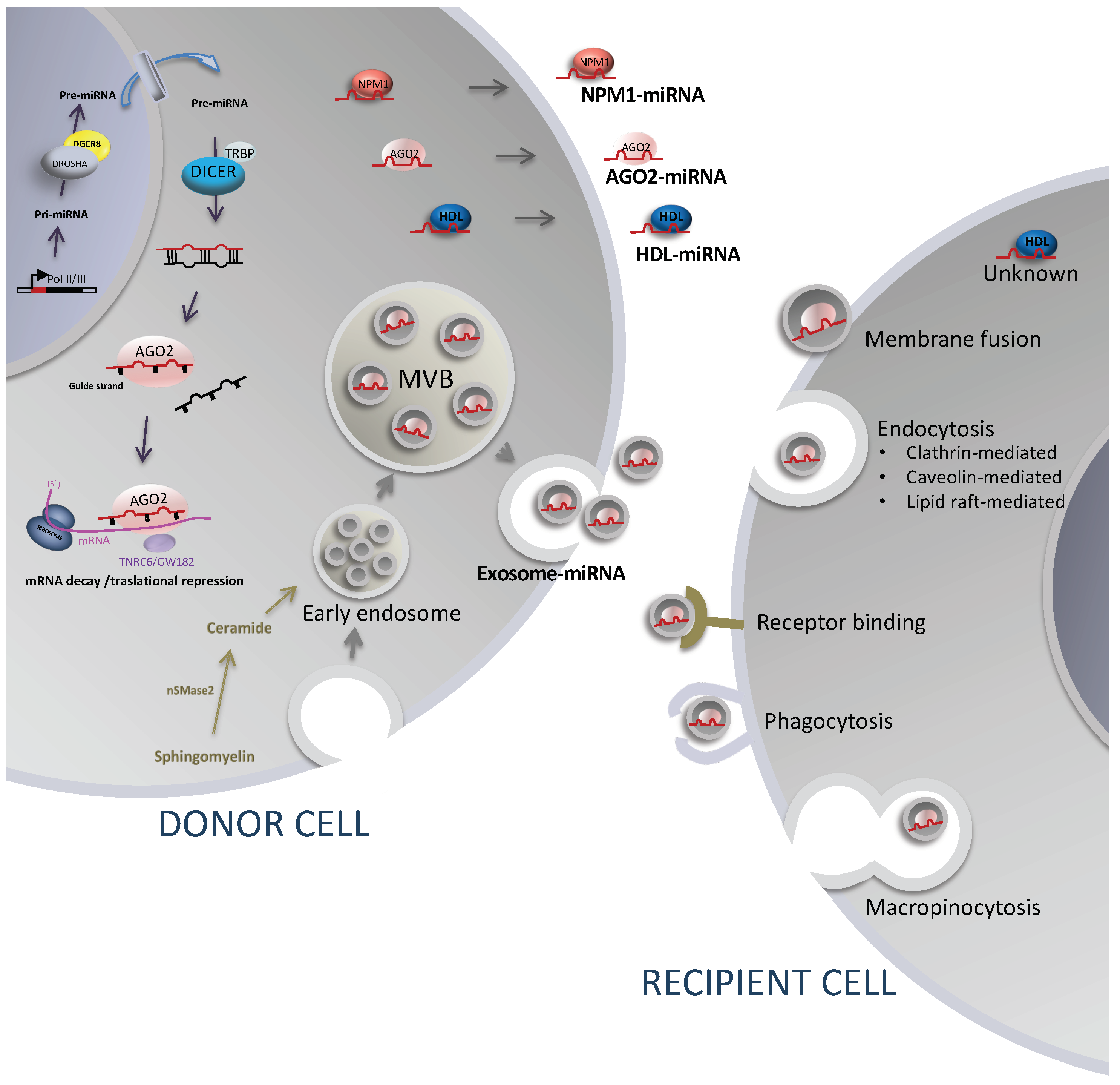
2.1. Biogenesis
2.2. Post-Transcriptional miRNA-Mediated Gene Silencing
2.3. Subcellular Compartmentalization
2.4. Functions
3. Extracellular MicroRNAs
3.1. Release Mechanism Involving Extracellular Vesicles, Sorting, and Uptake Mechanisms
3.2. Release Mechanisms Involving Protein and Lipid Carriers
4. Extracellular miRNAs as Cell-To-Cell Mediators in Cancer
4.1. Extracellular miRNAs and Epithelial–Mesenchymal Transition (EMT)
4.2. Extracellular miRNAs and Angiogenesis
4.3. Extracellular miRNAs and Evasion of Immune Response
4.4. Perspective on the Biological Relevance of Extracellular miRNAs
5. Extracellular miRNAs as Biomarker in Cancer
5.1. Perspective on Extracellular miRNAs as Cancer Biomarkers
5.1.1. Accurate and Complete Reporting
5.1.2. Pre-Analytical and Analytical Variables
5.1.3. Signatures or Panel of miRNAs
5.1.4. Size of the Cohorts
6. Conclusions
Funding
Conflicts of Interest
References
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Quintero, B. Cell-free microRNAs in blood and other body fluids, as cancer biomarkers. Cell Prolif. 2016, 49, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Fehlmann, T.; Ludwig, N.; Backes, C.; Meese, E.; Keller, A. Distribution of microRNA biomarker candidates in solid tissues and body fluids. RNA Biol. 2016, 13, 1084–1088. [Google Scholar] [CrossRef]
- Hanson, E.K.; Lubenow, H.; Ballantyne, J. Identification of forensically relevant body fluids using a panel of differentially expressed microRNAs. Anal. Biochem. 2009, 387, 303–314. [Google Scholar] [CrossRef]
- Endzelins, E.; Melne, V.; Kalnina, Z.; Lietuvietis, V.; Riekstina, U.; Llorente, A.; Line, A. Diagnostic, prognostic and predictive value of cell-free miRNAs in prostate cancer: A systematic review. Mol. Cancer 2016, 15, 41. [Google Scholar] [CrossRef]
- Martinez-Rivera, V.; Negrete-Garcia, M.C.; Avila-Moreno, F.; Ortiz-Quintero, B. Secreted and Tissue miRNAs as Diagnosis Biomarkers of Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2018, 19, 595. [Google Scholar] [CrossRef]
- Cui, L.; Zhang, X.; Ye, G.; Zheng, T.; Song, H.; Deng, H.; Xiao, B.; Xia, T.; Yu, X.; Le, Y.; et al. Gastric juice MicroRNAs as potential biomarkers for the screening of gastric cancer. Cancer 2013, 119, 1618–1626. [Google Scholar] [CrossRef]
- Park, N.J.; Zhou, H.; Elashoff, D.; Henson, B.S.; Kastratovic, D.A.; Abemayor, E.; Wong, D.T. Salivary microRNA: Discovery, characterization, and clinical utility for oral cancer detection. Clin. Cancer Res. 2009, 15, 5473–5477. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D.; et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012, 31, 3513–3523. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stahler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Guttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef]
- Hutvagner, G.; McLachlan, J.; Pasquinelli, A.E.; Balint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef]
- Knight, S.W.; Bass, B.L. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 2001, 293, 2269–2271. [Google Scholar] [CrossRef]
- Kawamata, T.; Tomari, Y. Making RISC. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Kobayashi, M.; Yoda, M.; Sakaguchi, Y.; Katsuma, S.; Suzuki, T.; Tomari, Y. Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Mol. Cell 2010, 39, 292–299. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Schirle, N.T.; Sheu-Gruttadauria, J.; MacRae, I.J. Structural basis for microRNA targeting. Science 2014, 346, 608–613. [Google Scholar] [CrossRef]
- Schirle, N.T.; MacRae, I.J. The crystal structure of human Argonaute2. Science 2012, 336, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Salomon, W.E.; Jolly, S.M.; Moore, M.J.; Zamore, P.D.; Serebrov, V. Single-Molecule Imaging Reveals that Argonaute Reshapes the Binding Properties of Its Nucleic Acid Guides. Cell 2015, 162, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Klum, S.M.; Chandradoss, S.D.; Schirle, N.T.; Joo, C.; MacRae, I.J. Helix-7 in Argonaute2 shapes the microRNA seed region for rapid target recognition. EMBO J. 2018, 37, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, J.; Hennig, J.; Herzog, F.; Aebersold, R.; Sattler, M.; Niessing, D.; Meister, G. Structural features of Argonaute-GW182 protein interactions. Proc. Natl. Acad. Sci. USA 2013, 110, E3770–E3779. [Google Scholar] [CrossRef]
- Lian, S.L.; Li, S.; Abadal, G.X.; Pauley, B.A.; Fritzler, M.J.; Chan, E.K. The C-terminal half of human Ago2 binds to multiple GW-rich regions of GW182 and requires GW182 to mediate silencing. RNA 2009, 15, 804–813. [Google Scholar] [CrossRef]
- Jinek, M.; Fabian, M.R.; Coyle, S.M.; Sonenberg, N.; Doudna, J.A. Structural insights into the human GW182-PABC interaction in microRNA-mediated deadenylation. Nat. Struct. Mol. Biol. 2010, 17, 238–240. [Google Scholar] [CrossRef]
- Zekri, L.; Huntzinger, E.; Heimstadt, S.; Izaurralde, E. The silencing domain of GW182 interacts with PABPC1 to promote translational repression and degradation of microRNA targets and is required for target release. Mol. Cell. Biol. 2009, 29, 6220–6231. [Google Scholar] [CrossRef]
- Moretti, F.; Kaiser, C.; Zdanowicz-Specht, A.; Hentze, M.W. PABP and the poly(A) tail augment microRNA repression by facilitated miRISC binding. Nat. Struct. Mol. Biol. 2012, 19, 603–608. [Google Scholar] [CrossRef]
- Kuzuoglu-Ozturk, D.; Huntzinger, E.; Schmidt, S.; Izaurralde, E. The Caenorhabditis elegans GW182 protein AIN-1 interacts with PAB-1 and subunits of the PAN2-PAN3 and CCR4-NOT deadenylase complexes. Nucleic Acids Res. 2012, 40, 5651–5665. [Google Scholar] [CrossRef]
- Fabian, M.R.; Cieplak, M.K.; Frank, F.; Morita, M.; Green, J.; Srikumar, T.; Nagar, B.; Yamamoto, T.; Raught, B.; Duchaine, T.F.; et al. miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat. Struct. Mol. Biol. 2011, 18, 1211–1217. [Google Scholar] [CrossRef]
- Wahle, E.; Winkler, G.S. RNA decay machines: Deadenylation by the Ccr4-not and Pan2-Pan3 complexes. Biochim. Biophys. Acta 2013, 1829, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Chang, T.C.; Yamashita, Y.; Zhu, W.; Zhong, Z.; Chen, C.Y.; Shyu, A.B. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat. Struct. Mol. Biol. 2005, 12, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev. 2013, 27, 2628–2641. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Basquin, J.; Ozgur, S.; Czarnocki-Cieciura, M.; Bonneau, F.; Aartse, A.; Dziembowski, A.; Nowotny, M.; Conti, E.; Filipowicz, W. Structural and biochemical insights to the role of the CCR4-NOT complex and DDX6 ATPase in microRNA repression. Mol. Cell 2014, 54, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.E.; Truffault, V.; Boland, A.; Huntzinger, E.; Chang, C.T.; Haas, G.; Weichenrieder, O.; Coles, M.; Izaurralde, E. A direct interaction between DCP1 and XRN1 couples mRNA decapping to 5′ exonucleolytic degradation. Nat. Struct. Mol. Biol. 2012, 19, 1324–1331. [Google Scholar] [CrossRef]
- Jinek, M.; Coyle, S.M.; Doudna, J.A. Coupled 5′ nucleotide recognition and processivity in Xrn1-mediated mRNA decay. Mol. Cell 2011, 41, 600–608. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- Kamenska, A.; Simpson, C.; Vindry, C.; Broomhead, H.; Benard, M.; Ernoult-Lange, M.; Lee, B.P.; Harries, L.W.; Weil, D.; Standart, N. The DDX6-4E-T interaction mediates translational repression and P-body assembly. Nucleic Acids Res. 2016, 44, 6318–6334. [Google Scholar] [CrossRef]
- Kamenska, A.; Lu, W.T.; Kubacka, D.; Broomhead, H.; Minshall, N.; Bushell, M.; Standart, N. Human 4E-T represses translation of bound mRNAs and enhances microRNA-mediated silencing. Nucleic Acids Res. 2014, 42, 3298–3313. [Google Scholar] [CrossRef]
- Ozgur, S.; Basquin, J.; Kamenska, A.; Filipowicz, W.; Standart, N.; Conti, E. Structure of a Human 4E-T/DDX6/CNOT1 Complex Reveals the Different Interplay of DDX6-Binding Proteins with the CCR4-NOT Complex. Cell Rep. 2015, 13, 703–711. [Google Scholar] [CrossRef]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.H.; Ghoshal, K.; Villen, J.; Bartel, D.P. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Valencia-Sanchez, M.A.; Hannon, G.J.; Parker, R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 2005, 7, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, L.; Zhuang, X.; Yu, Y.; Liu, X.; Cui, X.; Ji, L.; Pan, Z.; Cao, X.; Mo, B.; et al. MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell 2013, 153, 562–574. [Google Scholar] [CrossRef]
- Kim, Y.J.; Maizel, A.; Chen, X. Traffic into silence: Endomembranes and post-transcriptional RNA silencing. EMBO J. 2014, 33, 968–980. [Google Scholar] [CrossRef]
- Sripada, L.; Tomar, D.; Prajapati, P.; Singh, R.; Singh, A.K.; Singh, R. Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: Detailed analysis of mitochondrial associated miRNA. PLoS ONE 2012, 7, e44873. [Google Scholar] [CrossRef]
- Leung, A.K.L. The Whereabouts of microRNA Actions: Cytoplasm and Beyond. Trends Cell Biol. 2015, 25, 601–610. [Google Scholar] [CrossRef]
- Politz, J.C.; Zhang, F.; Pederson, T. MicroRNA-206 colocalizes with ribosome-rich regions in both the nucleolus and cytoplasm of rat myogenic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 18957–18962. [Google Scholar] [CrossRef]
- Hwang, H.W.; Wentzel, E.A.; Mendell, J.T. A hexanucleotide element directs microRNA nuclear import. Science 2007, 315, 97–100. [Google Scholar] [CrossRef]
- Jeffries, C.D.; Fried, H.M.; Perkins, D.O. Nuclear and cytoplasmic localization of neural stem cell microRNAs. RNA 2011, 17, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.T.; Li, L.; Chu, Y.; Janowski, B.A.; Corey, D.R. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, R.; Hicks, J.A.; Li, L.; Gagnon, K.T.; Sridhara, V.; Lemoff, A.; Mirzaei, H.; Corey, D.R. Stable association of RNAi machinery is conserved between the cytoplasm and nucleus of human cells. RNA 2016, 22, 1085–1098. [Google Scholar] [CrossRef]
- Pu, M.; Chen, J.; Tao, Z.; Miao, L.; Qi, X.; Wang, Y.; Ren, J. Regulatory network of miRNA on its target: Coordination between transcriptional and post-transcriptional regulation of gene expression. Cell. Mol. Life Sci. 2019, 76, 441–451. [Google Scholar] [CrossRef]
- Liu, H.; Lei, C.; He, Q.; Pan, Z.; Xiao, D.; Tao, Y. Nuclear functions of mammalian MicroRNAs in gene regulation, immunity and cancer. Mol. Cancer 2018, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Saetrom, P.; Snove, O., Jr.; Rossi, J.J. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16230–16235. [Google Scholar] [CrossRef]
- Miao, L.; Yao, H.; Li, C.; Pu, M.; Yao, X.; Yang, H.; Qi, X.; Ren, J.; Wang, Y. A dual inhibition: microRNA-552 suppresses both transcription and translation of cytochrome P450 2E1. Biochim. Biophys. Acta 2016, 1859, 650–662. [Google Scholar] [CrossRef]
- Xiao, M.; Li, J.; Li, W.; Wang, Y.; Wu, F.; Xi, Y.; Zhang, L.; Ding, C.; Luo, H.; Li, Y.; et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017, 14, 1326–1334. [Google Scholar] [CrossRef]
- Chen, J.F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef]
- Lynn, F.C.; Skewes-Cox, P.; Kosaka, Y.; McManus, M.T.; Harfe, B.D.; German, M.S. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes 2007, 56, 2938–2945. [Google Scholar] [CrossRef]
- Kawase-Koga, Y.; Otaegi, G.; Sun, T. Different timings of Dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev. Dyn. 2009, 238, 2800–2812. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.R.; Georges, S.A.; Seay, H.R.; Tapscott, S.J.; McManus, M.T.; Goldhamer, D.J.; Swanson, M.S.; Harfe, B.D. Essential role for Dicer during skeletal muscle development. Dev. Biol. 2007, 311, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Mancuso, M.R.; Hampton, J.; Stankunas, K.; Asano, T.; Chen, C.Z.; Kuo, C.J. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development 2008, 135, 3989–3993. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Walentek, P.; Sponer, N.; Klimke, A.; Lee, J.S.; Dixon, G.; Harland, R.; Wan, Y.; Lishko, P.; Lize, M.; et al. miR-34/449 miRNAs are required for motile ciliogenesis by repressing cp110. Nature 2014, 510, 115–120. [Google Scholar] [CrossRef]
- Rowe, R.G.; Wang, L.D.; Coma, S.; Han, A.; Mathieu, R.; Pearson, D.S.; Ross, S.; Sousa, P.; Nguyen, P.T.; Rodriguez, A.; et al. Developmental regulation of myeloerythroid progenitor function by the Lin28b-let-7-Hmga2 axis. J. Exp. Med. 2016, 213, 1497–1512. [Google Scholar] [CrossRef]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef]
- Bueno, M.J.; Malumbres, M. MicroRNAs and the cell cycle. Biochim. Biophys. Acta 2011, 1812, 592–601. [Google Scholar] [CrossRef]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef]
- Sherrard, R.; Luehr, S.; Holzkamp, H.; McJunkin, K.; Memar, N.; Conradt, B. miRNAs cooperate in apoptosis regulation during C. elegans development. Genes Dev. 2017, 31, 209–222. [Google Scholar] [CrossRef]
- Thompson, B.J.; Cohen, S.M. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell 2006, 126, 767–774. [Google Scholar] [CrossRef]
- Suarez, Y.; Sessa, W.C. MicroRNAs as novel regulators of angiogenesis. Circ. Res. 2009, 104, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.C.; Chuang, C.H.; Huang, W.C.; Weng, S.L.; Chen, C.H.; Chang, K.H.; Liao, K.W.; Huang, H.D. A panel of eight microRNAs is a good predictive parameter for triple-negative breast cancer relapse. Theranostics 2020, 10, 8771–8789. [Google Scholar] [CrossRef] [PubMed]
- Kanno, S.; Nosho, K.; Ishigami, K.; Yamamoto, I.; Koide, H.; Kurihara, H.; Mitsuhashi, K.; Shitani, M.; Motoya, M.; Sasaki, S.; et al. MicroRNA-196b is an independent prognostic biomarker in patients with pancreatic cancer. Carcinogenesis 2017, 38, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Rosignolo, F.; Memeo, L.; Monzani, F.; Colarossi, C.; Pecce, V.; Verrienti, A.; Durante, C.; Grani, G.; Lamartina, L.; Forte, S.; et al. MicroRNA-based molecular classification of papillary thyroid carcinoma. Int. J. Oncol. 2017, 50, 1767–1777. [Google Scholar] [CrossRef]
- Deng, L.; Tang, J.; Yang, H.; Cheng, C.; Lu, S.; Jiang, R.; Sun, B. MTA1 modulated by miR-30e contributes to epithelial-to-mesenchymal transition in hepatocellular carcinoma through an ErbB2-dependent pathway. Oncogene 2017, 36, 3976–3985. [Google Scholar] [CrossRef]
- Gururajan, M.; Josson, S.; Chu, G.C.; Lu, C.L.; Lu, Y.T.; Haga, C.L.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; et al. miR-154* and miR-379 in the DLK1-DIO3 MicroRNA Mega-Cluster Regulate Epithelial to Mesenchymal Transition and Bone Metastasis of Prostate Cancer. Clin. Cancer Res. 2014, 20, 6559–6569. [Google Scholar] [CrossRef]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef]
- Mukohyama, J.; Isobe, T.; Hu, Q.; Hayashi, T.; Watanabe, T.; Maeda, M.; Yanagi, H.; Qian, X.; Yamashita, K.; Minami, H.; et al. miR-221 Targets QKI to Enhance the Tumorigenic Capacity of Human Colorectal Cancer Stem Cells. Cancer Res. 2019, 79, 5151–5158. [Google Scholar] [CrossRef]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Tabet, F.; Vickers, K.C.; Cuesta Torres, L.F.; Wiese, C.B.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer - implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e418. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Gonzalez, S.; Sanchez-Cabo, F.; Gonzalez, M.A.; Bernad, A.; Sanchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Perez-Hernandez, D.; Vazquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sanchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef]
- Pigati, L.; Yaddanapudi, S.C.; Iyengar, R.; Kim, D.J.; Hearn, S.A.; Danforth, D.; Hastings, M.L.; Duelli, D.M. Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS ONE 2010, 5, e13515. [Google Scholar] [CrossRef]
- Cha, D.J.; Franklin, J.L.; Dou, Y.; Liu, Q.; Higginbotham, J.N.; Demory Beckler, M.; Weaver, A.M.; Vickers, K.; Prasad, N.; Levy, S.; et al. KRAS-dependent sorting of miRNA to exosomes. Elife 2015, 4, e07197. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, F.; Wang, B.; Li, H.; Xu, Y.; Liu, X.; Shi, L.; Lu, X.; Xu, W.; Lu, L.; et al. STAT3-regulated exosomal miR-21 promotes angiogenesis and is involved in neoplastic processes of transformed human bronchial epithelial cells. Cancer Lett. 2016, 370, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sai, B.; Wang, F.; Wang, L.; Wang, Y.; Zheng, L.; Li, G.; Tang, J.; Xiang, J. Hypoxic BMSC-derived exosomal miRNAs promote metastasis of lung cancer cells via STAT3-induced EMT. Mol. Cancer 2019, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Santangelo, L.; Giurato, G.; Cicchini, C.; Montaldo, C.; Mancone, C.; Tarallo, R.; Battistelli, C.; Alonzi, T.; Weisz, A.; Tripodi, M. The RNA-Binding Protein SYNCRIP Is a Component of the Hepatocyte Exosomal Machinery Controlling MicroRNA Sorting. Cell Rep. 2016, 17, 799–808. [Google Scholar] [CrossRef]
- Hobor, F.; Dallmann, A.; Ball, N.J.; Cicchini, C.; Battistelli, C.; Ogrodowicz, R.W.; Christodoulou, E.; Martin, S.R.; Castello, A.; Tripodi, M.; et al. A cryptic RNA-binding domain mediates Syncrip recognition and exosomal partitioning of miRNA targets. Nat. Commun. 2018, 9, 831. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, A.J.; Hoshino, D.; Hong, N.H.; Cha, D.J.; Franklin, J.L.; Coffey, R.J.; Patton, J.G.; Weaver, A.M. KRAS-MEK Signaling Controls Ago2 Sorting into Exosomes. Cell Rep. 2016, 15, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Shurtleff, M.J.; Temoche-Diaz, M.M.; Karfilis, K.V.; Ri, S.; Schekman, R. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Zeng, Z.; Song, Y.; Li, L.; Wu, Z.; Zhang, X.; Li, Z.; Ke, X.; Hu, X. YBX-1 mediated sorting of miR-133 into hypoxia/reoxygenation-induced EPC-derived exosomes to increase fibroblast angiogenesis and MEndoT. Stem Cell Res. Ther. 2019, 10, 263. [Google Scholar] [CrossRef]
- Lu, P.; Li, H.; Li, N.; Singh, R.N.; Bishop, C.E.; Chen, X.; Lu, B. MEX3C interacts with adaptor-related protein complex 2 and involves in miR-451a exosomal sorting. PLoS ONE 2017, 12, e0185992. [Google Scholar] [CrossRef]
- Teng, Y.; Ren, Y.; Hu, X.; Mu, J.; Samykutty, A.; Zhuang, X.; Deng, Z.; Kumar, A.; Zhang, L.; Merchant, M.L.; et al. MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nat. Commun. 2017, 8, 14448. [Google Scholar] [CrossRef]
- Temoche-Diaz, M.M.; Shurtleff, M.J.; Nottingham, R.M.; Yao, J.; Fadadu, R.P.; Lambowitz, A.M.; Schekman, R. Distinct mechanisms of microRNA sorting into cancer cell-derived extracellular vesicle subtypes. Elife 2019, 8. [Google Scholar] [CrossRef]
- Rana, S.; Yue, S.; Stadel, D.; Zoller, M. Toward tailored exosomes: The exosomal tetraspanin web contributes to target cell selection. Int. J. Biochem. Cell Biol. 2012, 44, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Barres, C.; Blanc, L.; Bette-Bobillo, P.; Andre, S.; Mamoun, R.; Gabius, H.J.; Vidal, M. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood 2010, 115, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Gaipl, U.S. The immune functions of phosphatidylserine in membranes of dying cells and microvesicles. Semin. Immunopathol. 2011, 33, 97–516. [Google Scholar] [CrossRef]
- Tian, T.; Zhu, Y.L.; Zhou, Y.Y.; Liang, G.F.; Wang, Y.Y.; Hu, F.H.; Xiao, Z.D. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef]
- Feng, D.; Zhao, W.L.; Ye, Y.Y.; Bai, X.C.; Liu, R.Q.; Chang, L.F.; Zhou, Q.; Sui, S.F. Cellular internalization of exosomes occurs through phagocytosis. Traffic 2010, 11, 675–687. [Google Scholar] [CrossRef]
- Laulagnier, K.; Javalet, C.; Hemming, F.J.; Chivet, M.; Lachenal, G.; Blot, B.; Chatellard, C.; Sadoul, R. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell. Mol. Life Sci. 2018, 75, 757–773. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Y.; Wang, H.; Zhu, Z.; Xiao, Z. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J. Cell. Biochem. 2010, 111, 488–496. [Google Scholar] [CrossRef]
- Zhang, H.; Lyden, D. Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat. Protoc. 2019, 14, 1027–1053. [Google Scholar] [CrossRef]
- Wu, A.Y.; Sung, Y.C.; Chen, Y.J.; Chou, S.T.; Guo, V.; Chien, J.C.; Ko, J.J.; Yang, A.L.; Huang, H.C.; Chuang, J.C.; et al. Multiresolution Imaging Using Bioluminescence Resonance Energy Transfer Identifies Distinct Biodistribution Profiles of Extracellular Vesicles and Exomeres with Redirected Tropism. Adv. Sci. 2020, 7, 2001467. [Google Scholar] [CrossRef]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e1018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Higginbotham, J.N.; Jeppesen, D.K.; Yang, Y.P.; Li, W.; McKinley, E.T.; Graves-Deal, R.; Ping, J.; Britain, C.M.; Dorsett, K.A.; et al. Transfer of Functional Cargo in Exomeres. Cell Rep. 2019, 27, 940–954.e946. [Google Scholar] [CrossRef] [PubMed]
- Prud’homme, G.J.; Glinka, Y.; Lichner, Z.; Yousef, G.M. Neuropilin-1 is a receptor for extracellular miRNA and AGO2/miRNA complexes and mediates the internalization of miRNAs that modulate cell function. Oncotarget 2016, 7, 68057–68071. [Google Scholar] [CrossRef] [PubMed]
- Fuji, T.; Umeda, Y.; Nyuya, A.; Taniguchi, F.; Kawai, T.; Yasui, K.; Toshima, T.; Yoshida, K.; Fujiwara, T.; Goel, A.; et al. Detection of circulating microRNAs with Ago2 complexes to monitor the tumor dynamics of colorectal cancer patients during chemotherapy. Int. J. Cancer 2019, 144, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Riwanto, M.; Besler, C.; Knau, A.; Fichtlscherer, S.; Roxe, T.; Zeiher, A.M.; Landmesser, U.; Dimmeler, S. Characterization of levels and cellular transfer of circulating lipoprotein-bound microRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1392–1400. [Google Scholar] [CrossRef]
- Scicali, R.; Di Pino, A.; Pavanello, C.; Ossoli, A.; Strazzella, A.; Alberti, A.; Di Mauro, S.; Scamporrino, A.; Urbano, F.; Filippello, A.; et al. Analysis of HDL-microRNA panel in heterozygous familial hypercholesterolemia subjects with LDL receptor null or defective mutation. Sci. Rep. 2019, 9, 20354. [Google Scholar] [CrossRef]
- Desgagne, V.; Guerin, R.; Guay, S.P.; Corbin, F.; Couture, P.; Lamarche, B.; Bouchard, L. Changes in high-density lipoprotein-carried miRNA contribution to the plasmatic pool after consumption of dietary trans fat in healthy men. Epigenomics 2017, 9, 669–688. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Barbazan, J.; Matic Vignjevic, D. Cancer associated fibroblasts: Is the force the path to the dark side? Curr. Opin. Cell Biol. 2019, 56, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Panadero, R.; Lucantoni, F.; Gamero-Sandemetrio, E.; Cruz-Merino, L.; Alvaro, T.; Noguera, R. The tumour microenvironment as an integrated framework to understand cancer biology. Cancer Lett. 2019, 461, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Sun, L.; Xu, F.; Liu, L.; Hu, F.; Song, D.; Hou, Z.; Wu, W.; Luo, X.; Wang, J.; et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res. 2019, 79, 146–158. [Google Scholar] [CrossRef]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Ning, Z.; Ma, L.; Liu, W.; Shao, C.; Shu, Y.; Shen, H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol. Cancer 2017, 16, 148. [Google Scholar] [CrossRef]
- Yang, M.; Chen, J.; Su, F.; Yu, B.; Su, F.; Lin, L.; Liu, Y.; Huang, J.D.; Song, E. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Nicoloso, M.S.; Spizzo, R.; Shimizu, M.; Rossi, S.; Calin, G.A. MicroRNAs-the micro steering wheel of tumour metastases. Nat. Rev. Cancer 2009, 9, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pochampally, R.; Watabe, K.; Lu, Z.; Mo, Y.Y. Exosome-mediated transfer of miR-10b promotes cell invasion in breast cancer. Mol. Cancer 2014, 13, 256. [Google Scholar] [CrossRef]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef]
- Shaham, L.; Binder, V.; Gefen, N.; Borkhardt, A.; Izraeli, S. MiR-125 in normal and malignant hematopoiesis. Leukemia 2012, 26, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Lin, K.Y.; Chen, Y.Q. Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 2013, 6, 6. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, L.X.; Wu, Q.N.; Du, Z.M.; Chen, J.; Liao, D.Z.; Huang, M.Y.; Hou, J.H.; Wu, Q.L.; Zeng, M.S.; et al. miR-125b is methylated and functions as a tumor suppressor by regulating the ETS1 proto-oncogene in human invasive breast cancer. Cancer Res. 2011, 71, 3552–3562. [Google Scholar] [CrossRef]
- Le, M.T.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Lamouille, S.; Subramanyam, D.; Blelloch, R.; Derynck, R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr. Opin. Cell Biol. 2013, 25, 200–207. [Google Scholar] [CrossRef]
- Legras, A.; Pecuchet, N.; Imbeaud, S.; Pallier, K.; Didelot, A.; Roussel, H.; Gibault, L.; Fabre, E.; Le Pimpec-Barthes, F.; Laurent-Puig, P.; et al. Epithelial-to-Mesenchymal Transition and MicroRNAs in Lung Cancer. Cancers 2017, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Musavi Shenas, M.H.; Eghbal-Fard, S.; Mehrisofiani, V.; Abd Yazdani, N.; Rahbar Farzam, O.; Marofi, F.; Yousefi, M. MicroRNAs and signaling networks involved in epithelial-mesenchymal transition. J. Cell. Physiol. 2019, 234, 5775–5785. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, X.; Zhu, Z.; Li, W.; Yu, G.; Jia, Z.; Wang, X. Prostate carcinoma cell-derived exosomal MicroRNA-26a modulates the metastasis and tumor growth of prostate carcinoma. Biomed. Pharmacother. 2019, 117, 109109. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, H.; Wang, J.; Li, L.; Chen, A.; Li, Z. MicroRNA-181d-5p-Containing Exosomes Derived from CAFs Promote EMT by Regulating CDX2/HOXA5 in Breast Cancer. Mol. Ther. Nucleic. Acids 2020, 19, 654–667. [Google Scholar] [CrossRef]
- Deng, T.; Zhang, H.; Yang, H.; Wang, H.; Bai, M.; Sun, W.; Wang, X.; Si, Y.; Ning, T.; Zhang, L.; et al. Exosome miR-155 Derived from Gastric Carcinoma Promotes Angiogenesis by Targeting the c-MYB/VEGF Axis of Endothelial Cells. Mol. Ther. Nucleic. Acids 2020, 19, 1449–1459. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Bai, M.; Li, J.; Yang, H.; Zhang, H.; Zhou, Z.; Deng, T.; Zhu, K.; Ning, T.; Fan, Q.; Ying, G.; et al. miR-135b Delivered by Gastric Tumor Exosomes Inhibits FOXO1 Expression in Endothelial Cells and Promotes Angiogenesis. Mol. Ther. 2019, 27, 1772–1783. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, H.; Ge, S.; Ning, T.; Bai, M.; Li, J.; Li, S.; Sun, W.; Deng, T.; Zhang, L.; et al. Exosome-Derived miR-130a Activates Angiogenesis in Gastric Cancer by Targeting C-MYB in Vascular Endothelial Cells. Mol. Ther. 2018, 26, 2466–2475. [Google Scholar] [CrossRef]
- Fang, J.H.; Zhang, Z.J.; Shang, L.R.; Luo, Y.W.; Lin, Y.F.; Yuan, Y.; Zhuang, S.M. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology 2018, 68, 1459–1475. [Google Scholar] [CrossRef]
- Lu, J.; Liu, Q.H.; Wang, F.; Tan, J.J.; Deng, Y.Q.; Peng, X.H.; Liu, X.; Zhang, B.; Xu, X.; Li, X.P. Exosomal miR-9 inhibits angiogenesis by targeting MDK and regulating PDK/AKT pathway in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 147. [Google Scholar] [CrossRef] [PubMed]
- Pakravan, K.; Babashah, S.; Sadeghizadeh, M.; Mowla, S.J.; Mossahebi-Mohammadi, M.; Ataei, F.; Dana, N.; Javan, M. MicroRNA-100 shuttled by mesenchymal stem cell-derived exosomes suppresses in vitro angiogenesis through modulating the mTOR/HIF-1alpha/VEGF signaling axis in breast cancer cells. Cell Oncol. 2017, 40, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bai, M.; Deng, T.; Liu, R.; Wang, X.; Qu, Y.; Duan, J.; Zhang, L.; Ning, T.; Ge, S.; et al. Cell-derived microvesicles mediate the delivery of miR-29a/c to suppress angiogenesis in gastric carcinoma. Cancer Lett. 2016, 375, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-kappaB pathways. Oncogene 2020, 39, 428–442. [Google Scholar] [CrossRef]
- Vignard, V.; Labbe, M.; Marec, N.; Andre-Gregoire, G.; Jouand, N.; Fonteneau, J.F.; Labarriere, N.; Fradin, D. MicroRNAs in Tumor Exosomes Drive Immune Escape in Melanoma. Cancer Immunol. Res. 2020, 8, 255–267. [Google Scholar] [CrossRef]
- Liu, J.; Fan, L.; Yu, H.; Zhang, J.; He, Y.; Feng, D.; Wang, F.; Li, X.; Liu, Q.; Li, Y.; et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology 2019, 70, 241–258. [Google Scholar] [CrossRef]
- Casadei, L.; Calore, F.; Creighton, C.J.; Guescini, M.; Batte, K.; Iwenofu, O.H.; Zewdu, A.; Braggio, D.A.; Bill, K.L.; Fadda, P.; et al. Exosome-Derived miR-25-3p and miR-92a-3p Stimulate Liposarcoma Progression. Cancer Res. 2017, 77, 3846–3856. [Google Scholar] [CrossRef]
- Ye, S.B.; Zhang, H.; Cai, T.T.; Liu, Y.N.; Ni, J.J.; He, J.; Peng, J.Y.; Chen, Q.Y.; Mo, H.Y.; Jun, C.; et al. Exosomal miR-24-3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J. Pathol. 2016, 240, 329–340. [Google Scholar] [CrossRef]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Zhou, Z.J.; Dai, Z.; Zhou, S.L.; Hu, Z.Q.; Chen, Q.; Zhao, Y.M.; Shi, Y.H.; Gao, Q.; Wu, W.Z.; Qiu, S.J.; et al. HNRNPAB induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by transcriptionally activating SNAIL. Cancer Res. 2014, 74, 2750–2762. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Poon, R.T.; Yuen, A.P.; Ling, M.T.; Kwok, W.K.; Wang, X.H.; Wong, Y.C.; Guan, X.Y.; Man, K.; Chau, K.L.; et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin. Cancer Res. 2006, 12, 5369–5376. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kgamma to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef]
- Ota, Y.; Takahashi, K.; Otake, S.; Tamaki, Y.; Okada, M.; Aso, K.; Makino, Y.; Fujii, S.; Ota, T.; Haneda, M. Extracellular vesicle-encapsulated miR-30e suppresses cholangiocarcinoma cell invasion and migration via inhibiting epithelial-mesenchymal transition. Oncotarget 2018, 9, 16400–16417. [Google Scholar] [CrossRef]
- Josson, S.; Gururajan, M.; Sung, S.Y.; Hu, P.; Shao, C.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; Li, Q.; et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 2015, 34, 2690–2699. [Google Scholar] [CrossRef]
- Li, J.; Yuan, H.; Xu, H.; Zhao, H.; Xiong, N. Hypoxic Cancer-Secreted Exosomal miR-182-5p Promotes Glioblastoma Angiogenesis by Targeting Kruppel-like Factor 2 and 4. Mol. Cancer Res. 2020, 18, 1218–1231. [Google Scholar] [CrossRef]
- Wang, Z.F.; Liao, F.; Wu, H.; Dai, J. Glioma stem cells-derived exosomal miR-26a promotes angiogenesis of microvessel endothelial cells in glioma. J. Exp. Clin. Cancer Res. 2019, 38, 201. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Liang, Y.; Li, J.; Zhao, J.M.; Wang, Z.N.; Lin, X.Y. Gastric Cancer Cell-Derived Exosomal microRNA-23a Promotes Angiogenesis by Targeting PTEN. Front Oncol. 2020, 10, 326. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef]
- Challagundla, K.B.; Wise, P.M.; Neviani, P.; Chava, H.; Murtadha, M.; Xu, T.; Kennedy, R.; Ivan, C.; Zhang, X.; Vannini, I.; et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef]
- Sunderland, N.; Skroblin, P.; Barwari, T.; Huntley, R.P.; Lu, R.; Joshi, A.; Lovering, R.C.; Mayr, M. MicroRNA Biomarkers and Platelet Reactivity: The Clot Thickens. Circ. Res. 2017, 120, 418–435. [Google Scholar] [CrossRef]
- Thomou, T.; Mori, M.A.; Dreyfuss, J.M.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 2017, 542, 450–455. [Google Scholar] [CrossRef]
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and other Tools) Resource Glossary. Available online: https://www.ncbi.nlm.nih.gov/books/NBK326791/ (accessed on 14 September 2020).
- Ying, L.; Du, L.; Zou, R.; Shi, L.; Zhang, N.; Jin, J.; Xu, C.; Zhang, F.; Zhu, C.; Wu, J.; et al. Development of a serum miRNA panel for detection of early stage non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 25036–25042. [Google Scholar] [CrossRef]
- Reis, P.P.; Drigo, S.A.; Carvalho, R.F.; Lopez Lapa, R.M.; Felix, T.F.; Patel, D.; Cheng, D.; Pintilie, M.; Liu, G.; Tsao, M.S. Circulating miR-16-5p, miR-92a-3p, and miR-451a in Plasma from Lung Cancer Patients: Potential Application in Early Detection and a Regulatory Role in Tumorigenesis Pathways. Cancers 2020, 12, 2071. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, L.; Lianidou, E.S. Liquid biopsy in ovarian cancer. Adv. Clin. Chem. 2020, 97, 13–71. [Google Scholar] [CrossRef] [PubMed]
- Staicu, C.E.; Predescu, D.V.; Rusu, C.M.; Radu, B.M.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. Role of microRNAs as Clinical Cancer Biomarkers for Ovarian Cancer: A Short Overview. Cells 2020, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, N.; Vernon, M.; Meryet-Figuiere, M.; Lambert, B.; Briand, M.; Louis, M.H.; Krieger, S.; Joly, F.; Lheureux, S.; Blanc-Fournier, C.; et al. Predictive Relevance of Circulating miR-622 in Patients with Newly Diagnosed and Recurrent High-Grade Serous Ovarian Carcinoma. Clin. Chem. 2020, 66, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, A.; Matsuzaki, J.; Yamamoto, Y.; Yoneoka, Y.; Takahashi, K.; Shimizu, H.; Uehara, T.; Ishikawa, M.; Ikeda, S.I.; Sonoda, T.; et al. Integrated extracellular microRNA profiling for ovarian cancer screening. Nat. Commun. 2018, 9, 4319. [Google Scholar] [CrossRef]
- Mello-Grand, M.; Gregnanin, I.; Sacchetto, L.; Ostano, P.; Zitella, A.; Bottoni, G.; Oderda, M.; Marra, G.; Munegato, S.; Pardini, B.; et al. Circulating microRNAs combined with PSA for accurate and non-invasive prostate cancer detection. Carcinogenesis 2019, 40, 246–253. [Google Scholar] [CrossRef]
- McDonald, A.C.; Vira, M.; Shen, J.; Sanda, M.; Raman, J.D.; Liao, J.; Patil, D.; Taioli, E. Circulating microRNAs in plasma as potential biomarkers for the early detection of prostate cancer. Prostate 2018, 78, 411–418. [Google Scholar] [CrossRef]
- Bidarra, D.; Constancio, V.; Barros-Silva, D.; Ramalho-Carvalho, J.; Moreira-Barbosa, C.; Antunes, L.; Mauricio, J.; Oliveira, J.; Henrique, R.; Jeronimo, C. Circulating MicroRNAs as Biomarkers for Prostate Cancer Detection and Metastasis Development Prediction. Front Oncol. 2019, 9, 900. [Google Scholar] [CrossRef]
- Urabe, F.; Matsuzaki, J.; Yamamoto, Y.; Kimura, T.; Hara, T.; Ichikawa, M.; Takizawa, S.; Aoki, Y.; Niida, S.; Sakamoto, H.; et al. Large-scale Circulating microRNA Profiling for the Liquid Biopsy of Prostate Cancer. Clin. Cancer Res. 2019, 25, 3016–3025. [Google Scholar] [CrossRef]
- Shiino, S.; Matsuzaki, J.; Shimomura, A.; Kawauchi, J.; Takizawa, S.; Sakamoto, H.; Aoki, Y.; Yoshida, M.; Tamura, K.; Kato, K.; et al. Serum miRNA-based Prediction of Axillary Lymph Node Metastasis in Breast Cancer. Clin. Cancer Res. 2019, 25, 1817–1827. [Google Scholar] [CrossRef]
- Li, H.; Liu, J.; Chen, J.; Wang, H.; Yang, L.; Chen, F.; Fan, S.; Wang, J.; Shao, B.; Yin, D.; et al. A serum microRNA signature predicts trastuzumab benefit in HER2-positive metastatic breast cancer patients. Nat. Commun. 2018, 9, 1614. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Zhu, S.; Chen, L.; Liu, X.; Wei, R.; Zhao, L.; Yang, Y.; Zhang, Z.; Kong, G.; Li, P.; et al. Evaluation of circulating small extracellular vesicles derived miRNAs as biomarkers of early colon cancer: A comparison with plasma total miRNAs. J. Extracell Vesicles 2019, 8, 1643670. [Google Scholar] [CrossRef] [PubMed]
- Abramovic, I.; Ulamec, M.; Katusic Bojanac, A.; Bulic-Jakus, F.; Jezek, D.; Sincic, N. miRNA in prostate cancer: Challenges toward translation. Epigenomics 2020, 12, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W. Circulating microRNA biomarker studies: Pitfalls and potential solutions. Clin. Chem. 2015, 61, 56–63. [Google Scholar] [CrossRef]
- McDonald, J.S.; Milosevic, D.; Reddi, H.V.; Grebe, S.K.; Algeciras-Schimnich, A. Analysis of circulating microRNA: Preanalytical and analytical challenges. Clin. Chem. 2011, 57, 833–840. [Google Scholar] [CrossRef]
- Rutjes, A.W.; Reitsma, J.B.; Di Nisio, M.; Smidt, N.; van Rijn, J.C.; Bossuyt, P.M. Evidence of bias and variation in diagnostic accuracy studies. CMAJ 2006, 174, 469–476. [Google Scholar] [CrossRef]
- Whiting, P.F.; Rutjes, A.W.; Westwood, M.E.; Mallett, S.; Group, Q.-S. A systematic review classifies sources of bias and variation in diagnostic test accuracy studies. J. Clin. Epidemiol. 2013, 66, 1093–1104. [Google Scholar] [CrossRef]
- Crowley, R.J.; Tan, Y.J.; Ioannidis, J.P.A. Empirical assessment of bias in machine learning diagnostic test accuracy studies. J. Am. Med. Inform. Assoc. 2020, 27, 1092–1101. [Google Scholar] [CrossRef]
- Lijmer, J.G.; Mol, B.W.; Heisterkamp, S.; Bonsel, G.J.; Prins, M.H.; van der Meulen, J.H.; Bossuyt, P.M. Empirical evidence of design-related bias in studies of diagnostic tests. JAMA 1999, 282, 1061–1066. [Google Scholar] [CrossRef]
- Bossuyt, P.M.; Reitsma, J.B.; Bruns, D.E.; Gatsonis, C.A.; Glasziou, P.P.; Irwig, L.; Lijmer, J.G.; Moher, D.; Rennie, D.; de Vet, H.C.; et al. STARD 2015: An updated list of essential items for reporting diagnostic accuracy studies. BMJ 2015, 351, h5527. [Google Scholar] [CrossRef]
- Sauerbrei, W.; Taube, S.E.; McShane, L.M.; Cavenagh, M.M.; Altman, D.G. Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): An Abridged Explanation and Elaboration. J. Natl. Cancer Inst. 2018, 110, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Sounderajah, V.; Ashrafian, H.; Aggarwal, R.; De Fauw, J.; Denniston, A.K.; Greaves, F.; Karthikesalingam, A.; King, D.; Liu, X.; Markar, S.R.; et al. Developing specific reporting guidelines for diagnostic accuracy studies assessing AI interventions: The STARD-AI Steering Group. Nat. Med. 2020, 26, 807–808. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, M.B.; Edelman, J.J.; Kao, S.C.; Vallely, M.P.; van Zandwijk, N.; Reid, G. The Impact of Hemolysis on Cell-Free microRNA Biomarkers. Front Genet. 2013, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.S.; Soon, P.S.; Marsh, D.J. Comparison of Methodologies to Detect Low Levels of Hemolysis in Serum for Accurate Assessment of Serum microRNAs. PLoS ONE 2016, 11, e0153200. [Google Scholar] [CrossRef] [PubMed]
- Blondal, T.; Jensby Nielsen, S.; Baker, A.; Andreasen, D.; Mouritzen, P.; Wrang Teilum, M.; Dahlsveen, I.K. Assessing sample and miRNA profile quality in serum and plasma or other biofluids. Methods 2013, 59, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.C. Phlebotomy site haemolysis rates vary inversely with workload. Clin. Chem. Lab. Med. 2010, 48, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuan, Y.; Cho, J.H.; McClarty, S.; Baxter, D.; Galas, D.J. Comparing the MicroRNA spectrum between serum and plasma. PLoS ONE 2012, 7, e41561. [Google Scholar] [CrossRef]
- Mestdagh, P.; Hartmann, N.; Baeriswyl, L.; Andreasen, D.; Bernard, N.; Chen, C.; Cheo, D.; D’Andrade, P.; DeMayo, M.; Dennis, L.; et al. Evaluation of quantitative miRNA expression platforms in the microRNA quality control (miRQC) study. Nat. Methods 2014, 11, 809–815. [Google Scholar] [CrossRef]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Campomenosi, P.; Gini, E.; Noonan, D.M.; Poli, A.; D’Antona, P.; Rotolo, N.; Dominioni, L.; Imperatori, A. A comparison between quantitative PCR and droplet digital PCR technologies for circulating microRNA quantification in human lung cancer. BMC Biotechnol. 2016, 16, 60. [Google Scholar] [CrossRef]
- Zhang, K.; Kang, D.K.; Ali, M.M.; Liu, L.; Labanieh, L.; Lu, M.; Riazifar, H.; Nguyen, T.N.; Zell, J.A.; Digman, M.A.; et al. Digital quantification of miRNA directly in plasma using integrated comprehensive droplet digital detection. Lab Chip 2015, 15, 4217–4226. [Google Scholar] [CrossRef] [PubMed]
- Marabita, F.; de Candia, P.; Torri, A.; Tegner, J.; Abrignani, S.; Rossi, R.L. Normalization of circulating microRNA expression data obtained by quantitative real-time RT-PCR. Brief Bioinform. 2016, 17, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Benz, F.; Roderburg, C.; Vargas Cardenas, D.; Vucur, M.; Gautheron, J.; Koch, A.; Zimmermann, H.; Janssen, J.; Nieuwenhuijsen, L.; Luedde, M.; et al. U6 is unsuitable for normalization of serum miRNA levels in patients with sepsis or liver fibrosis. Exp. Mol. Med. 2013, 45, e42. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; da Silva, A.M.; Calin, G.; Pantel, K. Data Normalization Strategies for MicroRNA Quantification. Clin. Chem. 2015, 61, 1333–1342. [Google Scholar] [CrossRef]
- Peltier, H.J.; Latham, G.J. Normalization of microRNA expression levels in quantitative RT-PCR assays: Identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 2008, 14, 844–852. [Google Scholar] [CrossRef]
- Mestdagh, P.; Van Vlierberghe, P.; De Weer, A.; Muth, D.; Westermann, F.; Speleman, F.; Vandesompele, J. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 2009, 10, R64. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| miRNAs | Donor Cell | Recipient Cell | Molecular Targets | Function | Tumor Type | Ref |
|---|---|---|---|---|---|---|
| miR-1247-3p | High-metastatic hepatocellular carcinoma cells (HCC) | Fibroblasts | B4GALT3, β1-integrin-NF-κB signaling | Induce the transformation of fibroblasts to CAFs. CAFs promote stemness, EMT, motility, and chemoresistance in recipient HCCs. | Liver cancer | Fang et al. [144] |
| miR-301a-3p | Hypoxic pancreatic cancer cells (PCC) | Macrophages | PTEN/PI3kγ signaling | Induce the M2 polarization of macrophages, which promotes the migration, invasion, and EMT of recipient PCCs. | Pancreatic cancer | Wang et al. [159] |
| miR-26a | Low-grade prostate carcinoma cell line LNCAP | Metastatic castration-resistant prostate carcinoma cell line PC-3 | ND | Inhibit the cell proliferation, migration, invasion, and EMT of PC-3. | Prostate cancer | Wang et al. [160] |
| miR-30e | Early-stage cholangiocarcinoma (CCA) cells | CCAs | Snail | Suppress EMT, cell invasion, and migration in recipient CCA. | Cholangiocarcinoma | Ota et al. [161] |
| miR-193a-3p, miR-210-3p, and miR-5100 | Bone-marrow-derived mesenchymal stem cells (BMSCs) | Lung cancer cells | STAT3 | Induce EMT and metastasis. | Lung cancer | Zhang et al. [112] |
| miR-181d-5p | CAFs | MCF-7 breast cancer cells | CDX2 and HOXA5 | Increased proliferation, invasion, and expression of EMT markers. | Breast cancer | Wang et al. [162] |
| miR-92a-3p | CAFs | Colorectal cancer cells (CRC) | FBXW7 and MOAP1 | Promote stemness, EMT, metastasis, and 5-FU/L-OHP resistance. | Colorectal cancer | Hu et al. [163] |
| miR-409 | Fibroblast | Prostate cancer cells | RSU1 and STAG2 | Induce cell proliferation and EMT. | Prostate cancer | Josson et al. [164] |
| miR-182-5p | Hypoxic glioblastoma cancer cells | Umbilical vascular endothelial cells (HUVEC) | KLF2 and KLF4 | Promote angiogenesis and inhibit tight-junction-related proteins. | Glioblastoma cancer | Li et al. [165] |
| miR-155 | SGC-7901 gastric cancer cells | HUVEC | c-MYB/VEGF | Promote angiogenesis. | Gastric cancer | Deng et al. [166] |
| miR-23a | HGC-27 gastric cancer cells | HUVEC | PTEN and TSP-1 | Promote angiogenesis. | Gastric cancer | Du et al. [167] |
| miR-135b | SGC7-901 gastric cancer celsl | HUVEC | FOXO1 | Promote angiogenesis. | Gastric cancer | Bai et al. [168] |
| miR-130a | SGC-7901 gastric cancer cells | HUVEC | c-MYB | Increase cell migration, proliferation, and ring formation in HUVEC. | Gastric cancer | Yang et al. [169] |
| miR-25-3p | SW480 CRC cells | HUVEC | KLF4 and KLF2 | Disrupt the integrity of junctions in epithelial cells. | Colorectal cancer | Zheng et al. [146] |
| mir-103 | QGY-7703 and HepG2 hepatocarcinoma cell lines | Endothelial cells | VE-Cad, p120, and zonula occludens 1. | Increase vascular permeability by abrogating junction integrity and promoting tumor metastasis. | Liver cancer | Fang et al. [170] |
| miR-9 | Nasopharyngeal cancer cells | HUVEC | MDK and PDK/AKT signaling | Inhibit angiogenesis. | Nasopharyngeal carcinoma | Lu et al. [171] |
| miR-100 | Mesenchymal stem cells (MSCs) | Breast cancer cells | mTOR/HIF-1a/VEGF | Decrease the proliferation, migration, and tube formation of HUVEC. | Breast cancer | Pakravan et al. [172] |
| miR-29a and miR-29c | SGC-7901 gastric cancer cells | Endothelial cells | VEGF | Decrease the proliferation, migration, and tube formation of endothelial cells. | Gastric cancer | Zhang et al. [173] |
| miR-1246 | Hypoxic glioma cells | Macrophages | STAT3 and NF-κB signaling | Induce M2 macrophages, which promote the glioma proliferation, migration, and invasion of glioma cells. | Glioma | Qian et al. [174] |
| miR-181a/b, miR-498 | Melanoma cells | CD8+ T cells | PTPRC (CD45) | Decrease TCR signaling, TNFα, and granzyme B secretion. | Melanoma | Vignard et al. [175] |
| miR-23a-3p | HCC cells under endoplasmic reticulum stress | Macrophages | PTEN, AKT, PD-L1 | Upregulation of the immunosuppressor molecule PD-L1. | Liver cancer | Liu et al. [176] |
| miR-24-3p miR-92a-3p | Liposarcoma cells (LSC) | Tumor-associated macrophages (TAMs) | TLR7/8, NF-κB pathway | Secretion of pro-inflammatory cytokine IL-6 by TAMs, which promote the proliferation, migration, and invasion capacity of LSCs. | Liposarcoma | Casadei et al. [177] |
| miR-24-3p | NPC cells | T cells | FGF11 | Inhibit the proliferation of T cells, inhibit the differentiation of Th1 and Th17 cells, and induce the differentiation of Treg cells. | Nasopharyngeal carcinoma | Ye et al. [178] |
| miR-29a-3p miR-21-5p | TAMs | CD4+ T cells | STAT3 | Treg/Th17 imbalance. | Epithelial ovarian cancer | Zhou et al. [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Quintero, B. Extracellular MicroRNAs as Intercellular Mediators and Noninvasive Biomarkers of Cancer. Cancers 2020, 12, 3455. https://doi.org/10.3390/cancers12113455
Ortiz-Quintero B. Extracellular MicroRNAs as Intercellular Mediators and Noninvasive Biomarkers of Cancer. Cancers. 2020; 12(11):3455. https://doi.org/10.3390/cancers12113455
Chicago/Turabian StyleOrtiz-Quintero, Blanca. 2020. "Extracellular MicroRNAs as Intercellular Mediators and Noninvasive Biomarkers of Cancer" Cancers 12, no. 11: 3455. https://doi.org/10.3390/cancers12113455
APA StyleOrtiz-Quintero, B. (2020). Extracellular MicroRNAs as Intercellular Mediators and Noninvasive Biomarkers of Cancer. Cancers, 12(11), 3455. https://doi.org/10.3390/cancers12113455