Serial Xenotransplantation in NSG Mice Promotes a Hybrid Epithelial/Mesenchymal Gene Expression Signature and Stemness in Rhabdomyosarcoma Cells

 , ,
, , 
Abstract
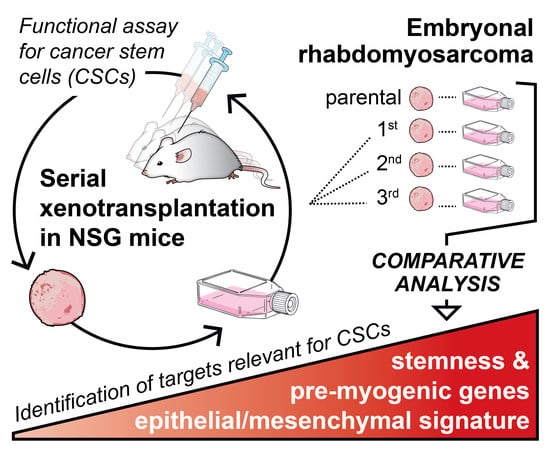
1. Introduction
2. Results
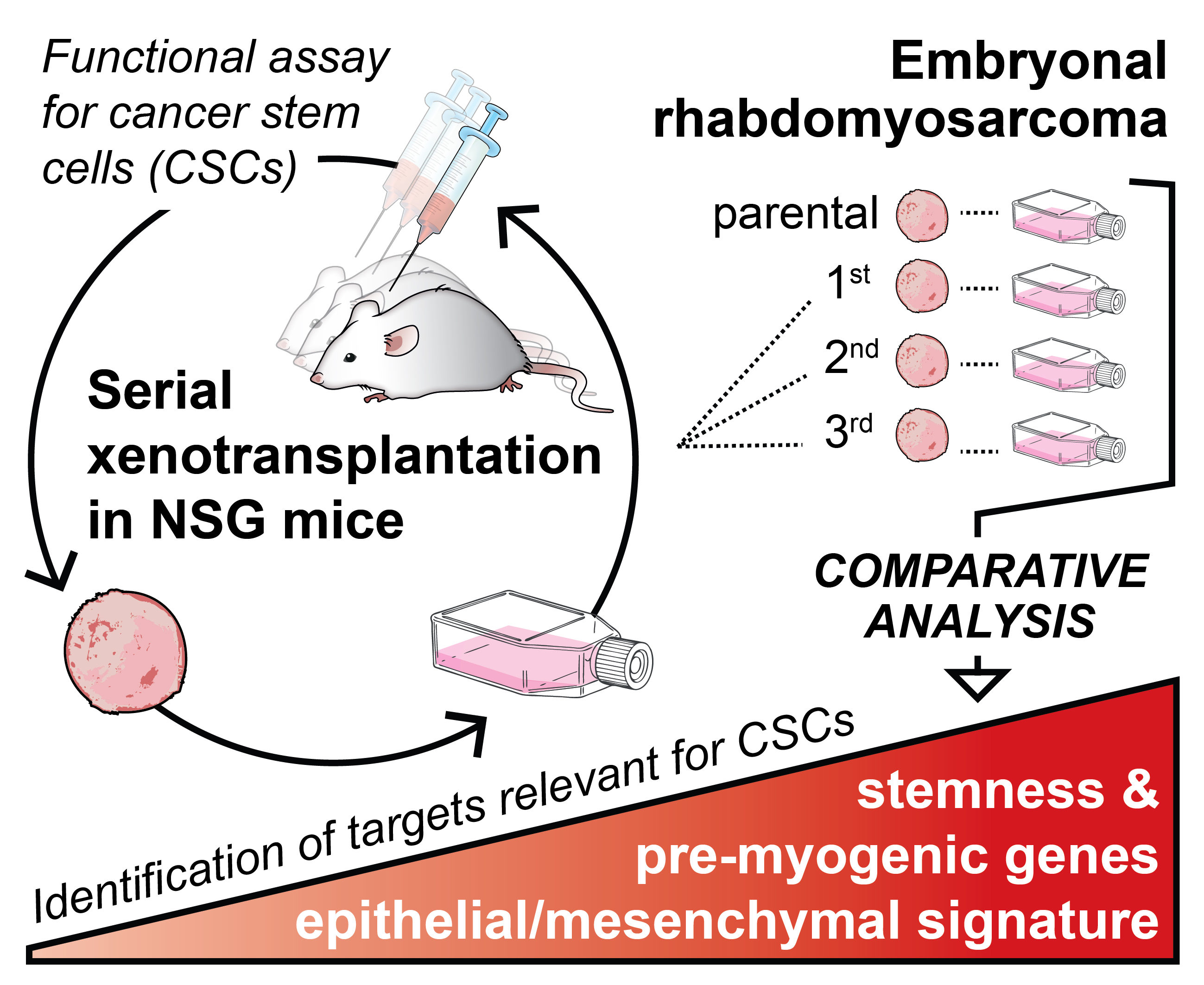
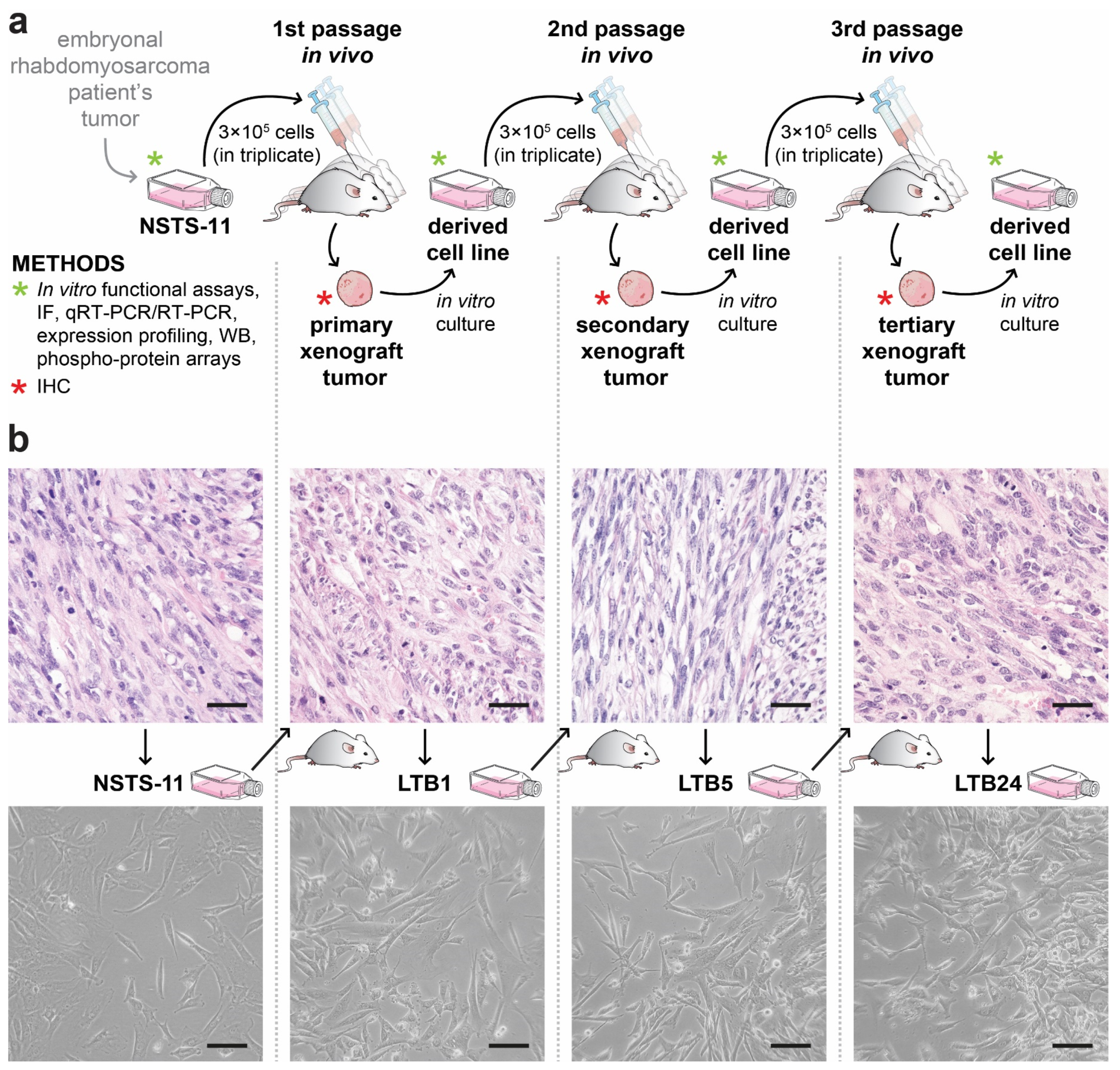
2.1. Tumorigenicity of NSTS-11 Rhabdomyosarcoma Cells Is Maintained during Serial Xenotransplantation in NSG Mice
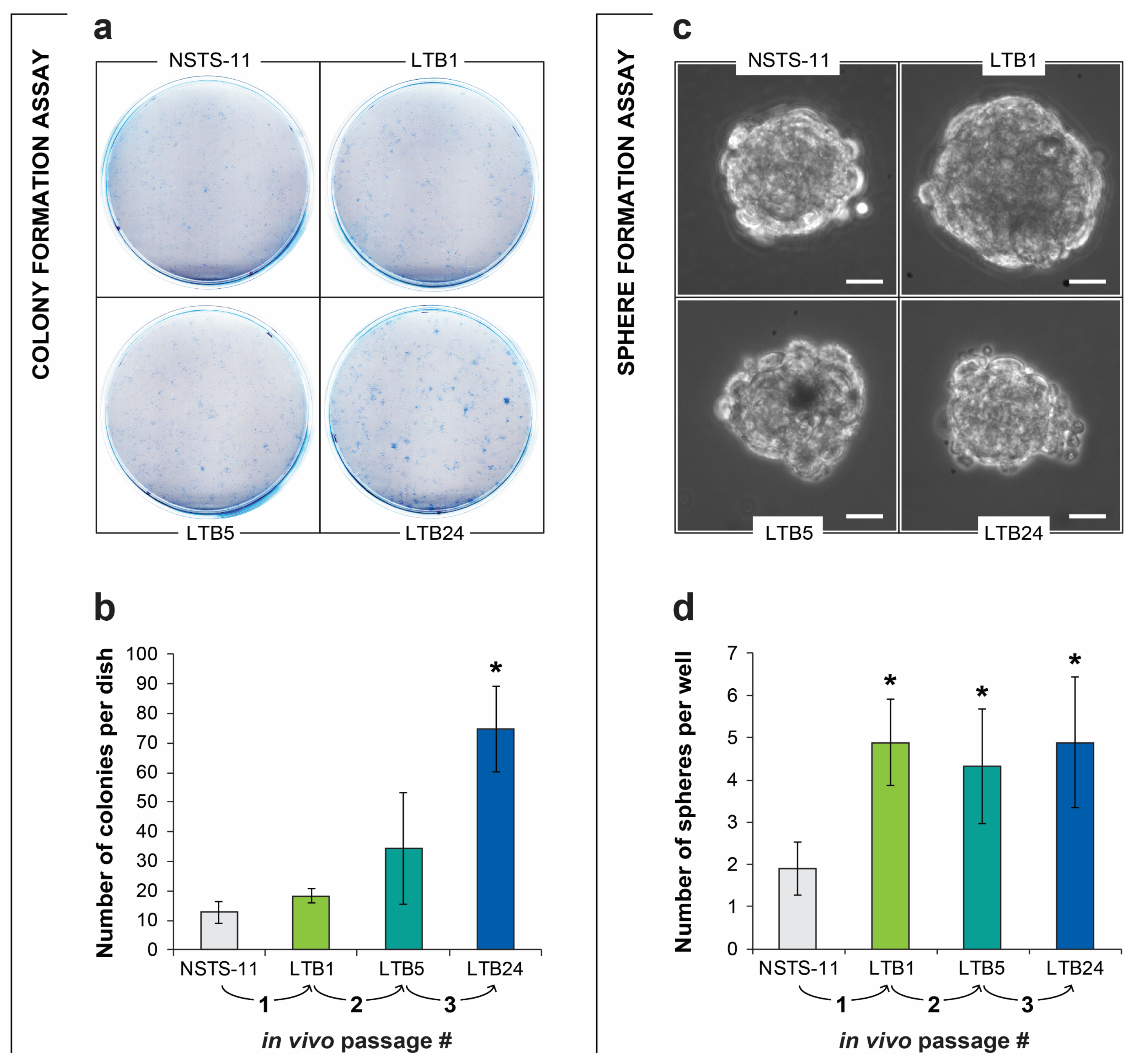
2.2. The Colony Formation and Sphere Formation Capacity of NSTS-11 Cells Is Enhanced after In Vivo Passages
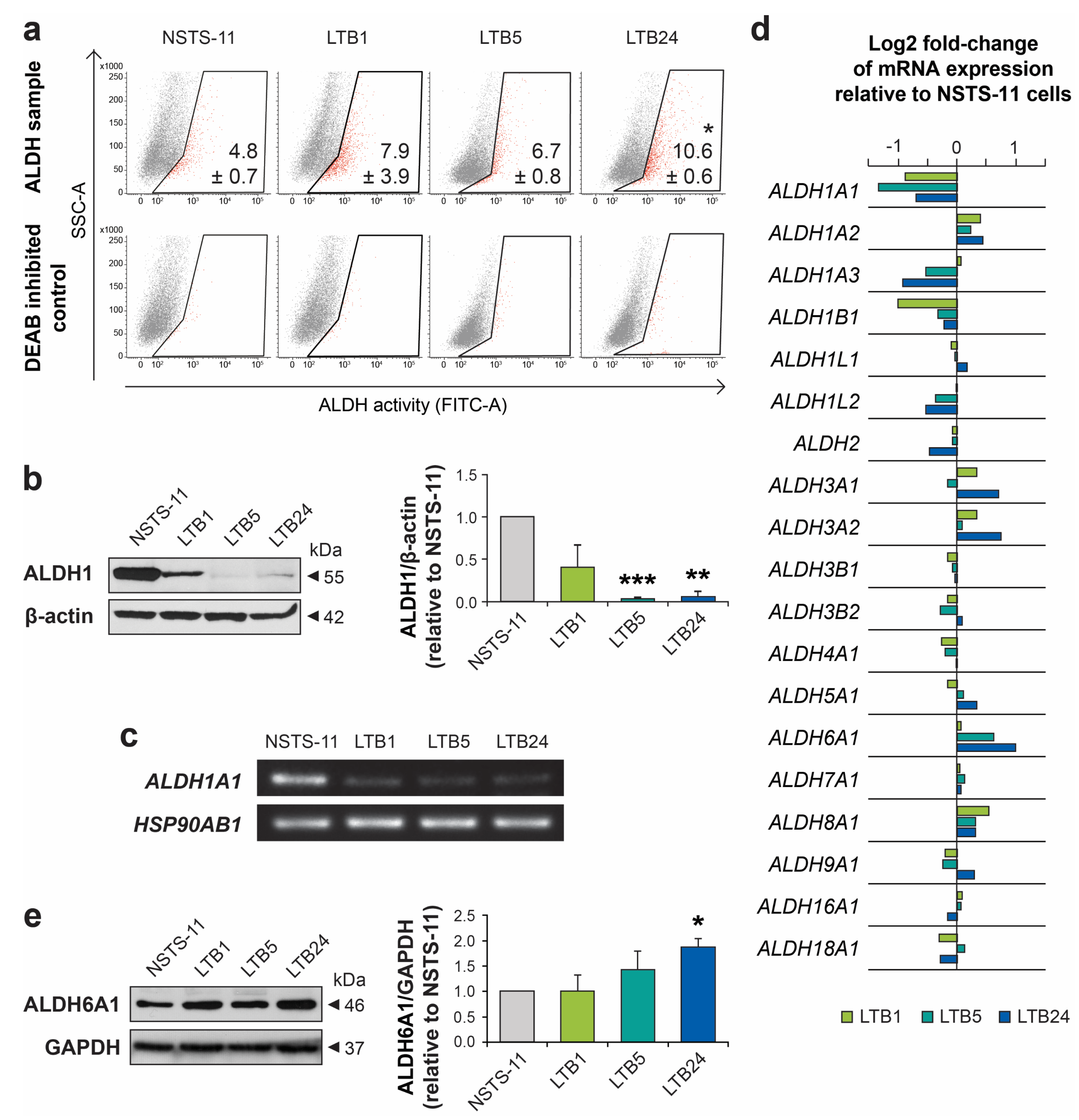
2.3. Serial Xenotransplantation in NSG Mice Increases Aldehyde Dehydrogenase Activity In Vitro
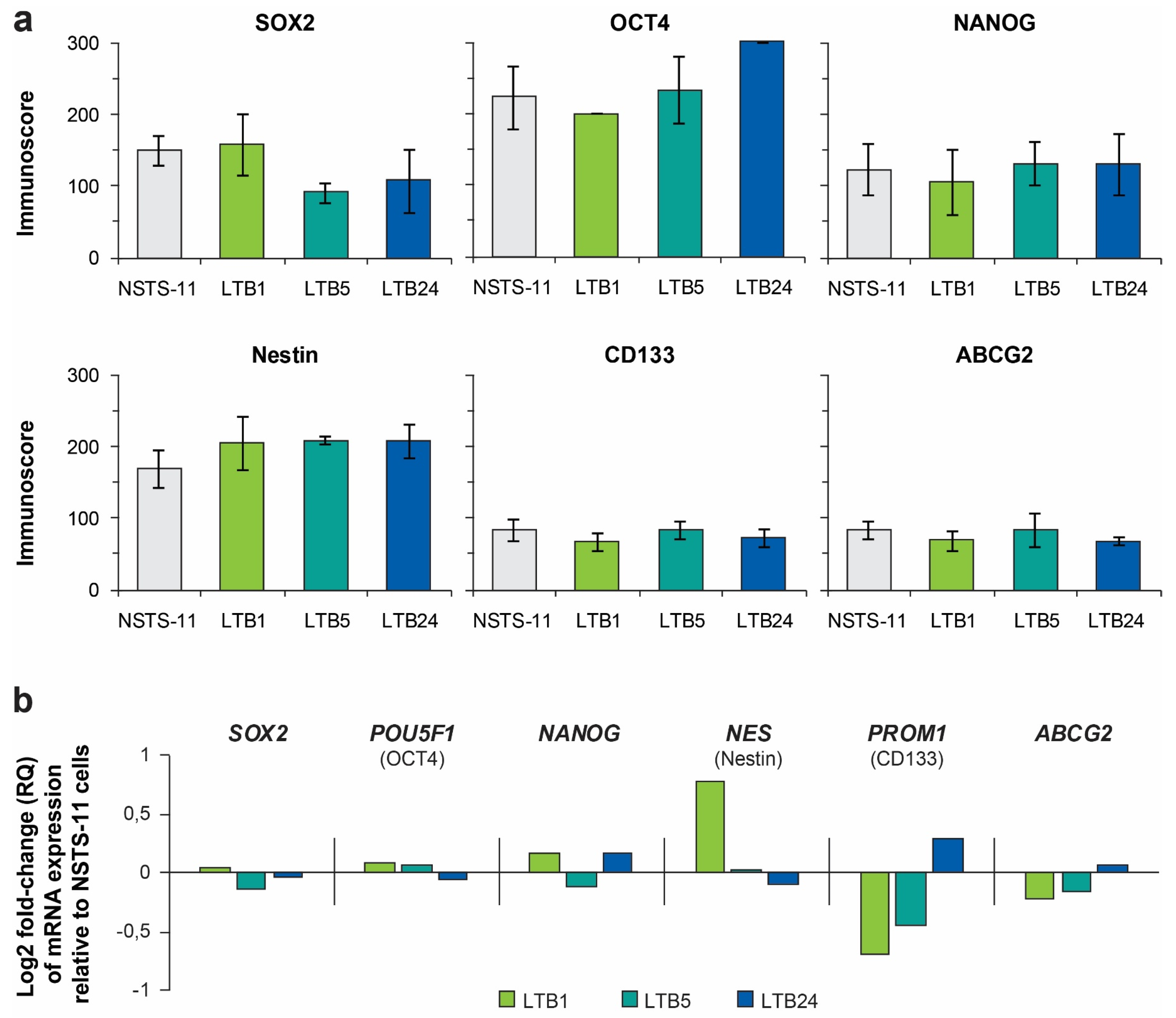
2.4. Expression Levels of Core Pluripotency Factors and Common CSC Markers Are Preserved Over the Passages In Vivo
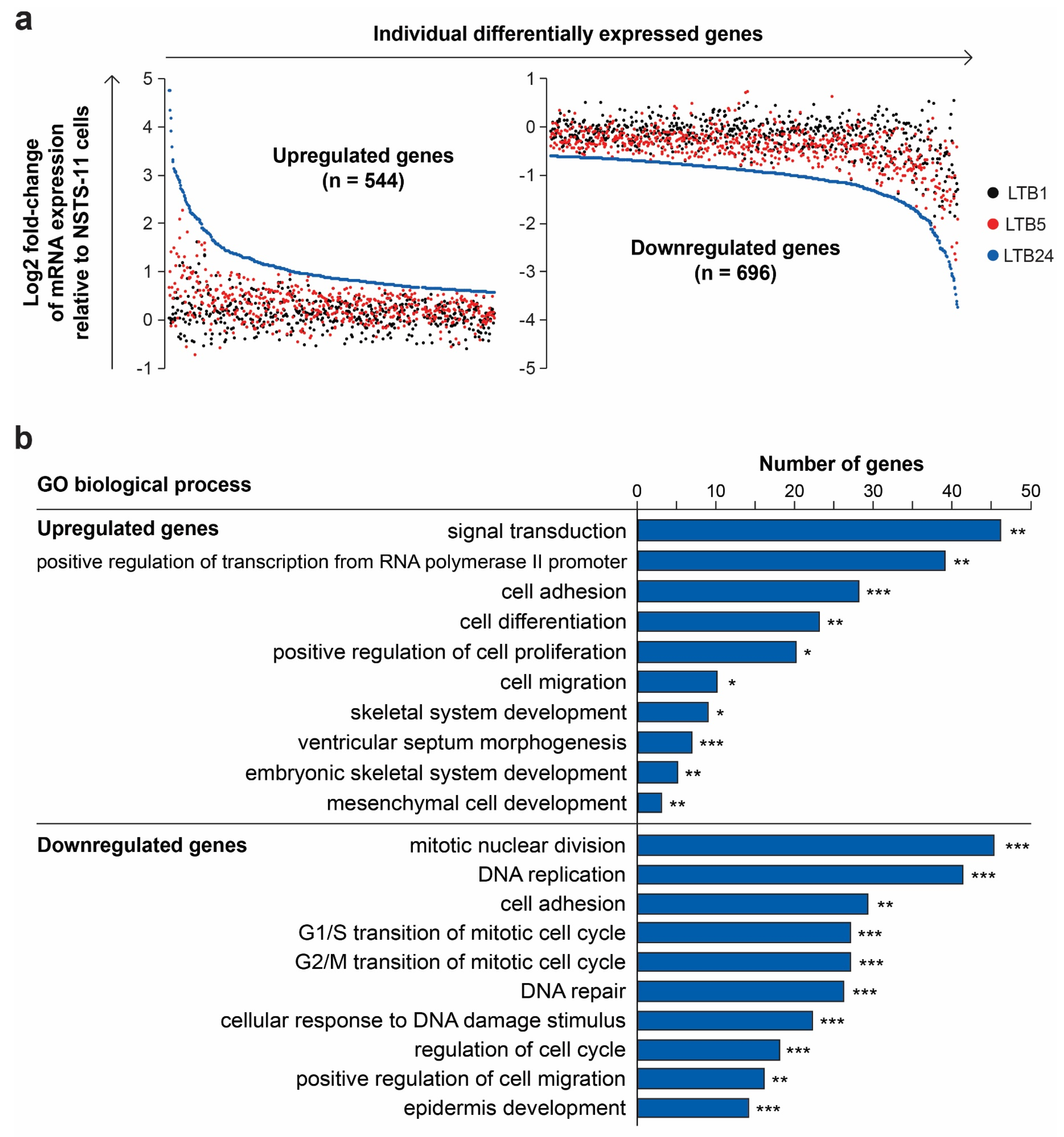
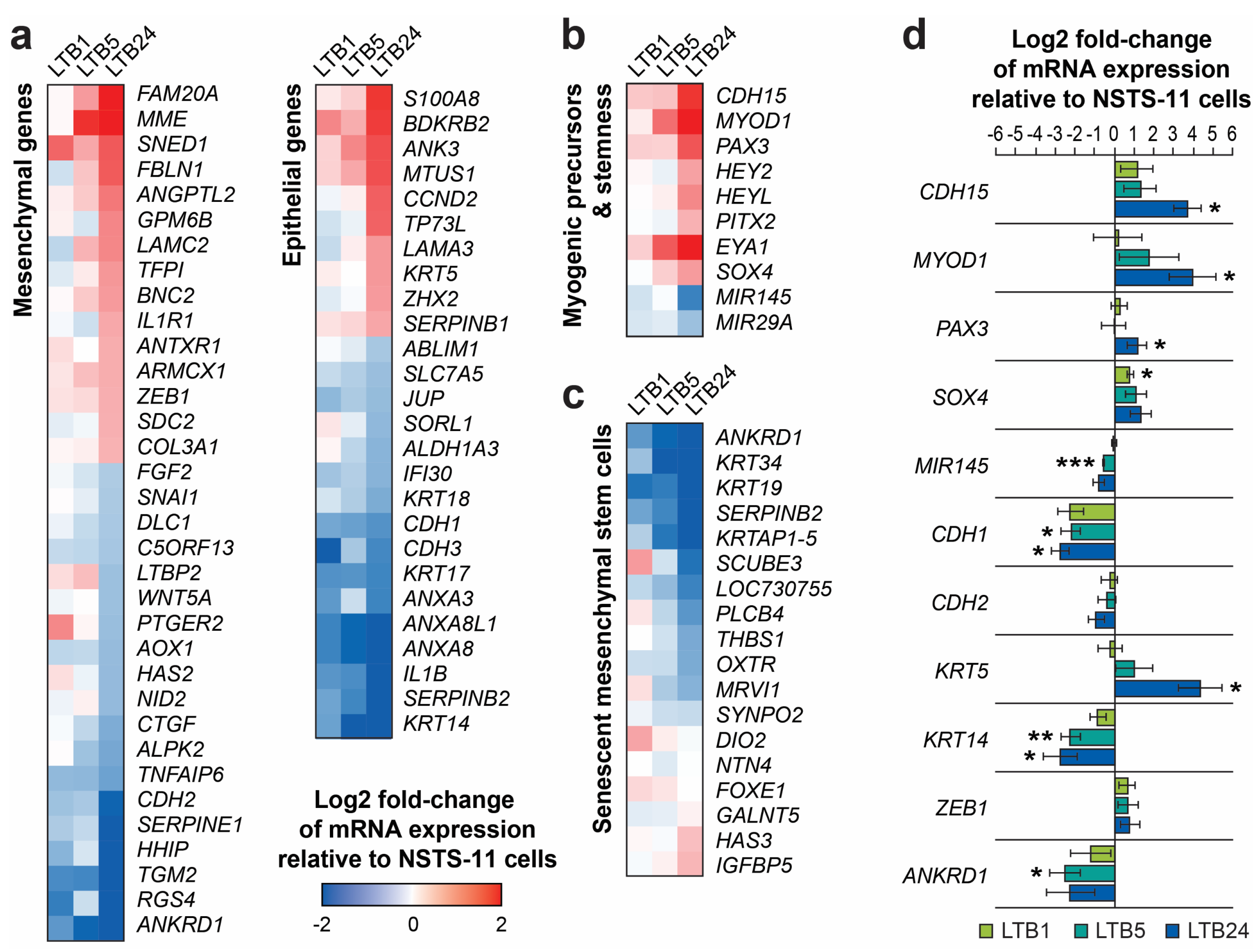
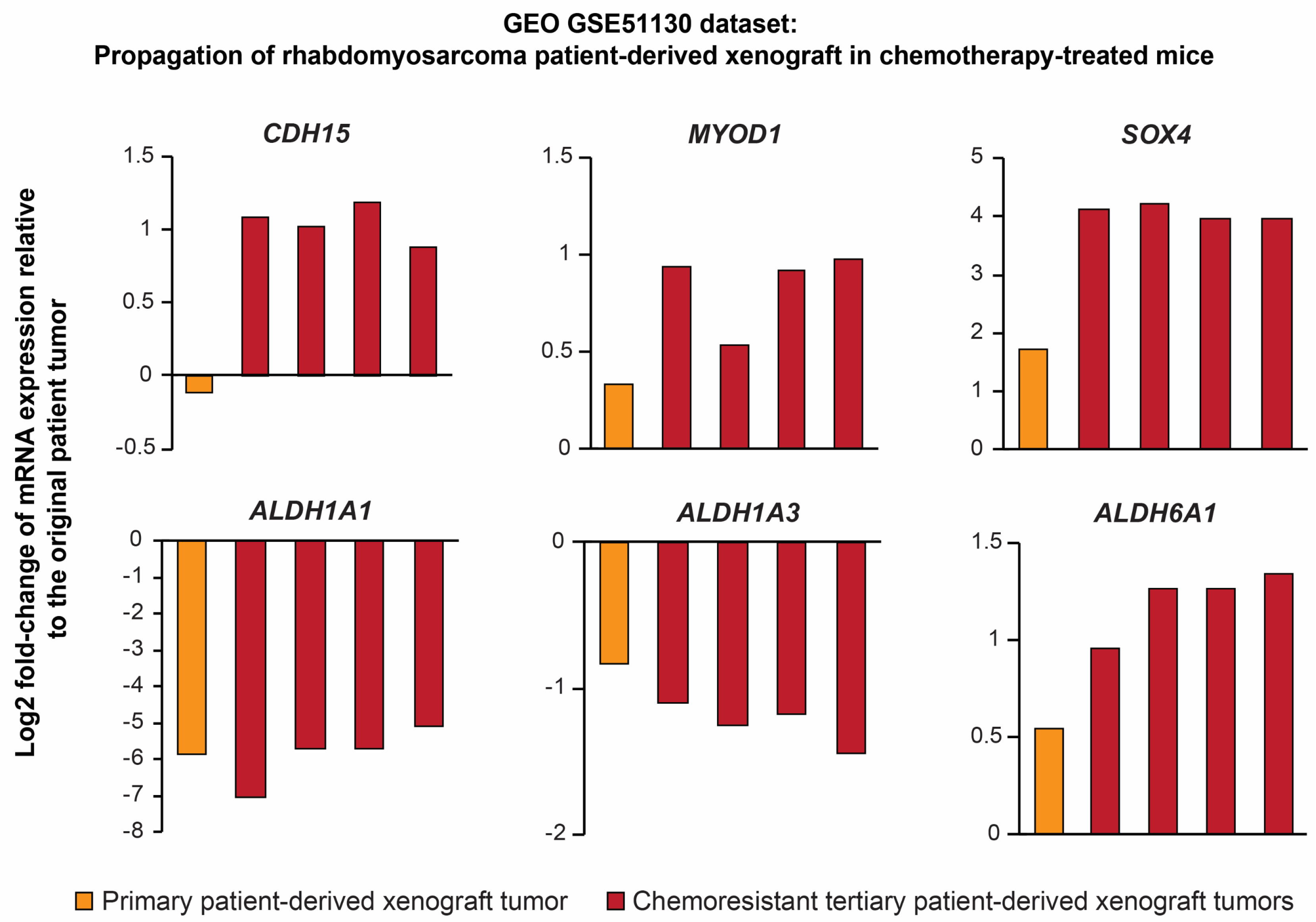
2.5. Serial Xenotransplantation of Rhabdomyosarcoma Cells Promotes an Expression Profile Associated with Muscle Progenitor Cells and a Hybrid Epithelial/Mesenchymal Phenotype
2.6. Serial Passaging In Vivo Enhanced the Phosphorylation of the Wnt Coreceptor RYK
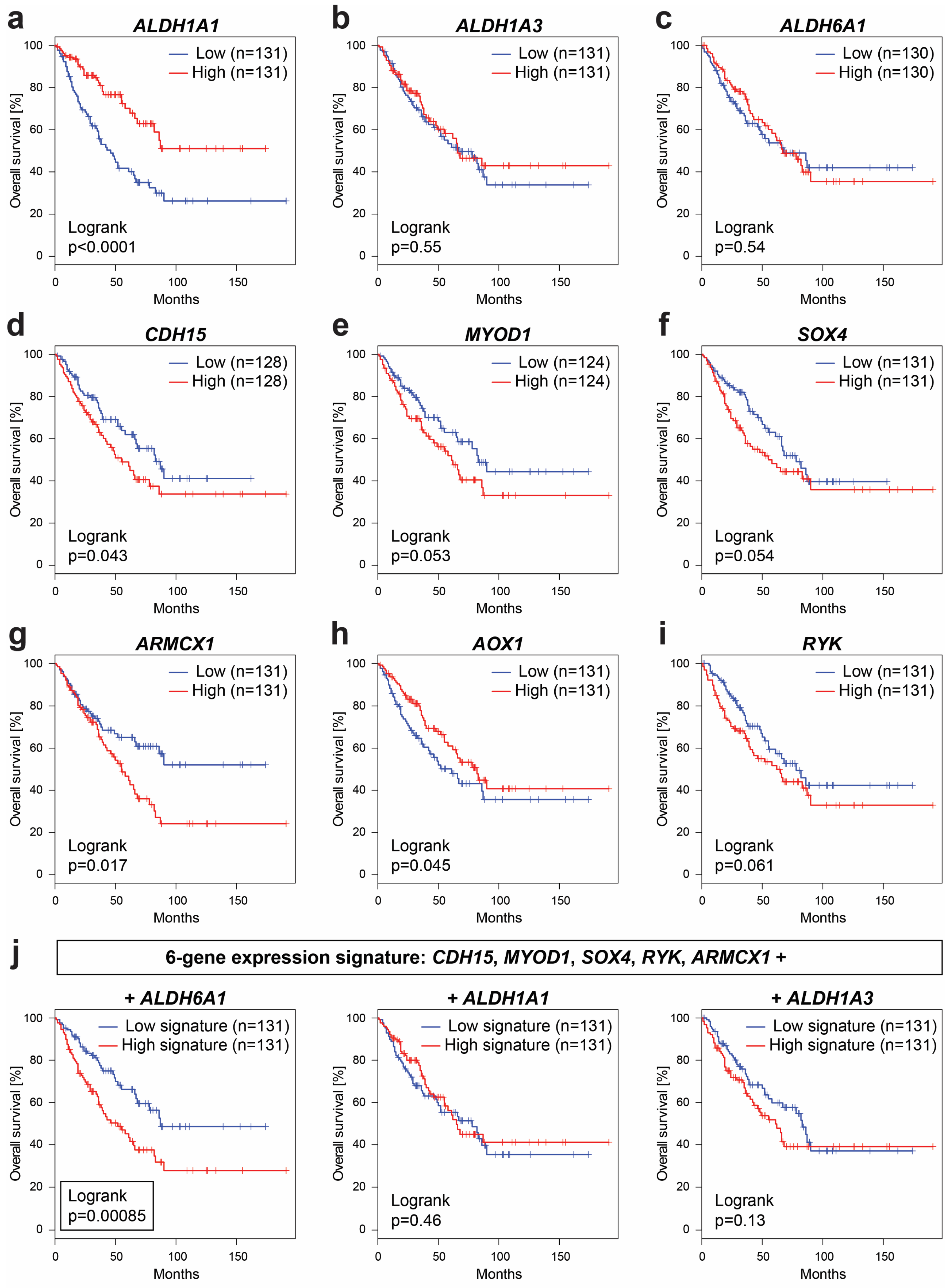
2.7. Expression of Several Identified Prospective CSC-Specific Target Genes Predicts Survival in Soft-Tissue Sarcomas
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Tumor Samples
4.2. In Vivo Tumorigenicity Assay
4.3. Colony Formation Assay
4.4. Sphere Formation Assay
4.5. Aldefluor Assay
4.6. Immunohistochemistry
4.7. Immunofluorescence
4.8. Western Blotting
4.9. RT-PCR and qRT-PCR
4.10. Expression Profiling
4.11. Phospho-Protein Arrays
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, L.; Takimoto, T.; Fujimoto, J. Prognostic model for predicting overall survival in children and adolescents with rhabdomyosarcoma. BMC Cancer 2014, 14, 654. [Google Scholar] [CrossRef]
- Winter, S.; Fasola, S.; Brisse, H.; Mosseri, V.; Orbach, D. Relapse after localized rhabdomyosarcoma: Evaluation of the efficacy of second-line chemotherapy. Pediatr. Blood Cancer 2015, 62, 1935–1941. [Google Scholar] [CrossRef]
- Sana, J.; Zambo, I.; Skoda, J.; Neradil, J.; Chlapek, P.; Hermanova, M.; Mudry, P.; Vasikova, A.; Zitterbart, K.; Hampl, A.; et al. CD133 expression and identification of CD133/nestin positive cells in rhabdomyosarcomas and rhabdomyosarcoma cell lines. Anal. Cell. Pathol. 2011, 34, 303–318. [Google Scholar] [CrossRef][Green Version]
- Walter, D.; Satheesha, S.; Albrecht, P.; Bornhauser, B.C.; D’Alessandro, V.; Oesch, S.M.; Rehrauer, H.; Leuschner, I.; Koscielniak, E.; Gengler, C.; et al. CD133 positive embryonal rhabdomyosarcoma stem-like cell population is enriched in rhabdospheres. PLoS ONE 2011, 6, e19506. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Nunukova, A.; Loja, T.; Zambo, I.; Neradil, J.; Mudry, P.; Zitterbart, K.; Hermanova, M.; Hampl, A.; Sterba, J.; et al. Cancer stem cell markers in pediatric sarcomas: Sox2 is associated with tumorigenicity in immunodeficient mice. Tumor Biol. 2016, 37, 9535–9548. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef]
- Skoda, J.; Veselska, R. Cancer stem cells in sarcomas: Getting to the stemness core. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2134–2139. [Google Scholar] [CrossRef]
- Nunukova, A.; Neradil, J.; Skoda, J.; Jaros, J.; Hampl, A.; Sterba, J.; Veselska, R. Atypical nuclear localization of CD133 plasma membrane glycoprotein in rhabdomyosarcoma cell lines. Int. J. Mol. Med. 2015, 36, 65–72. [Google Scholar] [CrossRef]
- Skoda, J.; Hermanova, M.; Loja, T.; Nemec, P.; Neradil, J.; Karasek, P.; Veselska, R. Co-Expression of Cancer Stem Cell Markers Corresponds to a Pro-Tumorigenic Expression Profile in Pancreatic Adenocarcinoma. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Duex, J.E.; Owens, C.; Chauca-Diaz, A.; Dancik, G.M.; Vanderlinden, L.A.; Ghosh, D.; Leivo, M.Z.; Hansel, D.E.; Theodorescu, D. Nuclear CD24 Drives Tumor Growth and Is Predictive of Poor Patient Prognosis. Cancer Res. 2017, 77, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Agliano, A.; Calvo, A.; Box, C. The challenge of targeting cancer stem cells to halt metastasis. Semin. Cancer Biol. 2017, 44, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.M.; Gumuzio, J.; Martin, A.G. Linking Pluripotency Reprogramming and Cancer. Stem Cells Transl. Med. 2017, 6, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Ying, M.; Liu, G.; Shimada, H.; Ding, W.; May, W.A.; He, Q.; Adams, G.B.; Wu, L. Human osteosarcoma CD49f−CD133+ cells: Impaired in osteogenic fate while gain of tumorigenicity. Oncogene 2013, 32, 4252–4263. [Google Scholar] [CrossRef][Green Version]
- Golan, H.; Shukrun, R.; Caspi, R.; Vax, E.; Pode-Shakked, N.; Goldberg, S.; Pleniceanu, O.; Bar-Lev, D.D.; Mark-Danieli, M.; Pri-Chen, S.; et al. In Vivo Expansion of Cancer Stemness Affords Novel Cancer Stem Cell Targets: Malignant Rhabdoid Tumor as an Example. Stem Cell Rep. 2018, 11, 795–810. [Google Scholar] [CrossRef]
- Nakahata, K.; Uehara, S.; Nishikawa, S.; Kawatsu, M.; Zenitani, M.; Oue, T.; Okuyama, H. Aldehyde Dehydrogenase 1 (ALDH1) Is a Potential Marker for Cancer Stem Cells in Embryonal Rhabdomyosarcoma. PLoS ONE 2015, 10, e0125454. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef]
- Groger, C.J.; Grubinger, M.; Waldhor, T.; Vierlinger, K.; Mikulits, W. Meta-analysis of gene expression signatures defining the epithelial to mesenchymal transition during cancer progression. PLoS ONE 2012, 7, e51136. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, M.; Schafer, B.W. Unpeaceful roles of mutant PAX proteins in cancer. Semin. Cell Dev. Biol. 2015, 44, 126–134. [Google Scholar] [CrossRef]
- Buckingham, M. Gene regulatory networks and cell lineages that underlie the formation of skeletal muscle. Proc. Natl. Acad. Sci. USA 2017, 114, 5830–5837. [Google Scholar] [CrossRef]
- Xu, N.; Papagiannakopoulos, T.; Pan, G.; Thomson, J.A.; Kosik, K.S. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 2009, 137, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Medeiros Tavares Marques, J.C.; Cornelio, D.A.; Nogueira Silbiger, V.; Ducati Luchessi, A.; de Souza, S.; Batistuzzo de Medeiros, S.R. Identification of new genes associated to senescent and tumorigenic phenotypes in mesenchymal stem cells. Sci. Rep. 2017, 7, 17837. [Google Scholar] [CrossRef] [PubMed]
- Katso, R.M.; Russell, R.B.; Ganesan, T.S. Functional analysis of H-Ryk, an atypical member of the receptor tyrosine kinase family. Mol. Cell. Biol. 1999, 19, 6427–6440. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.P.; Halford, M.M.; Stacker, S.A. The biochemistry, signalling and disease relevance of RYK and other WNT-binding receptor tyrosine kinases. Growth Factors 2018, 36, 15–40. [Google Scholar] [CrossRef]
- Cheyette, B.N. Ryk: Another heretical Wnt receptor defies the canon. Sci. STKE 2004, 2004, 54. [Google Scholar] [CrossRef]
- Grunberg, J.R.; Hoffmann, J.M.; Hedjazifar, S.; Nerstedt, A.; Jenndahl, L.; Elvin, J.; Castellot, J.; Wei, L.; Moverare-Skrtic, S.; Ohlsson, C.; et al. Overexpressing the novel autocrine/endocrine adipokine WISP2 induces hyperplasia of the heart, white and brown adipose tissues and prevents insulin resistance. Sci. Rep. 2017, 7, 43515. [Google Scholar] [CrossRef]
- Grunberg, J.R.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. The Novel Secreted Adipokine WNT1-inducible Signaling Pathway Protein 2 (WISP2) Is a Mesenchymal Cell Activator of Canonical WNT. J. Biol. Chem. 2014, 289, 6899–6907. [Google Scholar] [CrossRef] [PubMed]
- Davicioni, E.; Anderson, J.R.; Buckley, J.D.; Meyer, W.H.; Triche, T.J. Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: A report from the children’s oncology group. J. Clin. Oncol. 2010, 28, 1240–1246. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sheng, D.; Wang, D.; Ma, W.; Deng, Q.; Deng, L.; Liu, S. Identification of cancer-type specific expression patterns for active aldehyde dehydrogenase (ALDH) isoforms in ALDEFLUOR assay. Cell Biol. Toxicol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Levi, B.P.; Yilmaz, O.H.; Duester, G.; Morrison, S.J. Aldehyde dehydrogenase 1a1 is dispensable for stem cell function in the mouse hematopoietic and nervous systems. Blood 2009, 113, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Pan, D.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kang, S.; Kim, D.S.; Na, H.J.; Kim, Y.J.; Choi, Y.D.; Cho, N.H. HSP27, ALDH6A1 and Prohibitin Act as a Trio-biomarker to Predict Survival in Late Metastatic Prostate Cancer. Anticancer Res. 2018, 38, 6551–6560. [Google Scholar] [CrossRef]
- Martinez-Cruzado, L.; Tornin, J.; Santos, L.; Rodriguez, A.; Garcia-Castro, J.; Moris, F.; Rodriguez, R. Aldh1 Expression and Activity Increase During Tumor Evolution in Sarcoma Cancer Stem Cell Populations. Sci. Rep. 2016, 6, 27878. [Google Scholar] [CrossRef]
- Storer, N.Y.; White, R.M.; Uong, A.; Price, E.; Nielsen, G.P.; Langenau, D.M.; Zon, L.I. Zebrafish rhabdomyosarcoma reflects the developmental stage of oncogene expression during myogenesis. Development 2013, 140, 3040–3050. [Google Scholar] [CrossRef]
- Fukada, S.; Ma, Y.; Ohtani, T.; Watanabe, Y.; Murakami, S.; Yamaguchi, M. Isolation, characterization and molecular regulation of muscle stem cells. Front. Physiol. 2013, 4, 317. [Google Scholar] [CrossRef]
- Rubin, B.P.; Nishijo, K.; Chen, H.I.; Yi, X.; Schuetze, D.P.; Pal, R.; Prajapati, S.I.; Abraham, J.; Arenkiel, B.R.; Chen, Q.R.; et al. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell 2011, 19, 177–191. [Google Scholar] [CrossRef]
- Fukada, S.; Uezumi, A.; Ikemoto, M.; Masuda, S.; Segawa, M.; Tanimura, N.; Yamamoto, H.; Miyagoe-Suzuki, Y.; Takeda, S. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells 2007, 25, 2448–2459. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Hano, H.; Harada, T.; Takai, S.; Masui, F.; Ushigome, S. Evaluation of new monoclonal anti-MyoD1 and anti-myogenin antibodies for the diagnosis of rhabdomyosarcoma. Pathol. Int. 1999, 49, 62–68. [Google Scholar] [CrossRef]
- Tenente, I.M.; Hayes, M.N.; Ignatius, M.S.; McCarthy, K.; Yohe, M.; Sindiri, S.; Gryder, B.; Oliveira, M.L.; Ramakrishnan, A.; Tang, Q.; et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. Elife 2017, 6, e19214. [Google Scholar] [CrossRef]
- Gianakopoulos, P.J.; Mehta, V.; Voronova, A.; Cao, Y.; Yao, Z.; Coutu, J.; Wang, X.; Waddington, M.S.; Tapscott, S.J.; Skerjanc, I.S. MyoD directly up-regulates premyogenic mesoderm factors during induction of skeletal myogenesis in stem cells. J. Biol. Chem. 2011, 286, 2517–2525. [Google Scholar] [CrossRef]
- Bhattaram, P.; Penzo-Mendez, A.; Sock, E.; Colmenares, C.; Kaneko, K.J.; Vassilev, A.; Depamphilis, M.L.; Wegner, M.; Lefebvre, V. Organogenesis relies on SoxC transcription factors for the survival of neural and mesenchymal progenitors. Nat. Commun. 2010, 1, 9. [Google Scholar] [CrossRef]
- Lourenco, A.R.; Coffer, P.J. SOX4: Joining the Master Regulators of Epithelial-to-Mesenchymal Transition? Trends Cancer 2017, 3, 571–582. [Google Scholar] [CrossRef]
- Rajurkar, M.; Huang, H.; Cotton, J.L.; Brooks, J.K.; Sicklick, J.; McMahon, A.P.; Mao, J. Distinct cellular origin and genetic requirement of Hedgehog-Gli in postnatal rhabdomyosarcoma genesis. Oncogene 2014, 33, 5370–5378. [Google Scholar] [CrossRef]
- Wang, H.; Garzon, R.; Sun, H.; Ladner, K.J.; Singh, R.; Dahlman, J.; Cheng, A.; Hall, B.M.; Qualman, S.J.; Chandler, D.S.; et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008, 14, 369–381. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2018, 194, 161–184. [Google Scholar] [CrossRef]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the modules of EMT and stemness: A tunable stemness window model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef]
- Yang, J.; Du, X.; Wang, G.; Sun, Y.; Chen, K.; Zhu, X.; Lazar, A.J.; Hunt, K.K.; Pollock, R.E.; Zhang, W. Mesenchymal to epithelial transition in sarcomas. Eur. J. Cancer 2014, 50, 593–601. [Google Scholar] [CrossRef]
- Sannino, G.; Marchetto, A.; Kirchner, T.; Grunewald, T.G.P. Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transition in Mesenchymal Tumors: A Paradox in Sarcomas? Cancer Res. 2017, 77, 4556–4561. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef]
- Wiggan, O.; Fadel, M.P.; Hamel, P.A. Pax3 induces cell aggregation and regulates phenotypic mesenchymal-epithelial interconversion. J. Cell Sci. 2002, 115, 517–529. [Google Scholar]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef]
- Hoffding, M.K.; Hyttel, P. Ultrastructural visualization of the Mesenchymal-to-Epithelial Transition during reprogramming of human fibroblasts to induced pluripotent stem cells. Stem Cell Res. 2015, 14, 39–53. [Google Scholar] [CrossRef]
- Kashima, T.; Kawaguchi, J.; Takeshita, S.; Kuroda, M.; Takanashi, M.; Horiuchi, H.; Imamura, T.; Ishikawa, Y.; Ishida, T.; Mori, S.; et al. Anomalous cadherin expression in osteosarcoma. Possible relationships to metastasis and morphogenesis. Am. J. Pathol. 1999, 155, 1549–1555. [Google Scholar] [CrossRef]
- Kashima, T.; Nakamura, K.; Kawaguchi, J.; Takanashi, M.; Ishida, T.; Aburatani, H.; Kudo, A.; Fukayama, M.; Grigoriadis, A.E. Overexpression of cadherins suppresses pulmonary metastasis of osteosarcoma in vivo. Int. J. Cancer 2003, 104, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Camand, E.; Peglion, F.; Osmani, N.; Sanson, M.; Etienne-Manneville, S. N-cadherin expression level modulates integrin-mediated polarity and strongly impacts on the speed and directionality of glial cell migration. J. Cell Sci. 2012, 125, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Lammens, T.; Swerts, K.; Derycke, L.; De Craemer, A.; De Brouwer, S.; De Preter, K.; Van Roy, N.; Vandesompele, J.; Speleman, F.; Philippe, J.; et al. N-cadherin in neuroblastoma disease: Expression and clinical significance. PLoS ONE 2012, 7, e31206. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Takeichi, M. Neural crest emigration from the neural tube depends on regulated cadherin expression. Development 1998, 125, 2963–2971. [Google Scholar]
- Blindt, R.; Bosserhoff, A.K.; Dammers, J.; Krott, N.; Demircan, L.; Hoffmann, R.; Hanrath, P.; Weber, C.; Vogt, F. Downregulation of N-cadherin in the neointima stimulates migration of smooth muscle cells by RhoA deactivation. Cardiovasc. Res. 2004, 62, 212–222. [Google Scholar] [CrossRef]
- Tiwari, N.; Tiwari, V.K.; Waldmeier, L.; Balwierz, P.J.; Arnold, P.; Pachkov, M.; Meyer-Schaller, N.; Schubeler, D.; van Nimwegen, E.; Christofori, G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013, 23, 768–783. [Google Scholar] [CrossRef]
- Preussner, J.; Zhong, J.; Sreenivasan, K.; Gunther, S.; Engleitner, T.; Kunne, C.; Glatzel, M.; Rad, R.; Looso, M.; Braun, T.; et al. Oncogenic Amplification of Zygotic Dux Factors in Regenerating p53-Deficient Muscle Stem Cells Defines a Molecular Cancer Subtype. Cell Stem Cell 2018, 23, 794–805. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Furukawa, K. Application of coomassie brilliant blue staining to cultured hepatocytes. Cell Biol. Int. Rep. 1987, 11, 367–371. [Google Scholar] [CrossRef]
- Mikulenkova, E.; Neradil, J.; Zitterbart, K.; Sterba, J.; Veselska, R. Overexpression of the Np73 isoform is associated with centrosome amplification in brain tumor cell lines. Tumor Biol. 2015, 36, 7483–7491. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Skoda, J.; Neradil, J.; Zitterbart, K.; Sterba, J.; Veselska, R. EGFR signaling in the HGG-02 glioblastoma cell line with an unusual loss of EGFR gene copy. Oncol. Rep. 2014, 31, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Vivo Passage Number | Tumorigenic Efficiency | Mean Tumor Volume Increase (mm3/Day ± SEM) | |
|---|---|---|---|
| Mice with Tumors/Mice Injected | Tumors Formed in Total 1 | ||
| First | 3 of 3 (100%) | 3 | 2.89 ± 0.74 (n = 3) |
| Second | 6 of 9 (67%) | 9 | 4.37 ± 1.83 (n = 9) |
| Third | 27 of 27 (100%) | 33 | 5.66 ± 0.72 (n = 27) * |
| Antigen | Primary Tumor | Primary Xenografts 1 | Secondary Xenografts 1 | Tertiary Xenografts 1 | ||||
|---|---|---|---|---|---|---|---|---|
| %TC | IR | %TC | IR | %TC | IR | %TC | IR | |
| SOX2 | 2 | +++ | 3 | +++ | 3 | ++ | 3 | ++ |
| OCT4 | 1 | + | 1 | + | 1 | + | 1 | + |
| NANOG | 0 | - | 0 | - | 0 | - | 0 | - |
| Nestin | 4 | +++ | 4 | +++ | 4 | +++ | 4 | +++ |
| CD133 | 3 | + | 1 | + | 1 | + | 1 | + |
| ABCG2 | 1 | ++ | 1 | ++ | 1 | + | 1 | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoda, J.; Neradil, J.; Staniczkova Zambo, I.; Nunukova, A.; Macsek, P.; Borankova, K.; Dobrotkova, V.; Nemec, P.; Sterba, J.; Veselska, R. Serial Xenotransplantation in NSG Mice Promotes a Hybrid Epithelial/Mesenchymal Gene Expression Signature and Stemness in Rhabdomyosarcoma Cells. Cancers 2020, 12, 196. https://doi.org/10.3390/cancers12010196
Skoda J, Neradil J, Staniczkova Zambo I, Nunukova A, Macsek P, Borankova K, Dobrotkova V, Nemec P, Sterba J, Veselska R. Serial Xenotransplantation in NSG Mice Promotes a Hybrid Epithelial/Mesenchymal Gene Expression Signature and Stemness in Rhabdomyosarcoma Cells. Cancers. 2020; 12(1):196. https://doi.org/10.3390/cancers12010196
Chicago/Turabian StyleSkoda, Jan, Jakub Neradil, Iva Staniczkova Zambo, Alena Nunukova, Peter Macsek, Karolina Borankova, Viera Dobrotkova, Pavel Nemec, Jaroslav Sterba, and Renata Veselska. 2020. "Serial Xenotransplantation in NSG Mice Promotes a Hybrid Epithelial/Mesenchymal Gene Expression Signature and Stemness in Rhabdomyosarcoma Cells" Cancers 12, no. 1: 196. https://doi.org/10.3390/cancers12010196
APA StyleSkoda, J., Neradil, J., Staniczkova Zambo, I., Nunukova, A., Macsek, P., Borankova, K., Dobrotkova, V., Nemec, P., Sterba, J., & Veselska, R. (2020). Serial Xenotransplantation in NSG Mice Promotes a Hybrid Epithelial/Mesenchymal Gene Expression Signature and Stemness in Rhabdomyosarcoma Cells. Cancers, 12(1), 196. https://doi.org/10.3390/cancers12010196