A Tyrosine Kinase Expression Signature Predicts the Post-Operative Clinical Outcome in Triple Negative Breast Cancers

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Patient Population
2.2. Identification of a Robust Prognostic Tyrosine Kinase Signature
2.3. Correlations of Our 13-Gene Classification with Clinicopathological and Molecular Features
2.4. Univariate and Multivariate Prognostic Analyses for DFS
2.5. Univariate and Multivariate Prognostic Analyses for Overall Survival
2.6. Biological Processes Associated to Our 13-Gene Classification
2.7. Prognostic and/or Predictive Value of Our13-Gene Classifier
3. Discussion
4. Materials and Methods
4.1. Tumor Samples and Kinase Genes
4.2. Gene Expression Data Processing
4.3. Gene Expression Data Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heymach, J.; Krilov, L.; Alberg, A.; Baxter, N.; Chang, S.M.; Corcoran, R.B.; Dale, W.; DeMichele, A.; Magid Diefenbach, C.S.; Dreicer, R. Clinical cancer advances 2018: Annual report on progress against cancer from the American Society of Clinical Oncology. J. Clin. Oncol. 2018, 36, 1020–1044. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Mamessier, E.; Adélaïde, J.; Debono, S.; Houvenaeghel, G.; Maraninchi, D.; Viens, P.; et al. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res. 2006, 66, 4636–4644. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. JCO 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Prat, A.; Adamo, B.; Cheang, M.C.U.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular Characterization of Basal-Like and Non-Basal-Like Triple-Negative Breast Cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef]
- de Nonneville, A.; Gonçalves, A.; Zemmour, C.; Cohen, M.; Classe, J.M.; Reyal, F.; Colombo, P.E.; Jouve, E.; Giard, S.; Barranger, E.; et al. Adjuvant chemotherapy in pT1ab node-negative triple-negative breast carcinomas: Results of a national multi-institutional retrospective study. Eur. J. Cancer 2017, 84, 34–43. [Google Scholar] [CrossRef]
- Sabatier, R.; Finetti, P.; Mamessier, E.; Raynaud, S.; Cervera, N.; Lambaudie, E.; Jacquemier, J.; Viens, P.; Birnbaum, D.; Bertucci, F. Kinome expression profiling and prognosis of basal breast cancers. Mol. Cancer 2011, 10, 86. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Viens, P.; Birnbaum, D. Difference in Therapeutic Response Between Basal and Nonbasal Triple-Negative Breast Cancers. Oncologist 2013, 18, 1060–1061. [Google Scholar] [CrossRef][Green Version]
- Huang, S.; Murphy, L.; Xu, W. Genes and functions from breast cancer signatures. BMC Cancer 2018, 18. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Miremadi, A.; Pinder, S.E.; Ellis, I.O.; Caldas, C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007, 8, R157. [Google Scholar] [CrossRef]
- Rody, A.; Holtrich, U.; Pusztai, L.; Liedtke, C.; Gaetje, R.; Ruckhaeberle, E.; Solbach, C.; Hanker, L.; Ahr, A.; Metzler, D.; et al. T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res. 2009, 11, R15. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Simeone, I.; Hendrickx, W.; Wang, E.; Marincola, F.M.; Viens, P.; Mamessier, E.; Ceccarelli, M.; Birnbaum, D.; et al. The immunologic constant of rejection classification refines the prognostic value of conventional prognostic signatures in breast cancer. Br. J. Cancer 2018, 119, 1383. [Google Scholar] [CrossRef]
- Stovgaard, E.S.; Nielsen, D.; Hogdall, E.; Balslev, E. Triple negative breast cancer—Prognostic role of immune-related factors: A systematic review. Acta Oncol. 2018, 57, 74–82. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.-Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef]
- Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Chabannon, C.; Charpin, C.; Chaffanet, M.; Jacquemier, J.; Viens, P.; Birnbaum, D.; Bertucci, F. Sixteen-kinase gene expression identifies luminal breast cancers with poor prognosis. Cancer Res. 2008, 68, 767–776. [Google Scholar] [CrossRef]
- Speers, C.; Tsimelzon, A.; Sexton, K.; Herrick, A.M.; Gutierrez, C.; Culhane, A.; Quackenbush, J.; Hilsenbeck, S.; Chang, J.; Brown, P. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin. Cancer Res. 2009, 15, 6327–6340. [Google Scholar] [CrossRef]
- Nair, A.; Chung, H.-C.; Sun, T.; Tyagi, S.; Dobrolecki, L.E.; Dominguez-Vidana, R.; Kurley, S.J.; Orellana, M.; Renwick, A.; Henke, D.M.; et al. Combinatorial inhibition of PTPN12-regulated receptors leads to a broadly effective therapeutic strategy in triple-negative breast cancer. Nat. Med. 2018, 24, 505–511. [Google Scholar] [CrossRef]
- Gaule, P.; Mukherjee, N.; Corkery, B.; Eustace, A.J.; Gately, K.; Roche, S.; O’Connor, R.; O’Byrne, K.J.; Walsh, N.; Duffy, M.J.; et al. Dasatinib Treatment Increases Sensitivity to c-Met Inhibition in Triple-Negative Breast Cancer Cells. Cancers 2019, 11, 548. [Google Scholar] [CrossRef]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef]
- Gatza, M.L.; Lucas, J.E.; Barry, W.T.; Kim, J.W.; Wang, Q.; Crawford, M.D.; Datto, M.B.; Kelley, M.; Mathey-Prevot, B.; Potti, A.; et al. A pathway-based classification of human breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6994–6999. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- van de Vijver, M.J.; He, Y.D.; van’t Veer, L.J.; Dai, H.; Hart, A.A.M.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A Multigene Assay to Predict Recurrence of Tamoxifen-Treated, Node-Negative Breast Cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Powers, C.; Aigner, A.; Stoica, G.E.; McDonnell, K.; Wellstein, A. Pleiotrophin signaling through anaplastic lymphoma kinase is rate-limiting for glioblastoma growth. J. Biol. Chem. 2002, 277, 14153–14158. [Google Scholar] [CrossRef]
- Verma, N.; Müller, A.-K.; Kothari, C.; Panayotopoulou, E.; Kedan, A.; Selitrennik, M.; Mills, G.B.; Nguyen, L.K.; Shin, S.; Karn, T.; et al. Targeting of PYK2 Synergizes with EGFR Antagonists in Basal-like TNBC and Circumvents HER3-Associated Resistance via the NEDD4–NDRG1 Axis. Cancer Res. 2017, 77, 86–99. [Google Scholar] [CrossRef]
- Robertson, F.M.; Petricoin III, E.F.; Van Laere, S.J.; Bertucci, F.; Chu, K.; Fernandez, S.V.; Mu, Z.; Alpaugh, K.; Pei, J.; Circo, R.; et al. Presence of anaplastic lymphoma kinase in inflammatory breast cancer. SpringerPlus 2013, 2, 497. [Google Scholar] [CrossRef]
- Siraj, A.K.; Beg, S.; Jehan, Z.; Prabhakaran, S.; Ahmed, M.; Hussain, A.R.; Al-Dayel, F.; Tulbah, A.; Ajarim, D.; Al-Kuraya, K.S. ALK alteration is a frequent event in aggressive breast cancers. Breast Cancer Res. 2015, 17, 127. [Google Scholar] [CrossRef]
- Mylona, E.; Alexandrou, P.; Mpakali, A.; Giannopoulou, I.; Liapis, G.; Markaki, S.; Keramopoulos, A.; Nakopoulou, L. Clinicopathological and prognostic significance of vascular endothelial growth factors (VEGF)-C and -D and VEGF receptor 3 in invasive breast carcinoma. Eur. J. Surg. Oncol. 2007, 33, 294–300. [Google Scholar] [CrossRef]
- Golfmann, K.; Meder, L.; Koker, M.; Volz, C.; Borchmann, S.; Tharun, L.; Dietlein, F.; Malchers, F.; Florin, A.; Büttner, R.; et al. Synergistic anti-angiogenic treatment effects by dual FGFR1 and VEGFR1 inhibition in FGFR1-amplified breast cancer. Oncogene 2018, 37, 5682–5693. [Google Scholar] [CrossRef]
- Nikas, I.; Ryu, H.S.; Theocharis, S. Viewing the Eph receptors with a focus on breast cancer heterogeneity. Cancer Lett. 2018, 434, 160–171. [Google Scholar] [CrossRef]
- Hachim, I.Y.; Villatoro, M.; Canaff, L.; Hachim, M.Y.; Boudreault, J.; Haiub, H.; Ali, S.; Lebrun, J.-J. Transforming Growth Factor-beta Regulation of Ephrin Type-A Receptor 4 Signaling in Breast Cancer Cellular Migration. Sci. Rep. 2017, 7, 14976. [Google Scholar] [CrossRef]
- Hochgräfe, F.; Zhang, L.; O’Toole, S.A.; Browne, B.C.; Pinese, M.; Cubas, A.P.; Lehrbach, G.M.; Croucher, D.R.; Rickwood, D.; Boulghourjian, A.; et al. Tyrosine Phosphorylation Profiling Reveals the Signaling Network Characteristics of Basal Breast Cancer Cells. Cancer Res. 2010, 70, 9391–9401. [Google Scholar] [CrossRef]
- Bhushan, L.; Tavitian, N.; Dey, D.; Tumur, Z.; Parsa, C.; Kandpal, R.P. Modulation of liver-intestine cadherin (Cadherin 17) expression, ERK phosphorylation and WNT signaling in EPHB6 receptor-expressing MDA-MB-231 cells. Cancer Genom. Proteomics 2014, 11, 239–249. [Google Scholar]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Andreotti, A.H.; Schwartzberg, P.L.; Joseph, R.E.; Berg, L.J. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb. Perspect. Biol. 2010, 2, a002287. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Developmental cell biology: Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 2005, 6, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Truitt, L.; Freywald, A. Dancing with the dead: Eph receptors and their kinase-null partnersThis paper is one of a selection of papers published in a Special Issue entitled CSBMCB 53rd Annual Meeting—Membrane Proteins in Health and Disease, and has undergone the Journal’s usual peer review process. Biochem. Cell Biol. 2011, 89, 115–129. [Google Scholar] [PubMed]
- Herath, N.I.; Doecke, J.; Spanevello, M.D.; Leggett, B.A.; Boyd, A.W. Epigenetic silencing of EphA1 expression in colorectal cancer is correlated with poor survival. Br. J. Cancer 2009, 100, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, J.; Sheng, Z.; Li, G.; Ma, H.; Wang, X.; Zhang, R.; Lu, G.; Hu, Q.; Sugimura, H.; et al. Downregulation of EphA1 in colorectal carcinomas correlates with invasion and metastasis. Mod. Pathol. 2009, 22, 151–160. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, B.O.; Jiang, W.G.; Zhou, D.; Cui, Y.-X. Knockdown of EPHA1 by CRISPR/CAS9 Promotes Adhesion and Motility of HRT18 Colorectal Carcinoma Cells. Anticancer Res. 2016, 36, 1211–1219. [Google Scholar] [PubMed]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph Receptor Signaling and Ephrins. Cold Spring Harb. Perspect. Biol. 2013, 5, a009159. [Google Scholar] [CrossRef]
- Yeatman, T.J. A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef]
- Qian, X.-L.; Zhang, J.; Li, P.-Z.; Lang, R.-G.; Li, W.-D.; Sun, H.; Liu, F.-F.; Guo, X.-J.; Gu, F.; Fu, L. Dasatinib inhibits c-src phosphorylation and prevents the proliferation of Triple-Negative Breast Cancer (TNBC) cells which overexpress Syndecan-Binding Protein (SDCBP). PLoS ONE 2017, 12, e0171169. [Google Scholar] [CrossRef]
- Morgan, L.; Gee, J.; Pumford, S.; Farrow, L.; Finlay, P.; Robertson, J.; Ellis, I.; Kawakatsu, H.; Nicholson, R.; Hiscox, S. Elevated Src kinase activity attenuates tamoxifen response in vitro and is associated with poor prognosis clinically. Cancer Biol. Ther. 2009, 8, 1550–1558. [Google Scholar] [CrossRef]
- Finn, R.S. Targeting Src in breast cancer. Ann. Oncol. 2008, 19, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Qayyum, T.; Fyffe, G.; Duncan, M.; McArdle, P.A.; Hilmy, M.; Orange, C.; Halbert, G.; Seywright, M.; Horgan, P.G.; Underwood, M.A.; et al. The interrelationships between Src, Cav-1 and RhoGD12 in transitional cell carcinoma of the bladder. Br. J. Cancer 2012, 106, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Paul, W.E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Dolezal, J.M.; Wang, H.; Jackson, L.; Lu, J.; Frodey, B.P.; Dosunmu-Ogunbi, A.; Li, Y.; Fromherz, M.; Kang, A.; et al. Ribosomopathy-like properties of murine and human cancers. PLoS ONE 2017, 12, e0182705. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.W.; Jin, Z.; Macdonald, D.; Wei, W.; Qian, X.J.; Choi, W.S.; He, R.; Sun, X.; Chan, G. Prolonged mitotic arrest induced by Wee1 inhibition sensitizes breast cancer cells to paclitaxel. Oncotarget 2017, 8, 73705–73722. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef]
- Broude, E.V.; Győrffy, B.; Chumanevich, A.A.; Chen, M.; McDermott, M.S.J.; Shtutman, M.; Catroppo, J.F.; Roninson, I.B. Expression of CDK8 and CDK8-interacting Genes as Potential Biomarkers in Breast Cancer. Curr. Cancer Drug Targets 2015, 15, 739–749. [Google Scholar] [CrossRef]
- Philip, S.; Kumarasiri, M.; Teo, T.; Yu, M.; Wang, S. Cyclin-Dependent Kinase 8: A New Hope in Targeted Cancer Therapy? J. Med. Chem. 2018, 61, 5073–5092. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Theillet, C.; Adelaide, J.; Louason, G.; Bonnet-Dorion, F.; Jacquemier, J.; Adnane, J.; Longy, M.; Katsaros, D.; Sismondi, P.; Gaudray, P. FGFRI and PLAT genes and DNA amplification at 8p12 in breast and ovarian cancers. Genes Chromosomes Cancer 1993, 7, 219–226. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Staaf, J.; Ringnér, M.; Vallon-Christersson, J.; Jönsson, G.; Bendahl, P.-O.; Holm, K.; Arason, A.; Gunnarsson, H.; Hegardt, C.; Agnarsson, B.A.; et al. Identification of subtypes in human epidermal growth factor receptor 2—Positive breast cancer reveals a gene signature prognostic of outcome. J. Clin. Oncol. 2010, 28, 1813–1820. [Google Scholar] [CrossRef]
- Venet, D.; Dumont, J.E.; Detours, V. Most Random Gene Expression Signatures Are Significantly Associated with Breast Cancer Outcome. PLoS Comput. Biol. 2011, 7, e1002240. [Google Scholar] [CrossRef]
- Boutros, P.C.; Lau, S.K.; Pintilie, M.; Liu, N.; Shepherd, F.A.; Der, S.D.; Tsao, M.-S.; Penn, L.Z.; Jurisica, I. Prognostic gene signatures for non-small-cell lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 2824–2828. [Google Scholar] [CrossRef]
- Palmer, C.; Diehn, M.; Alizadeh, A.A.; Brown, P.O. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genom. 2006, 7, 115. [Google Scholar]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef]
- Hochberg, Y.; Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M. Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK). JNCI J. Natl. Cancer Inst. 2005, 97, 1180–1184. [Google Scholar] [CrossRef]
- Disis, M.L.; Park, K.H. Immunomodulation of Breast Cancer via Tumor Antigen Specific Th1. Cancer Res. Treat. 2009, 41, 117–121. [Google Scholar] [CrossRef][Green Version]
- Bertucci, F.; Gonçalves, A. Immunotherapy in Breast Cancer: The Emerging Role of PD-1 and PD-L1. Curr. Oncol. Rep. 2017, 19, 64. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | N (%) |
|---|---|
| Patients’ age | |
| ≤50 years | 484 (45%) |
| >50 years | 584 (55%) |
| Pathological grade | |
| 1 | 23 (3%) |
| 2 | 144 (17%) |
| 3 | 690 (81%) |
| Pathological axillary lymph nodes status (pN) | |
| Negative | 504 (59%) |
| Positive | 347 (41%) |
| Pathological tumor size (pT) | |
| pT1 | 277 (34%) |
| pT2 | 459 (57%) |
| pT3 | 67 (8%) |
| Pathological type | |
| Ductal | 555 (82%) |
| Lobular | 25 (4%) |
| Other | 98 (14%) |
| Lehmann’s TNBC subtypes | |
| Basal-like 1 | 216 (18%) |
| Basal-like 2 | 90 (7%) |
| Immunomodulatory | 259 (21%) |
| Luminal Androgen Receptor | 189 (15%) |
| Mesenchymal | 301 (25%) |
| Mesenchymal stem-like | 171 (14%) |
| Median follow-up, months (range) | 44 (1–286) |
| DFS events | 410 (33%) |
| 5-year DFS | 63% (95%CI 60–66) |
| OS events | 215 (25%) |
| 5-year OS | 73% (95%CI 70–76) |
| Characteristics | N | 13-Genes Classification | p-Value | ||
|---|---|---|---|---|---|
| Low-Risk | High-Risk | ||||
| Patients’ age | 0.149 | ||||
| ≤50 years | 484 | 242 (43%) | 242 (48%) | ||
| >50 years | 584 | 319 (57%) | 265 (52%) | ||
| Pathological grade | 4.4 × 10−2 | ||||
| 1 | 23 | 14 (3%) | 9 (2%) | ||
| 2 | 144 | 69 (14%) | 75 (20%) | ||
| 3 | 690 | 408 (83%) | 282 (77%) | ||
| Pathological axillary lymph nodes status (pN) | 8.3 × 10−5 | ||||
| Negative | 504 | 251 (53%) | 253 (67%) | ||
| Positive | 347 | 221 (47%) | 126 (33%) | ||
| Pathological tumor size (pT) | 2.4 × 10−2 | ||||
| pT1 | 277 | 173 (39%) | 104 (29%) | ||
| pT2 | 459 | 242 (54%) | 217 (61%) | ||
| pT3 | 67 | 34 (8%) | 33 (9%) | ||
| Pathological type | 0.541 | ||||
| Ductal | 555 | 317 (82%) | 238 (82%) | ||
| Lobular | 25 | 12 (3%) | 13 (4%) | ||
| Other | 98 | 59 (15%) | 39 (13%) | ||
| Lehmann’s TNBC subtype [17] | 3.4 × 10−17 | ||||
| Basal−like 1 | 216 | 110 (18%) | 106 (17%) | ||
| Basal−like 2 | 90 | 36 (6%) | 54 (9%) | ||
| Immunomodulatory | 259 | 188 (31%) | 71 (12%) | ||
| Luminal Androgen Receptor | 189 | 92 (15%) | 97 (16%) | ||
| Mesenchymal | 301 | 103 (17%) | 198 (32%) | ||
| Mesenchymal stem-like | 171 | 85 (14%) | 86 (14%) | ||
| 70-gene signature [27] | 0.118 | ||||
| Low-risk | 15 | 4 (1%) | 11 (2%) | ||
| High-risk | 1211 | 610 (99%) | 601 (98%) | ||
| Recurrence Score [28] | 0.397 | ||||
| Low-risk | 5 | 1 (0%) | 4 (1%) | ||
| High-risk | 1176 | 591 (96%) | 585 (96%) | ||
| Intermediate-risk | 45 | 22 (4%) | 23 (4%) | ||
| ROR-P signature [6] | 0.61 | ||||
| Low-risk | 77 | 36 (6%) | 41 (7%) | ||
| High-risk | 1005 | 510 (83%) | 495 (81%) | ||
| Intermediate-risk | 144 | 68 (11%) | 76 (12%) | ||
| Immune 28-kinase [9] | 1.4 × 10−32 | ||||
| Low-risk | 298 | 239 (39%) | 59 (10%) | ||
| High-risk | 928 | 375 (61%) | 553 (90%) | ||
| IR signature [12] | 6.5 × 10−5 | ||||
| Low-risk | 602 | 335 (56%) | 267 (44%) | ||
| High-risk | 609 | 268 (44%) | 341 (56%) | ||
| LCK signature [13] | 2.9 × 10−29 | ||||
| Low-risk | 710 | 258 (42%) | 452 (74%) | ||
| High-risk | 516 | 356 (58%) | 160 (26%) | ||
| Gatza’s molecular pathways activation score [25] | |||||
| IFN alpha | 1226 | 0.68 (0.01−0.99) | 0.51 (0−0.99) | 3.4 × 10−17 | |
| IFN gamma | 1226 | 0.72 (0–1) | 0.56 (0–0.99) | 2.1 × 10−16 | |
| TGF beta | 1226 | 0.45 (0.01–1) | 0.51 (0.01–1) | 7.9 × 10−5 | |
| Lymphocyte infiltration (%) | 199 | 8.93 (0–100) | 6.99 (0–100) | 0.459 | |
| Lymphocyte infiltration | 1.00 | ||||
| ≤10% | 167 | 58 (84%) | 109 (84%) | ||
| >10% | 32 | 11 (16%) | 21 (16%) | ||
| Bindea’s signatures [29] | |||||
| B cells | 1226 | 0.49 (−0.62–3.4) | 0.14 (−0.89–1.74) | 1.3 × 10−26 | |
| T cells | 1226 | 0.6 (−2.26–4.21) | 0.01 (−1.6–2.09) | 2.9 × 10−37 | |
| T helper cells | 1226 | 0.11 (−1.71–1.49) | 0.02 (−1.11–1.61) | 3.4 × 10−5 | |
| Tcm | 1226 | 0.07 (−1.28–2.19) | −0.02 (−1.09–0.82) | 1.8 × 10−6 | |
| Tem | 1226 | 0.09 (−0.82–1.17) | 0 (−1.07–0.88) | 1.8 × 10−7 | |
| Th1 cells | 1226 | 0.27 (−0.69–1.46) | 0.1 (−0.98–0.93) | 4.5 × 10−20 | |
| Th2 cells | 1226 | 0.09 (−0.86–0.85) | 0.12 (−0.76–1.18) | 0.0734 | |
| TFH | 1226 | 0.03 (−1.16–1.18) | −0.06 (−0.77–0.81) | 7.7 × 10−10 | |
| Th17 cells | 1226 | −0.08 (−1.98–3.17) | −0.16 (−1.87–4.09) | 0.0293 | |
| TReg | 1226 | 0.15 (−3.69–6.7) | 0.08 (−2.35–4.42) | 0.246 | |
| CD8 T cells | 1226 | 0.02 (−0.88–1.65) | −0.1 (−0.93–0.57) | 7.5 × 10−13 | |
| Tgd | 1226 | 0.24 (−1.38–5.5) | 0.02 (−2.09–2.63) | 9.2 × 10−10 | |
| Cytotoxic cells | 1226 | 0.38 (−1.21–2.5) | −0.07 (−1.61–1.71) | 1.9 × 10−35 | |
| NK cells | 1226 | −0.05 (−0.96–0.89) | −0.04 (−1–0.88) | 0.607 | |
| NK CD56dim cells | 1226 | 0.3 (−0.97–3.2) | 0.08 (−1.16–2.06) | 2.0 × 10−16 | |
| NK CD56bright cells | 1226 | −0.17 (−1.49–2.9) | −0.25 (−1.69–1.25) | 2.6 × 10−4 | |
| DC | 1226 | 0.31 (−1.11–3.29) | 0.1 (−1.92–2.32) | 3.5 × 10−9 | |
| iDC | 1226 | 0.06 (−0.89–1.61) | −0.06 (−1.26–1.58) | 7.3 × 10−9 | |
| aDC | 1226 | 0.65 (−1.11–3.07) | 0.24 (−1.74–4.72) | 1.6 × 10−22 | |
| pDC | 1226 | 0.33 (−3.27–6.34) | −0.04 (−2.91–4.26) | 2.6 × 10−9 | |
| Eosinophils | 1226 | −0.11 (−0.84–0.73) | -0.15 (−1.01–0.55) | 0.00498 | |
| Macrophages | 1226 | 0.24 (−1.27–2.79) | 0.12 (-1.17–2.57) | 1.4 × 10−5 | |
| Mast cells | 1226 | −0.17 (-0.93–1.2) | -0.17 (-1.32–1.29) | 0.96 | |
| Neutrophils | 1226 | 0.16 (−1.19–2.77) | 0.06 (−1.22–2.19) | 6.1 × 10−5 | |
| SW480 cancer cells | 1226 | 0.13 (−1.32–1.42) | 0.16 (−0.77–1.59) | 0.085 | |
| Normal mucosa | 1226 | −0.2 (−1.4–0.96) | −0.11 (−1.59–1.31) | 2.6 × 10−4 | |
| Blood vessels | 1226 | −0.31 (−4.44–4.93) | −0.23 (−3.39−3.66) | 0.169 | |
| Lymph vessels | 1226 | −0.06 (−2.06–2.75) | −0.13 (−2.46−2.55) | 0.0924 | |
| Cytolytic activity score [26] | 1226 | 0.41 (−2.42–4.66) | 0.05 (−1.95–3.75) | 7.87 × 10−10 | |
| Median follow-up (months) | 1226 | 53 (1–286) | 31 (1–229) | 9.8 × 10−17 | |
| DFS events (months) | 1226 | 157 (74%) | 253 (41%) | 5.6 × 10−9 | |
| 5-year DFS | 1226 | 73% (69–77) | 53% (48–58) | 1.3 × 10−15 | |
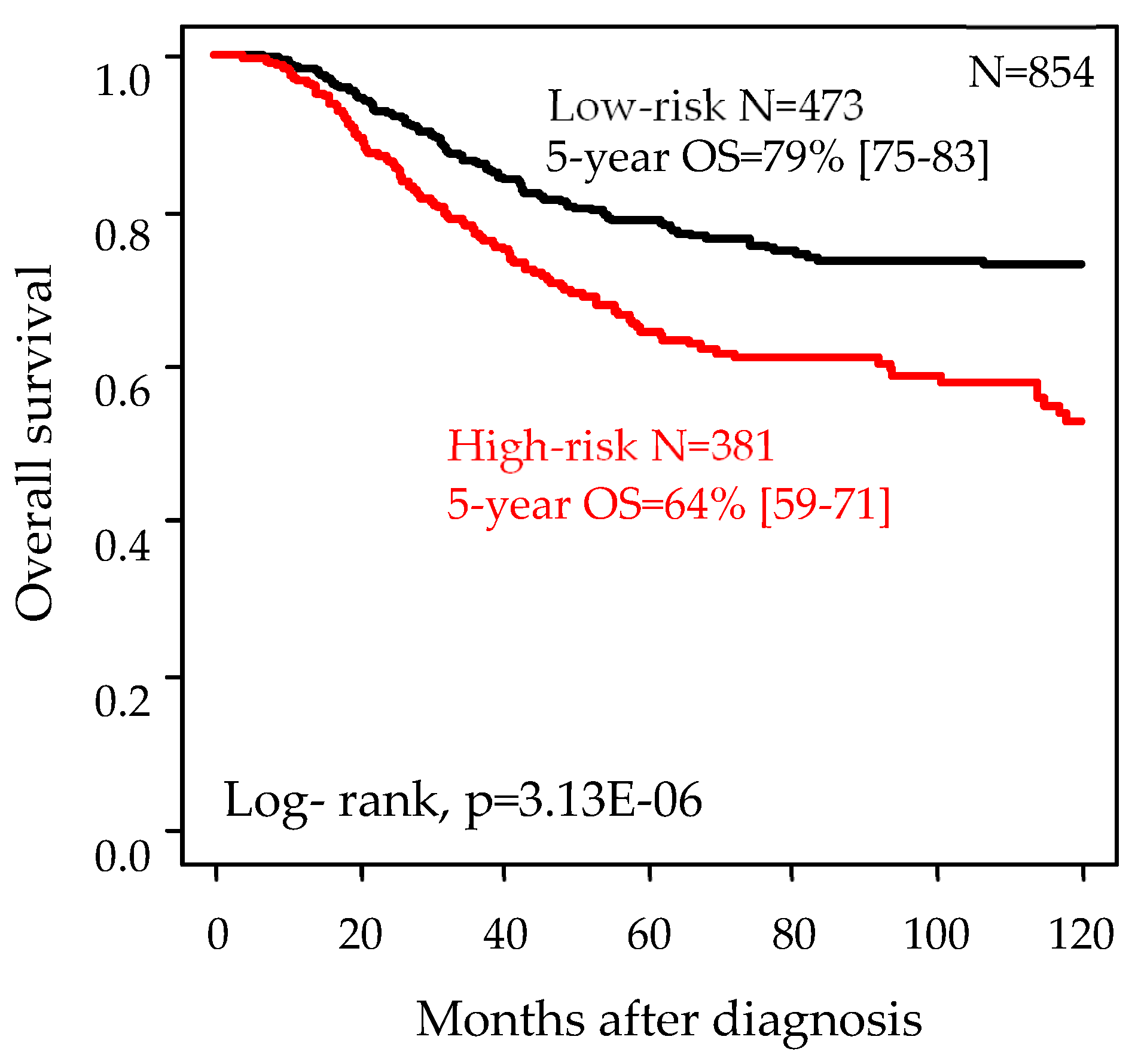
| OS events (months) | 1226 | 101 (21%) | 114 (30%) | 4.3 × 10−3 | |
| 5-year OS | 1226 | 79% (75–83) | 64% (59–71) | 3.0 × 10−6 | |
| Variable | Test | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|---|
| N | HR (95% CI) | p-Value | N | HR (95%CI) | p-Value | ||
| Patients’ age | >50 vs. ≤50 years | 351 | 0.88 (0.60−1.31) | 0.540 | |||
| Pathological grade | 2 vs. 1 | 275 | 18841300 (0–Inf) | 0.445 | |||
| 3 vs. 1 | 27280092 (0–Inf) | ||||||
| Pathological axillary lymph node status (pN) | Positive vs. negative | 273 | 1.35 (0.87–2.09) | 0.177 | |||
| Pathological tumor size (pT) | pT2 vs. pT1 | 254 | 1.35 (0.81–2.25) | 0.505 | |||
| pT3 vs. pT1 | 1.32 (0.50–3.50) | ||||||
| Pathological type | Lobular vs. ductal | 219 | 2.12 (0.76–5.92) | 0.337 | |||
| Other vs. ductal | 1.19 (0.60–2.35) | ||||||
| Lehmann’s TNBC subtype [17] | Basal-like 2 vs. Basal-like 1 | 401 | 2.44 (1.27–4.69) | 3.79 × 10−2 | 394 | 2.62 (1.36–5.08) | 4.22 × 10−3 |
| Immunomodulatory vs. Basal-like 1 | 0.98 (0.57–1.67) | 394 | 1.48 (0.82–2.67) | 0.198 | |||
| Luminal AR vs. Basal-like 1 | 1.15 (0.65–2.04) | 394 | 1.30 (0.72–2.33) | 0.385 | |||
| Mesenchymal vs. Basal-like 1 | 0.88 (0.52–1.52) | 394 | 0.75 (0.43–1.29) | 0.292 | |||
| Mesenchymal stem-like vs. Basal-like 1 | 0.85 (0.47–1.55) | 394 | 1.16 (0.61–2.19) | 0.650 | |||
| 70-gene signature [27] | High- vs. low-risk | 401 | 2.88 (0.40–20.6) | 0.292 | |||
| Recurrence Score [28] | High- vs. low-risk | 401 | 1233058 (0–Inf) | 0.635 | |||
| Intermediate- vs. low-risk | 624862 (0–Inf) | ||||||
| PAM50 and ROR-P [6] | High- vs. low-risk | 401 | 6.88 (0.96–49.3) | 0.152 | |||
| Median vs. low-risk | 6.30 (0.84–47.3) | ||||||
| 28-gene Immune Kinase [9] | High- vs. low-risk | 401 | 1.22 (0.83–1.79) | 0.313 | |||
| Immune response [12] | High- vs. low-risk | 394 | 1.41 (1.00–1.98) | 0.052 | 394 | 1.33 (0.93–1.90) | 0.125 |
| Lymphocyte-specific kinase [13] | High- vs. low-risk | 401 | 0.68 (0.48–0.97) | 0.032 | 394 | 0.83 (0.52–1.33) | 0.445 |
| Bindea’s CD8 T-cells signature | 401 | 0.47 (0.25–0.90) | 2.18 × 10−2 | 394 | 0.51 (0.24–1.08) | 0.080 | |
| 13-tyrosine kinase genes | High- vs. low-risk | 401 | 1.72 (1.22–2.44) | 2.09 × 10−3 | 394 | 1.61 (1.11−2.36) | 1.30 × 10−2 |
| Variable | Test | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|---|
| N | HR (95%CI) | p-Value | N | HR (95% CI) | p-Value | ||
| Patients’ age | >50 vs. ≤50 years | 830 | 0.84 (0.64–1.10) | 0.209 | |||
| Pathological grade | 2 vs. 1 | 641 | 2.36 (0.56–9.93) | 0.068 | 532 | 2.14 (0.5–9.24) | 0.308 |
| 3 vs. 1 | 3.33 (0.83–13.5) | 532 | 2.84 (0.67–12.1) | 0.158 | |||
| Pathological axillary lymph node status (pN) | Positive vs. negative | 742 | 2.00 (1.50–2.67) | 2.26 × 10−6 | 532 | 1.93 (1.39–2.68) | 8.04 × 10−5 |
| Pathological tumor size (pT) | pT2 vs. pT1 | 744 | 1.59 (1.16–2.19) | 3.77 × 10−3 | 532 | 1.34 (0.94–1.91) | 0.106 |
| pT3 vs. pT1 | 2.04 (1.22–3.39) | 532 | 1.37 (0.75–2.51) | 0.299 | |||
| Pathological type | Lobular vs. ductal | 677 | 0.91 (0.42–1.94) | 0.144 | |||
| Other vs. ductal | 0.60 (0.37–1.00) | ||||||
| Lehmann’s TNBC subtype [17] | Basal-like 2 vs. Basal-like 1 | 854 | 1.49 (0.83–2.68) | 1.55 × 10−3 | 532 | 1.99 (1.02–3.9) | 4.47 × 10−2 |
| Immunomodulatory vs. Basal-like 1 | 0.54 (0.34–0.87) | 532 | 1.04 (0.57–1.91) | 0.892 | |||
| Luminal AR vs. Basal-like 1 | 1.12 (0.73–1.70) | 532 | 1.67 (1.00–2.77) | 4.91 × 10−2 | |||
| Mesenchymal vs. Basal-like 1 | 1.33 (0.91–1.94) | 532 | 1.72 (1.08–2.76) | 2.33 × 10−2 | |||
| Mesenchymal stem-like vs. Basal-like 1 | 0.79 (0.47–1.31) | 532 | 1.15 (0.57–2.30) | 0.699 | |||
| 70-gene signature [27] | High- vs. low-risk | 854 | 3.03 (0.42–21.6) | 0.269 | |||
| Recurrence Score [28] | High- vs. low-risk | 854 | 1236427 (0–Inf) | 0.42 | |||
| Intermediate- vs. low-risk | 330274 (0–Inf) | ||||||
| PAM50 and ROR-P [6] | High- vs. low-risk | 854 | 1.90 (0.84–4.28) | 0.217 | |||
| Median vs. low-risk | 1.54 (0.62–3.84) | ||||||
| 28-gene Immune Kinase [9] | High- vs. low-risk | 854 | 1.83 (1.31–2.55) | 4.14 × 10−4 | 532 | 0.96 (0.53–1.71) | 0.881 |
| Immune response [12] | High- vs. low-risk | 840 | 1.16 (0.89–1.52) | 0.28 | |||
| Lymphocyte-specific kinase [13] | High- vs. low-risk | 854 | 0.58 (0.44–0.77) | 1.37 × 10−4 | 532 | 0.92 (0.57–1.47) | 0.713 |
| Bindea’s CD8 T-cells signature | 854 | 0.45 (0.27–0.75) | 2.16 × 10−3 | 532 | 0.94 (0.45–1.97) | 0.87 | |
| 13-tyrosine kinase genes | High- vs. low-risk | 854 | 1.88 (1.43–2.46) | 4.48 × 10−6 | 532 | 1.86 (1.32–2.62) | 4.18 × 10−4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Nonneville, A.; Finetti, P.; Adelaide, J.; Lambaudie, É.; Viens, P.; Gonçalves, A.; Birnbaum, D.; Mamessier, E.; Bertucci, F. A Tyrosine Kinase Expression Signature Predicts the Post-Operative Clinical Outcome in Triple Negative Breast Cancers. Cancers 2019, 11, 1158. https://doi.org/10.3390/cancers11081158
de Nonneville A, Finetti P, Adelaide J, Lambaudie É, Viens P, Gonçalves A, Birnbaum D, Mamessier E, Bertucci F. A Tyrosine Kinase Expression Signature Predicts the Post-Operative Clinical Outcome in Triple Negative Breast Cancers. Cancers. 2019; 11(8):1158. https://doi.org/10.3390/cancers11081158
Chicago/Turabian Stylede Nonneville, Alexandre, Pascal Finetti, José Adelaide, Éric Lambaudie, Patrice Viens, Anthony Gonçalves, Daniel Birnbaum, Emilie Mamessier, and François Bertucci. 2019. "A Tyrosine Kinase Expression Signature Predicts the Post-Operative Clinical Outcome in Triple Negative Breast Cancers" Cancers 11, no. 8: 1158. https://doi.org/10.3390/cancers11081158
APA Stylede Nonneville, A., Finetti, P., Adelaide, J., Lambaudie, É., Viens, P., Gonçalves, A., Birnbaum, D., Mamessier, E., & Bertucci, F. (2019). A Tyrosine Kinase Expression Signature Predicts the Post-Operative Clinical Outcome in Triple Negative Breast Cancers. Cancers, 11(8), 1158. https://doi.org/10.3390/cancers11081158