1. Introduction
Breast cancer is the most common malignancy in women and the principal cause of cancer death worldwide [
1]. FOXO proteins are Forkhead Box (class O) transcription factors that are evolutionarily conserved from invertebrates to mammals, with invertebrates having only one
FOXO gene and mammals four-
FOXO1, FOXO3, FOXO4 and
FOXO6 [
2]. FOXO3 is a tumor suppressor often deregulated in cancers, including glioblastoma, leukemia and breast and prostate cancer, due to its role in restricting cell proliferation and promoting cell death [
3]. Once activated, FOXO3 proteins bind to the conserved sequence motif GTAAA(C/T)A to induce transcription of their target genes [
4,
5]. The biological activity of FOXO3 is predominantly regulated by post-translational modifications (PTMs) in response to different environmental and intracellular stimuli [
6]. These PTMs include phosphorylation, acetylation, methylation, ubiquitination and glycosylation, which subsequently control FOXO3 protein expression, subcellular localization, DNA binding ability, transcriptional activity and stability [
6,
7].
Among all FOXO PTMs, phosphorylation is the best studied. Upon activation of phosphatidylinositol 3-kinase-protein B kinase (PI3K-PKB/AKT) signaling pathway, phosphorylation of human FOXO3 at three conserved sites (T32, S253 and S315) by AKT creates binding sites for the scaffolding protein 14-3-3, which in turn sequesters the phosphorylated protein in the cytoplasm and ultimately leads to their proteasomal degradation [
3,
8,
9].
Another important PTM of FOXO3 but as yet not well-studied, is acetylation. Protein acetylation is regulated by the opposing actions of the acetyl-transferases [e.g., EP300 or (cAMP response element binding (CREB) binding protein (CBP)] and the de-acetylases, such as sirtuins (SIRTs). Oxidative stress and the formation of reactive oxygen species (ROS) induce FOXO3 acetylation on lysine residues K242, K245 and K259 [
10], which are found within the DNA binding domain of the FOXO protein. In turn, acetylation can affect the DNA binding ability and transcriptional activity of FOXO3. However, it is still under debate as to whether this modification causes an increase or a decrease in the affinity of FOXO3 for DNA [
2,
11]. Nonetheless, acetylation has also been suggested to switch target gene expression from cell cycle arrest to apoptosis and increase the nuclear concentration of FOXO proteins [
7,
12]. Conversely, de-acetylation of FOXO3 has been suggested to promote expression of genes implicated in cell cycle arrest and stress resistance [
8].
Lapatinib is a small molecule inhibitor administered to patients with metastatic breast cancer over-expressing the human epidermal growth factor receptor 1/2 (EGFR1/HER2) [
13]. It is a dual tyrosine kinase inhibitor (TKI) that inhibits downstream signaling initiated by the epidermal growth factor (EGF) receptor (EGFR, also known as
HER1) and HER2 [
13,
14]. However, resistance to lapatinib often arises through mechanisms, including increased drug efflux, enhanced pro-survival pathways and over-activation of PI3K-AKT signaling pathway [
4]. In this study, we used the
HER2 over-expressing breast cancer cell line, BT474 and a lapatinib resistant derivative (BT474 Lap
R), in an effort to investigate the functional outcome of FOXO3 acetylation as well as the role of FOXO3 acetylation in modulating lapatinib response.
3. Discussion
Overexpression of
HER2 gene occurs in ~20–25% of primary breast cancers and is associated with poor clinical outcomes in the metastatic patients [
27]. Monoclonal antibodies and tyrosine-kinase inhibitors are two major targeted therapies for HER2-positive breast cancer but these cancer cells often quickly develop adaptive responses to these HER2-targeted therapies [
28]. Previously, we and others have shown FOXO3 to be an important regulator in the antiproliferative potency of the tyrosine kinase inhibitors Gefitinib and lapatinib [
16,
17,
18,
19,
20]. To investigate further the role and regulation of FOXO3 in HER2-targeted therapies, a resistant derivative (BT474-Lap
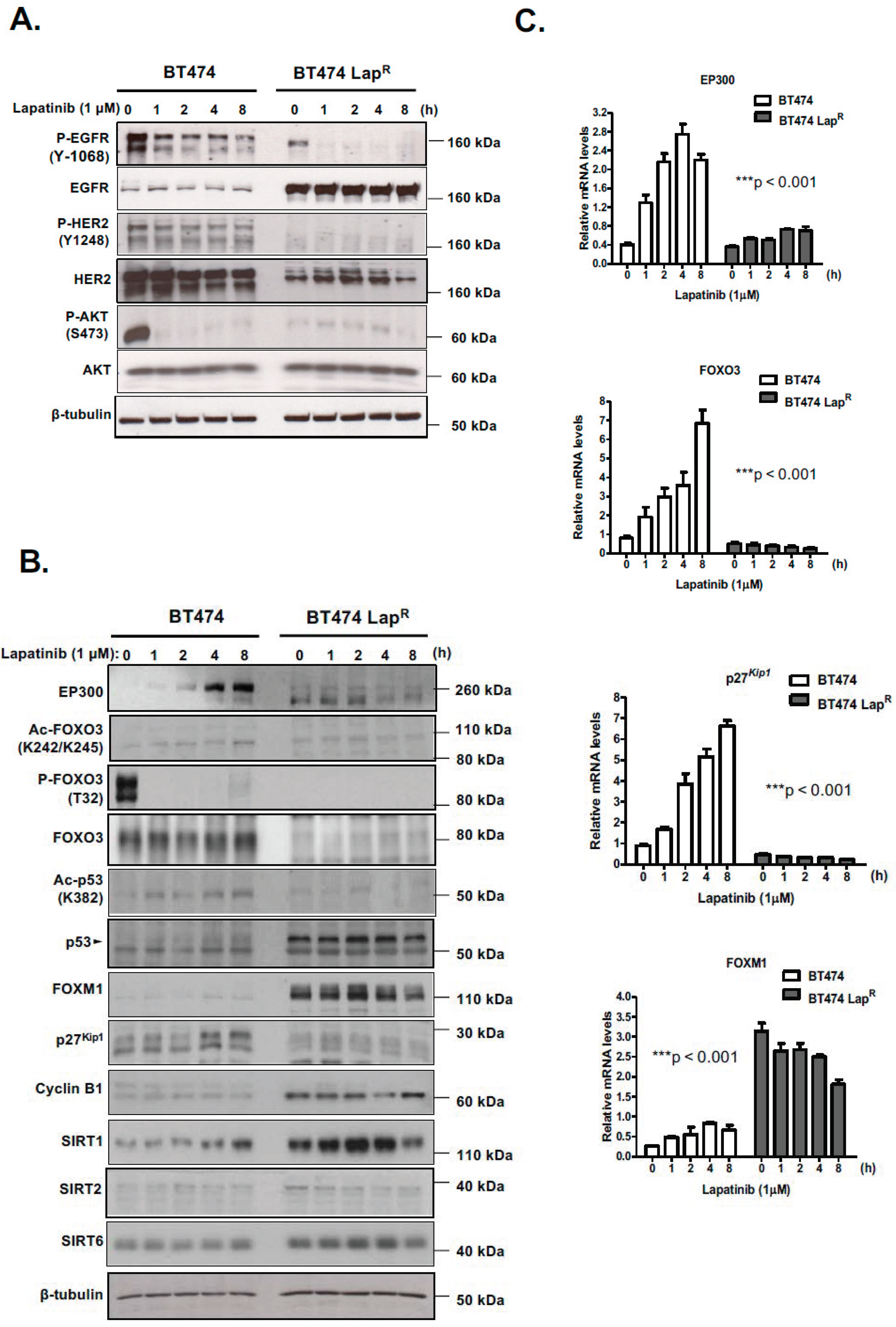
R) of the lapatinib-sensitive BT474 breast cancer cell line has been established. Lapatinib treatment causes dephosphorylation of EGFR, HER2 and AKT and subsequently dephosphorylation and activation of FOXO3. Moreover, EP300, FOXO3 and p27
Kip1 expression is increased upon lapatinib treatment in sensitive BT474 cells but not in resistant BT474-Lap
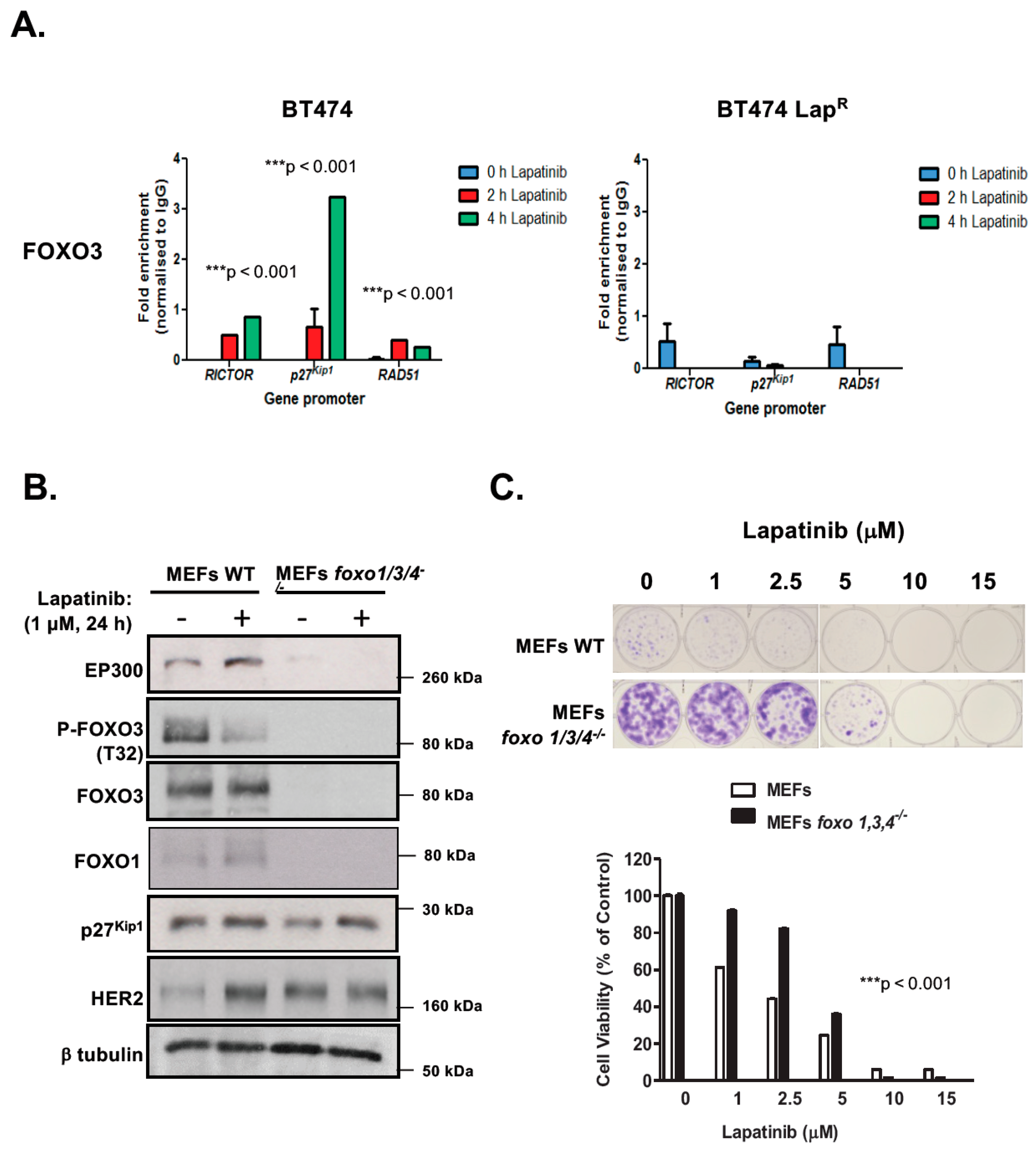
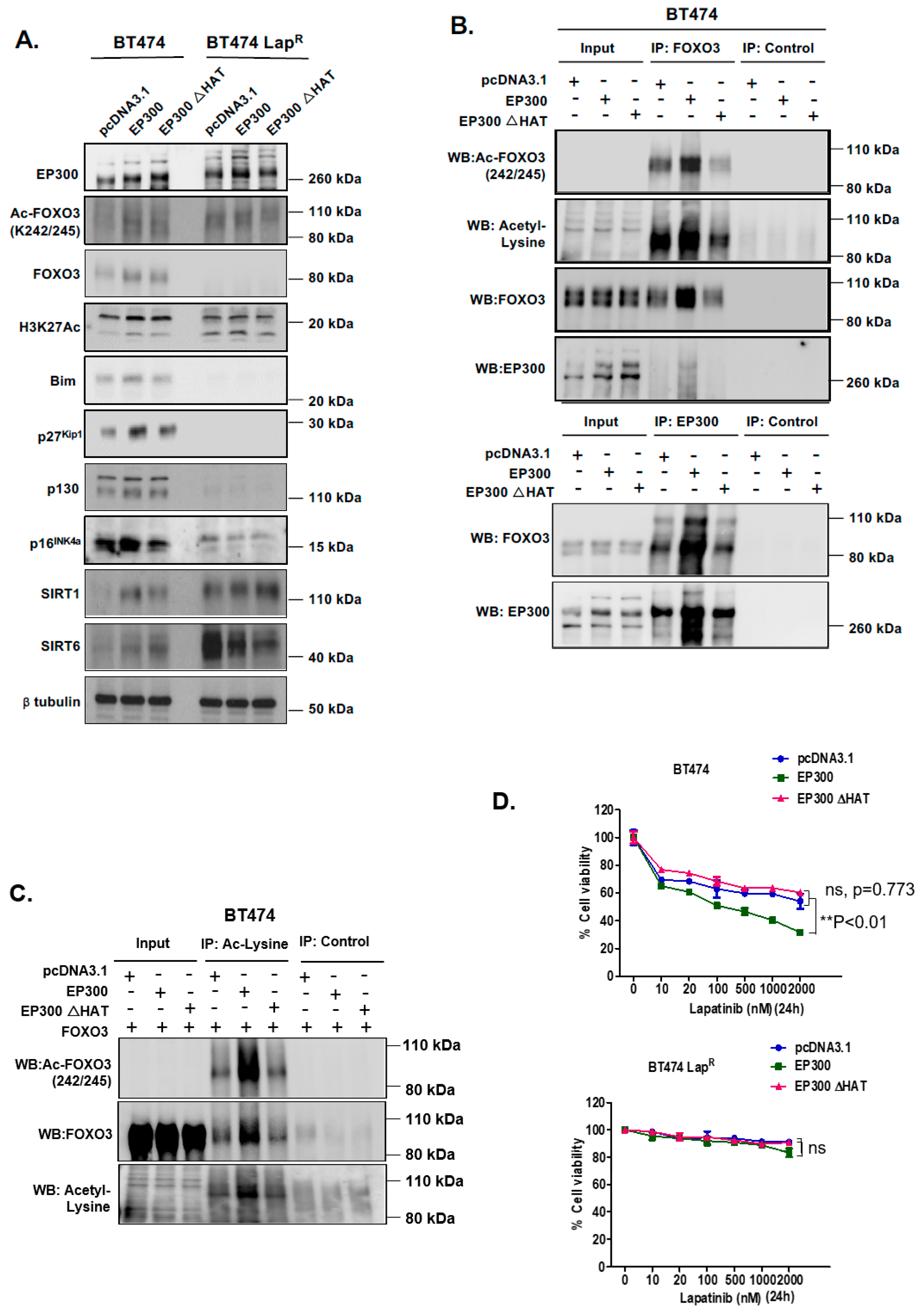
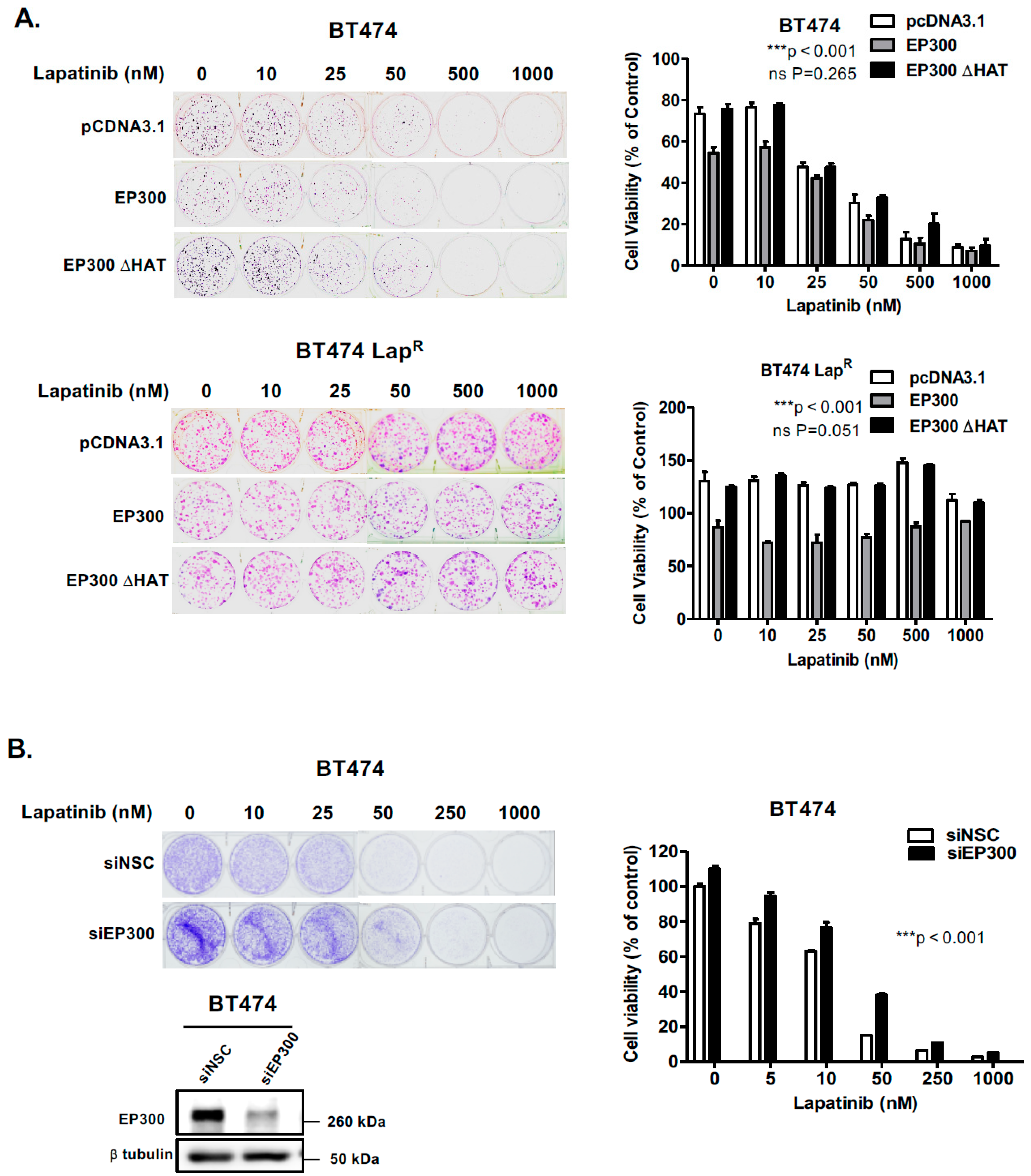
R cells, suggesting that EP300-mediated acetylation increases FOXO3 activity and expression in response to lapatinib in sensitive but not the resistant BT474 cells. In addition, Duolink PLA assays confirm that lapatinib induces interactions between EP300 and FOXO3 and the associated FOXO3 acetylation. All together the results suggest that effective lapatinib response involves EP300 and FOXO3 interaction which results in FOXO3 acetylation and subsequent antiproliferative target gene expression in BT474 sensitive cells. Notably, this EP300-mediated FOXO3 acetylation requires the acetyl-transferase activity of EP300 as the acetyl-transferase deficient HAΤ Δ1472–1522 mutant fails to cause FOXO3 acetylation in BT474 cells.
To evaluate the effect of FOXO3 acetylation more thoroughly, transient EP300 over-expression has been performed in both BT474 and BT474-LapR cells. The results show that EP300 overexpression induces FOXO3 expression and acetylation, which promotes the expression of the anti-proliferative target genes, including p27Kip1 and Bim in the sensitive BT474 cells. However, FOXO3 is deregulated and downstream targets uncoupled from FOXO3 in the resistant cells. In agreement, overexpression of FOXO3 increases both short and long-term lapatinib sensitivity in BT474 but not in BT474-LapR cells. ChIP assays provide further evidence that the insensitivity of the resistant cells to lapatinib treatment could be due to the failure of FOXO3 to be recruited to its target genes. Together, these results suggest a significant role of EP300-mediated acetylation of FOXO3 in regulating HER2-positive breast cancer cell survival and their lapatinib sensitivity.
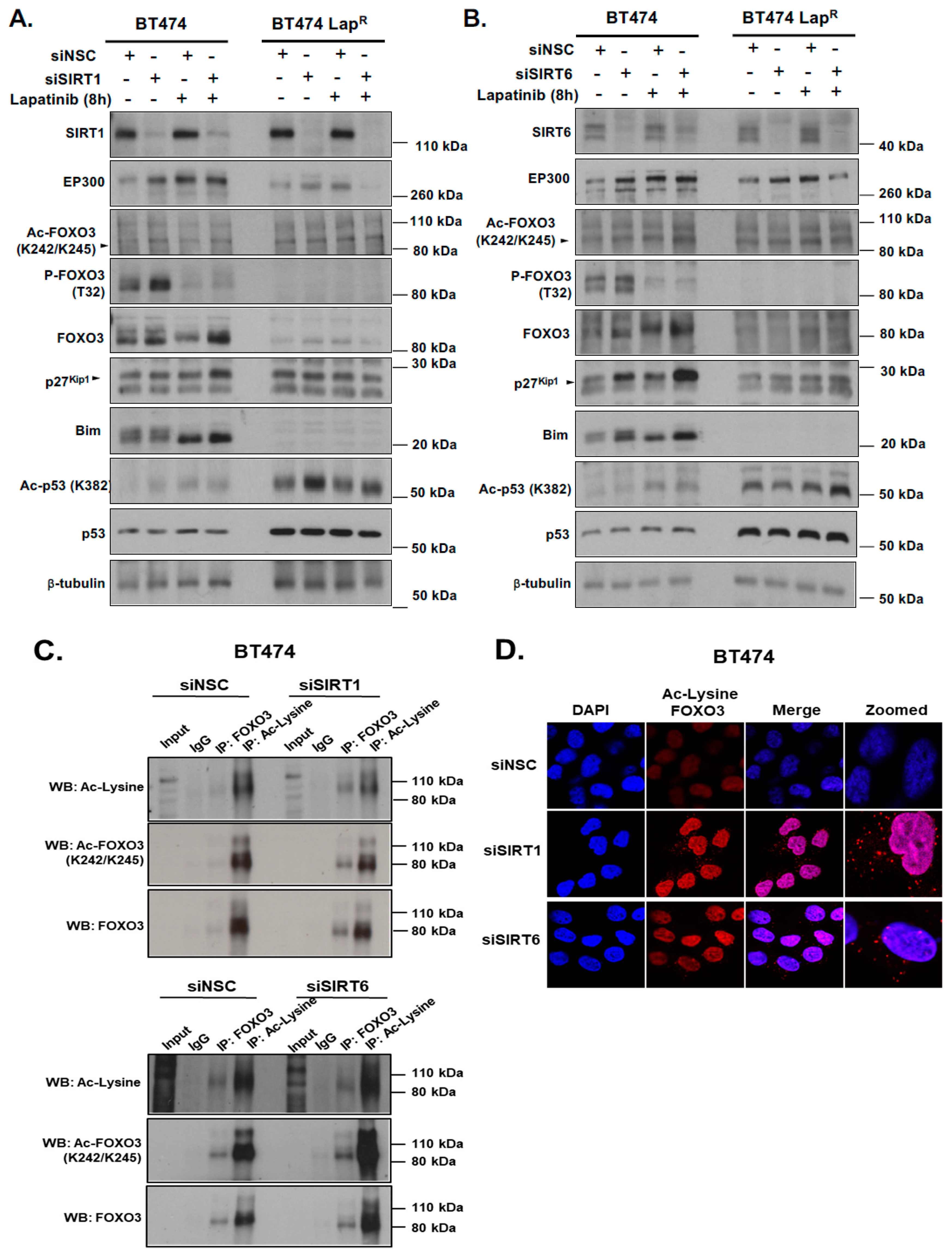
Notably, consistent low levels of EP300 and high levels of nuclear sirtuins (SIRT1 and SIRT6) are found in BT474-Lap
R cells. This suggests that the de-acetylation of FOXO3 by these nuclear SIRTs might also contribute to lapatinib resistance. Therefore, FOXO3 acetylation, regulated coordinately by EP300 and sirtuins, is an important PTM which regulates FOXO3 expression and transcriptional activity to modulate lapatinib sensitivity in breast cancer (
Supplementary Figure S6). Consistently, sirtuin-mediated deacetylation of FOXO3 has been reported to restrict EP300-mediated transactivation of FOXO3 and promote stress resistance and longevity [
2,
8]. Our data show that SIRT1 and SIRT6 knockdown in combination with lapatinib treatment causes an upregulation of FOXO3 expression and acetylation. Enhanced FOXO3 acetylation mediated by SIRT-depletion in BT474 cells also induces the expression of FOXO3 downstream target genes, such as
p27Kip1 and
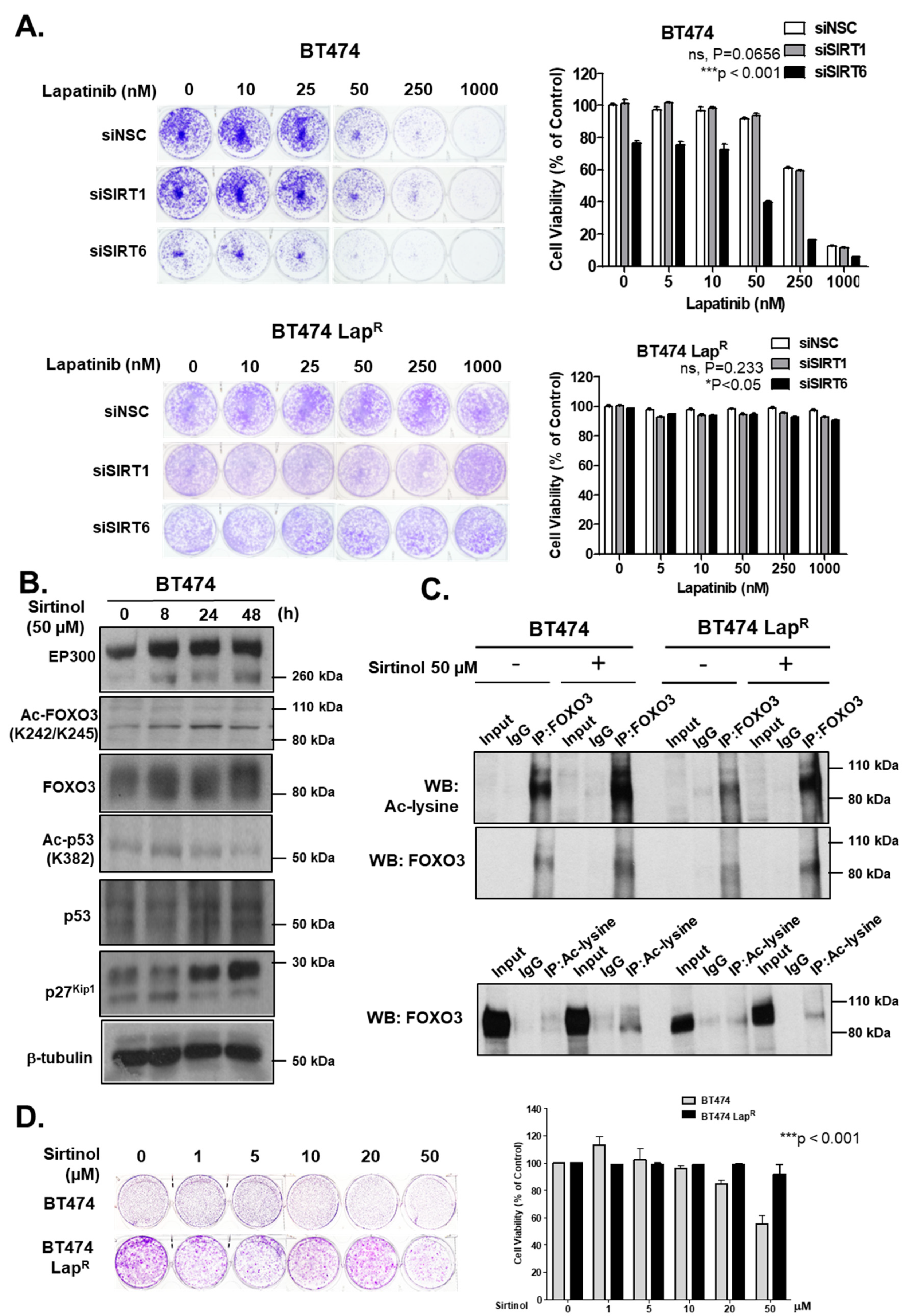
Bim. Moreover, knockdown of SIRT6 reduces the cell proliferation and increases lapatinib sensitivity in BT474 cells, although silencing of SIRT1 does not result in any significant effects on drug sensitivity. This could be due to the fact that SIRT6 plays a more significant role in the inhibition of FOXO3 acetylation in these cells and/or that SIRT6 can functionally compensate for the loss of SIRT1. In agreement, the pan-sirtuin inhibitor, sirtinol which can target both SIRT1 and SIRT6 increases p27
Kip1 expression in the BT474 cells.
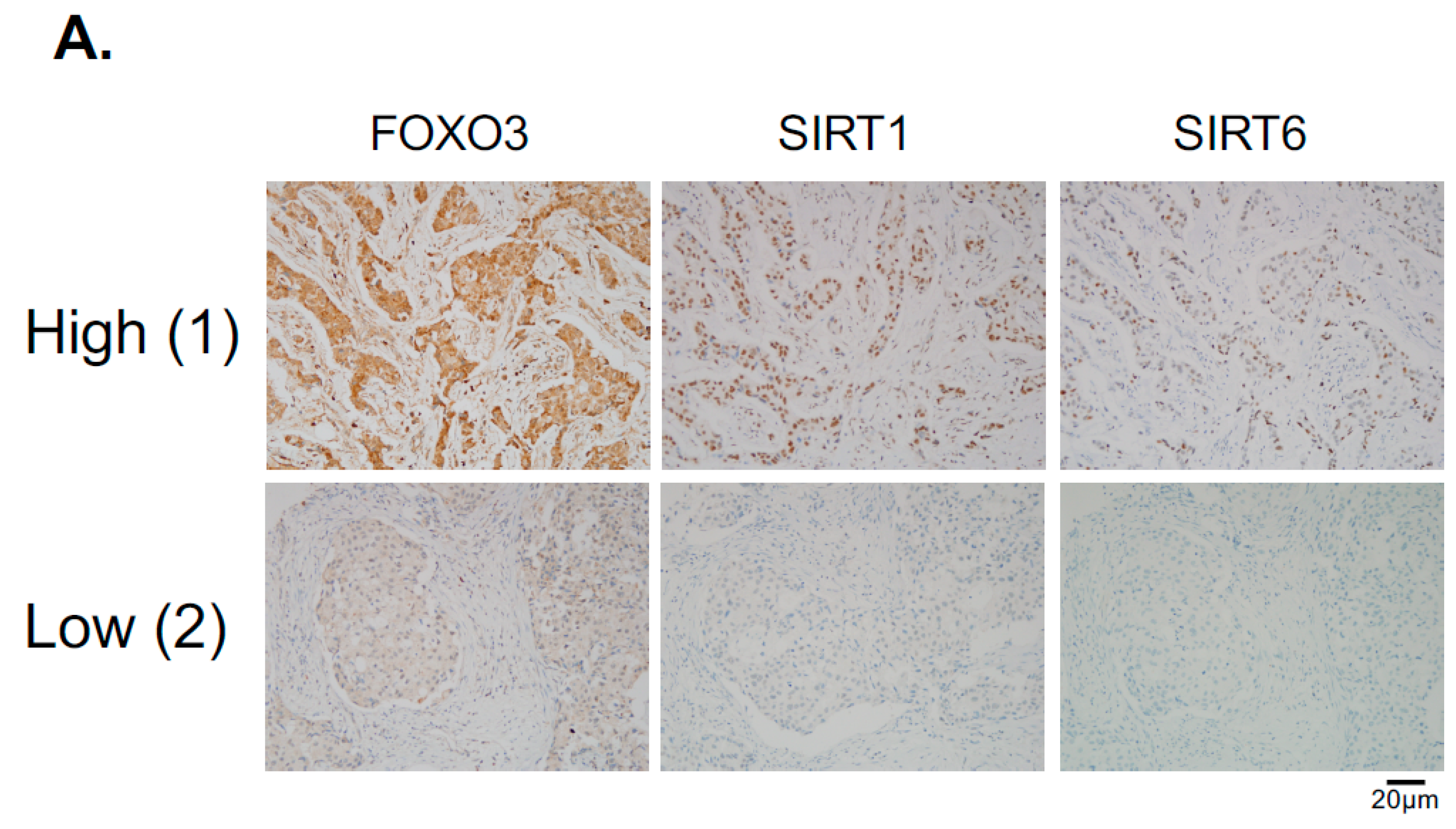
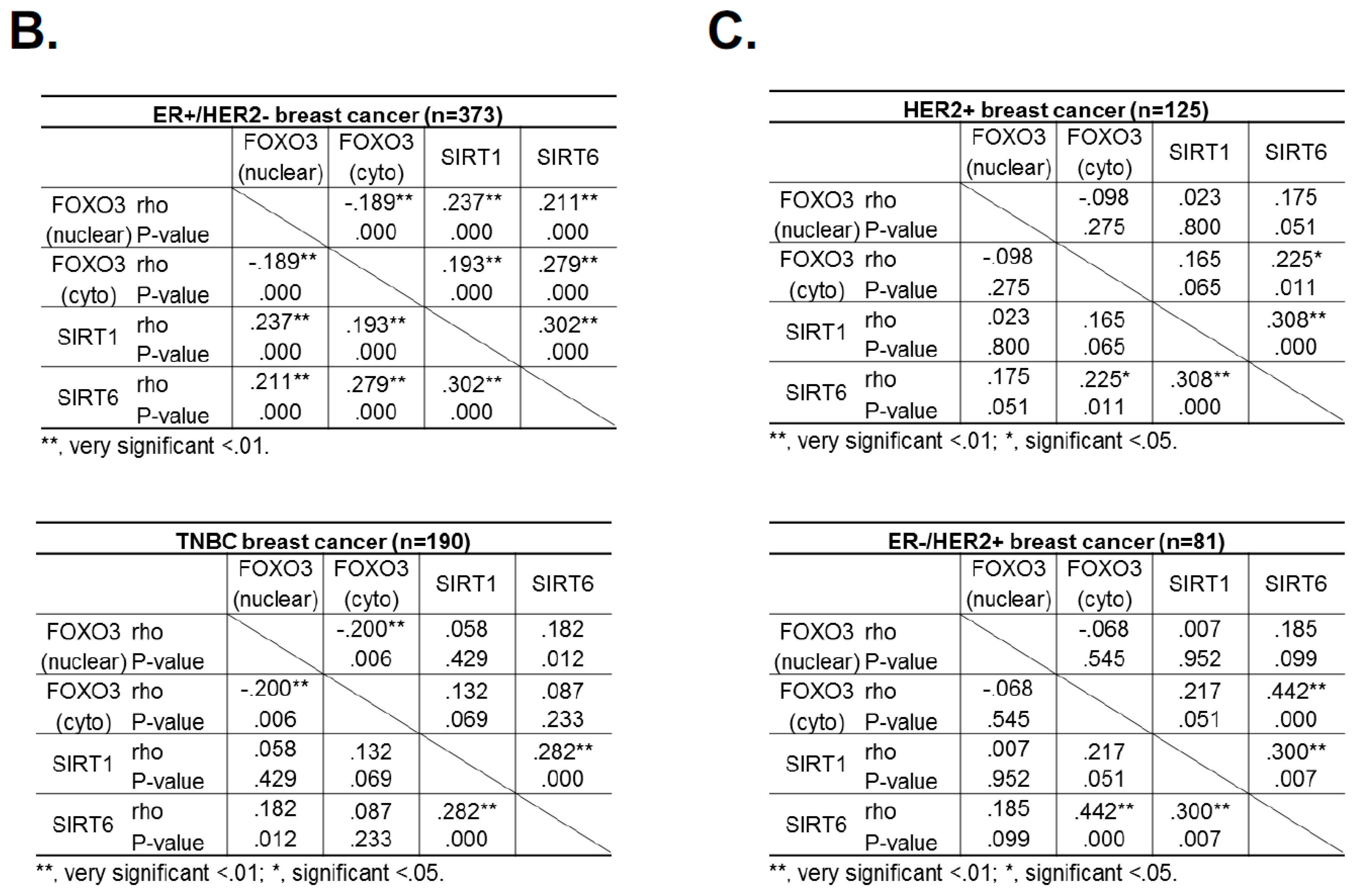
Since the levels of SIRT1 and SIRT6 do not change in response to lapatinib treatment, they are unlikely to have a direct role in modulating lapatinib signaling but may instead play a part in fine-tuning FOXO3 acetylation and therefore lapatinib sensitivity by setting the baseline for the de-acetylation activity in both lapatinib sensitive and resistant cancer cells. The finding that inhibition of SIRT1/6 by sirtinol does not increase lapatinib sensitivity in the resistant cells is likely due to the fact that EP300, which plays a critical part in FOXO3 acetylation, is not induced in the lapatinib resistant cells. This further suggests that SIRT1 and SIRT6 cooperate to set a threshold for FOXO3 acetylation in the cancer cells. Immunohistochemical staining results also show that FOXO3 expression correlates positively with SIRT6/1 levels in different breast cancer subtypes. As high FOXO3 expression, particularly in the nucleus, predicts FOXO3 deregulation/inactivation and poor prognosis in patients [
25,
26], these IHC results provide further evidence that SIRT1 and SIRT6 cooperate to deregulate FOXO3 activity.
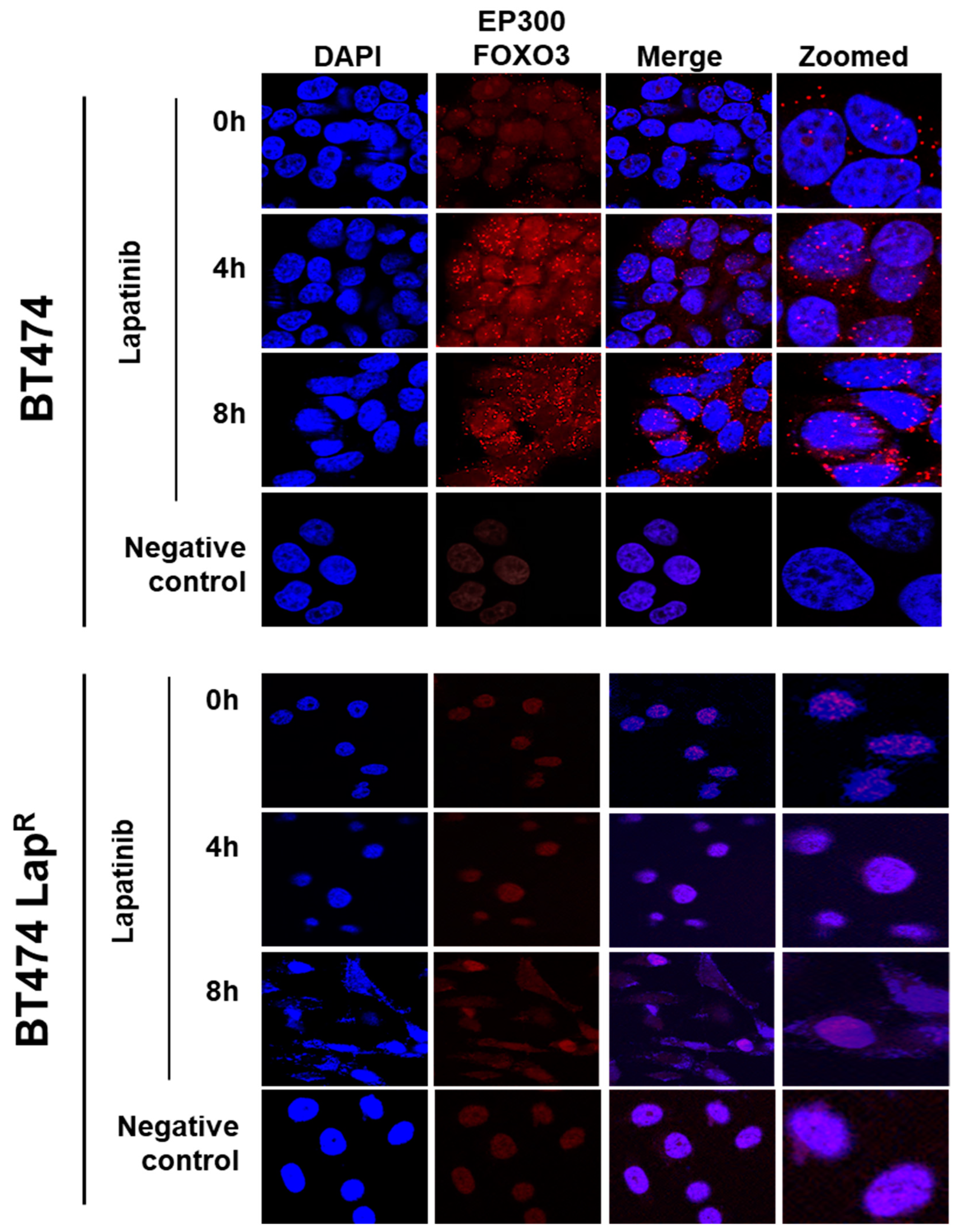
Lapatinib treatment is correlated with EP300 expression, FOXO3 acetylation and dephosphorylation (T32) and an increase in FOXO3 expression as well as cytoplasm to nucleus translocation in the sensitive BT474 cells. This suggests that lapatinib-induced FOXO3 acetylation via EP300 might drive FOXO3 dephosphorylation (T32), nuclear translocation and activation. Consistently, overexpression of wild-type EP300 alone can also induce FOXO3 acetylation, dephosphorylation (T32) and transcriptional activation in the sensitive BT474 cells. Interestingly, EP300 has previously been shown to mediate FOXO3 acetylation to antagonize its antiproliferative activity through restricting Bim expression [
29]. In contrast, we show in here that EP300 promotes the antiproliferative function of FOXO3 through mediating its acetylation. In agreement with our findings, FOXO3 acetylation has been shown to mediate the antiproliferative functions of glucocorticoid in B acute lymphoblastic leukemia (B-ALL) [
30]. Notably, we found that both HER2 and P-AKT are downregulated while EGFR is upregulated in the Lapatinib resistant cells and this is at odds with a previous study showing that both HER2 and P-AKT were upregulated in the Lapatinib resistant cells [
1]. The reason for the discrepancy is unclear. However, the HER2-inhibitor resistant cells used in the other study were primarily generated by retrovirally transducing hotspot PIK3CA mutations into HER2-amplified cells [
31]. The transduced PIK3CA mutations would maintain the high HER2 expression and high P-Akt activities as the PIK3CA mutations function downstream of HER2 will directly activate AKT activity. We have previously found that FOXO3 can control Akt activity (P-Akt) in a negative feedback mechanism [
32]. The Lapatinib-resistant cells have low FOXO3 expression as an adaptive response during the development of lapatinib resistance and as a result Akt activity (P-Akt) is also lower in the resistant cells.
In summary, in the present study we identify a role of EP300-mediated FOXO3 acetylation in the regulation of lapatinib sensitivity in breast cancer. Our findings also suggest that SIRT1 and SIRT6 also play a coordinate role in fine-tuning FOXO3 acetylation and therefore lapatinib sensitivity by setting a threshold for the de-acetylation activity in both lapatinib sensitive and resistant cancer cells. Our data suggest that FOXO3 acetylation by EP300 could be a potential marker for the identification of tumors that are likely to be sensitive to these drugs and indicate a novel therapeutic strategy to overcome lapatinib resistance in HER2-positive breast cancers.
4. Materials and Methods
4.1. Cell Lines, Cell Culture and Treatments
The BT474 cell line was originally obtained from the American Type Culture Collection (ATCC). Human breast cancer cell line, MCF-7 (Michigan Cancer Foundation-7) was acquired from the Cell Culture Service of Cancer Research UK, where they were tested and authenticated. lapatinib resistant BT474 cells (denoted as BT474 Lap
R) were derived from parental BT474 cells that were continuously exposed to 1 µM lapatinib for over 3 months. BT474-LapR cells were routinely maintained with 1 µM lapatinib in the culture medium. Wild type mouse embryonic fibroblasts (MEFs) and triple knock-out
foxo1/3/4-/- MEFs were kind gifts from Prof. Boudewijn Burgering, UMC, Utrecht, the Netherlands and have been described previously [
33]. All cells were cultured in Dulbecco’s modified eagle’s medium (DMEM) (Sigma Aldrich, Poole, UK) and supplemented with 10% (
v/
v) fetal calf serum (FCS) (First Link Ltd., Birmingham, UK), 100 Unit/mL penicillin/streptomycin (Sigma-Aldrich, UK) and 2 mM glutamine and maintained at 37 °C in a humidified atmosphere containing 10% CO
2. Cultured cells were also maintained by serial passaging using 25 g/L trypsin solution (Sigma Aldrich, Poole, UK) in 0.02% ethylene diamine tetra acetic acid (EDTA) (Sigma Aldrich, Poole, UK) when 70–80% confluence was reached.
4.2. Plasmids
Plasmid pcDNA3.1-p300 (plasmid #23252) and the acetyl-transferase mutant pcDNA3.1-p300 (HAT-) (plasmid #23254) were obtained from Addgene (Addgene, MA, USA). Empty vector pcDNA3.1 was purchased from Invitrogen. All plasmids were cloned into E. coli DH5α and the Purelink® HiPure Plasmid Maxiprep Kit (Thermo Fisher Scientific, Hemel Hampstead, UK) was used to isolate plasmid DNA as indicated in the manufacturer’s instructions. This kit uses an alkaline lysis protocol to isolate plasmid DNA that is bound to a positively charged resin before elution under high salt conditions. For gene silencing.
4.3. Breast Cancer Cell Transfections
BT474 and BT474-LapR cell lines were seeded to achieve approximately 60–70% confluency in T75 flasks before transfection. The plasmid-encoding human EP300, pcDNA3.1-EP300, the acetyl-transferase mutant, pcDNA3.1-p300 (HAT-) and the empty vector were transfected into cells using XtremeGene (Sigma-Aldrich). Transfection was performed at a 3:1 XtremeGene: DNA ratio using 8 µg of plasmid following manufacturer’s instruction. Transfected cells were counted and used for any subsequent assays 24 h post-transfection. For gene silencing, cells were plated in at 60–70% densities. The following day, cells were transfected with ON-TARGET plus siRNAs (GE Dharmacon, Horizon Discovery LTD, Cambridge, United Kingdom) targeting EP300 (L-003486-00-0005), SIRT1 (L-003540-00-0005) or SIRT6 (L-013306-00-0005) using oligofectamine (Invitrogen, Thermo Fisher Scientific, UK) according to the manufacturer’s protocol. Non-Targeting siRNA pool (GE Dharmacon; D-001210-01-05) was used as transfection control.
4.4. Duolink In Situ PLA In Situ
The analysis was carried out according to the manufacturer’s instructions [
34]. This assay is used to detect, visualize and quantify protein expression, protein interactions and specific post-translational protein modification [
34]. In the cases of detecting protein interactions and post-translational modification, two primary antibodies are required which are raised from different species. The secondary antibodies called PLA probes (PLA probe MINUS and PLA probe PLUS) and each one is conjugated with a unique oligonucleotide, were obtained from Sigma-Aldrich (DUO92008 SIGMA; POOLE UK). When the two PLA probes are in close proximity (<40 nm), the oligonucleotides will hybridize and join to a closed circle by adding enzymatic ligation solution. The ligated circle as a template, is then amplified through rolling circle amplification and generating several-hundredfold repeated sequence product subsequently. Since the amplification solution consists of nucleotides, polymerase as well as fluorescently labelled oligonucleotides, the replicated product can be easily visible as distinct fluorescent spot when viewed with a fluorescence microscopy.
4.5. Protein Extraction and Quantification
Whole cell extracts were prepared by harvesting cells by using 1×trypin-EDTA. Once detached, medium was added to inactivate the trypsin and samples were spun in 2000× rpm for 5 min. The supernatant was discarded and cell pellets were washed in PBS and spun for an additional 5 min (1200× g). The supernatant was discarded and the cell pellet was frozen at −80 °C until lysis was performed. Frozen pellets were lysed in lysis buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 5 mM EDTA, 5 mM Dithiothreitol (DTT), 1% NP-40 (or IGEPAL) and 1 tablet of protease inhibitors (“Complete” protease inhibitor mixture, as instructed by the manufacturer, Roche Applied Science; Burgess Hill, UK) supplemented with phosphatase inhibitors (1 mM sodium fluoride (NaF), 1 mM sodium orthovanadate (Na3VO4) and 1 mM PMSF as protease inhibitor. The samples were kept on ice for 10 min, vortexing every 3 min to disrupt the cell membranes. The supernatant (protein extract) was finally collected by centrifugation at 800× g for 10 min at 4 °C. Protein concentration was determined using the Pierce BCA Protein Assay Reagents A and B (Thermo Scientific) according to manufacturer’s instruction. Absorbance was read at 562 nm using the Sunrise spectrophotometer (Tecan; Reading, UK). Protein concentrations were determined by the equation of absorbance × 25 = µg/µL.
4.6. Western Blotting
SDS-PAGE gels are consisting of an upper stacking and a lower resolving gel. The resolving gel was made with varying amounts of bis-acrylamide solution, according to the molecular size of the protein being isolated with percentages of gels used varying from 7 to 14. Lower percentage gels allowed better examination of proteins of higher molecular weight (MW) and vice versa. The bis-acrylamide with 25% ammonium persulfate (APS) and tetramethylethylenediamine (TEMED) were used as the catalysts for gel polymerization.
To separate proteins, 20 μg of protein lysate was added with 2 × SDS loading buffer (200 mM Tris-HCl pH 6.8, 6% sodium dodecyl sulfate (SDS), 2 mM EDTA, 10% 2-mercaptoethanol, 10% glycerol, 0.02% bromophenol blue) and boiled for 5 min at 100 °C. Samples were then spun at 800× g for 1 min and the appropriate volume of sample was loaded into the stacking wells. SDS-PAGE gels were run in running buffer (25 mM Tris-Base, 250 mM glycine, 0.1% (w/v) SDS) at 60 V through the stacking gel and then 100 V through the resolving gel.
Once the proteins had been separated by SDS-PAGE, proteins were electro-transferred on to 0.45 μm Protran nitrocellulose membranes (Schleicher and Schuell, Whatman, Brentford, UK) using a wet tank blotting system (Bio-Rad Laboratories Trans-Blot Cell) in transfer buffer (25 mM Tris, 190 mM glycine and 20% (v/v) ethanol) for 90 min at 90 V. Tanks were packed in ice to avoid overheating.
Membranes were then blocked in 5% (w/v) bovine serum albumin (BSA) in Tris-buffered solution with 0.05% Tween-20 (TBST, pH 7.5) for 1 h at room temperature. Primary antibody incubation was performed with antibodies diluted in a 5% BSA-TBST solution overnight at 4 °C. The membranes were washed four times with TBST, every 5 min with 50 mL TBST at room temperature. Membranes were then incubated in their respective secondary antibodies (anti-rabbit or anti-mouse; Santa-Cruz Biotechnologies; Santa Cruz, CA, USA) coupled with horseradish peroxidase (Dako, Ely, UK) at a 1:2000 dilution for 1 h at room temperature. After that, membranes were washed for five times in every 5 min with 50 mL TBST at room temperature to remove excess secondary antibodies. Proteins were then visualized using enhanced chemiluminescence detection system (GE Healthcare, Cambridge, United Kingdom).
Primary antibody against Ac-FOXO3 (K242/K245) developed by Professor Eric lam and produced in PickCell Laboratories. Other primary antibodies used were P-EGFR (Cell signaling, #2234), EGFR (Cell signaling, #4267), P-HER2 (Cell signaling, #2244), HER2 (Cell signaling, #2242), P-AKT (S473) (Cell signaling, #4060), AKT (Cell signaling, #4685), β-tubulin (Santa Cruz Biotechnology, sc9104), EP300 (Abcam, ab10485), P-FOXO3 (T32) (Cell signaling, #9464), FOXO3 (cell signaling, #2497), Ac-p53 (K382) (Cell signaling, #2525), p53 (Santa Cruz Biotechnology, #sc98), FOXM1 (Santa Cruz Biotechnology, sc502), p27
Kip1 (Santa Cruz biotechnology, sc-528), SIRT1 (Abcam, Ab32441), SIRT2 (Santa Cruz Biotechnology, sc20966), SIRT6 (Cell Signaling, 2590 and Abcam, ab62739), HER3 (Cell signaling #4754), H3K27Ac (Abcam, #ab4729), Bim (Cell signaling, #2933), p130 (BD Biosciences, #610272), p16
INK4a (BD Biosciences, #554079), Acetylated lysine (Cell signaling, #9441). The original blots (
Supplementary Figure S7) were scanned and densitometry readings/intensity ratio of each band calculated for analysis (
Supplementary Figure S8).
4.7. Real-Time Quantitative PCR
Cells were harvested by centrifugation (1200× g) and cell pellets were stored at −80° until RNA extraction. Total mRNA was obtained from frozen cell pellets using the RNeasy Mini Kit (Qiagen, Manchester, United Kingdom) according to manufacturer’s instructions. mRNA purity and concentration were determined by measuring the spectrophotometric absorption at 260 nm and 280 nm on NanoDrop ND-1000.
Two µg of extracted total RNA was used as a template for first strand cDNA synthesis reaction and the resulting first-strand cDNA was used as template in the real-time PCR. The initial reaction mix to reverse transcribe the total RNA contains random primers, 10 µM of each dNTP and Superscript III reverse transcriptase (Invitrogen). The initial mixture was heated for 65 °C (5 min) for denaturation and cooled on ice. 5× First-Strand Buffer, 0.1 M DDT and RNaseOUT RNase Inhibitor (40 units/µL) were added to the denatured samples and were returned to the PCR machine programmed to incubate at 25 °C (5 min), 50 °C (60 min) for annealing and a final deactivation by heating at 70 °C for (15 min). By pooling equal amounts of all diluted cDNA samples, a cDNA standard was obtained. Four serial 1:4 dilutions (1/4, 1/16, 1/64 and 1/256) of the standard in water were used for generation of standard curve points. The nonregulated ribosomal housekeeping gene L19 was used as an internal control to normalize gene expression between samples. All amplifications were performed in triplicate.
Real time quantitative PCR (RT-qPCR) was performed using 100 ng cDNA which was added to SYBR-Green Master Mix (Applied BioSystems). All RT-qPCR assays were assayed in 96-well plates in the ABI PRISM® 7900 HT Fast Real-time PCR System (Applied BioSystems) on a cycling program of 90 °C for 10 min for enzyme activation followed by 40 cycles of denaturation and primer annealing/extension consisting of 95 °C for 10 s and 60 °C for 30 s, respectively.
Primers used for RT-qPCR are summarized in
Table 1.
4.8. Co-Immunoprecipitation Assay (Co-IP)
BT474 and BT474-LapR cells were harvested as already described for protein extraction. Cell pellets were lysed in IP lysis buffer [1% Nonidet P-40, 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 5 mM EDTA) supplemented with fresh 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, 1 mM DTT and one EDTA-free protease inhibitor tablet (Complete protease inhibitor cocktail; Roche, Lewes, UK). The samples were incubated for 20 min on ice, vortexing every 5 min to disrupt cell membrane and then centrifuged at 800× g for 10 min at 4 °C to collect protein extract in new Eppendorf tube. After protein quantification using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA), 10% of the total protein (25–50 µg) from each sample was kept as an input control. Subsequently, 250–500 µg of each lysate was rotated with 20 µL of pre-washed protein agarose Dynabeads A (Novex, Life |Technologies, Carlsbad, CA, USA) for 4 h at 4 °C in order to reduce non-specific binding. After the pre-clearing step, the lysates were transferred to new Eppendorf tubes containing new beads that have been previously washed three times with IP lysis buffer. Samples were incubated overnight with 0.5 µg of primary antibody-FOXO3a (sc-11351, Santa Cruz), Anti-acetyl lysine (Cell-Signaling technology) or IgG negative control (2729, Cell-Signaling technology) at 4 °C on a rotator. The supernatant was discarded on the following day and beads were washed three times with IP lysis buffer. Samples were boiled with 2 × SDS sample buffer for 5 min at 100 °C and were analyzed by SDS-PAGE followed by western blotting.
4.9. Chromatin Immunoprecipitation (ChIP)
BT474 cells were either left untreated or treated with 1 μM Lapatinib for 2 and 4 h, before harvesting. DNA fragments were purified using phenol chloroform (Sigma Aldrich, St Louis, MO, USA). For subsequent qPCR analysis, the 7900HT Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) was used. The first stage was held at 95 °C for 10 min and subsequent amplification was carried out by 40 cycles of the following three steps: 95 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s. A dissociation stage consisting of three steps (95 °C, 60 °C and 95 °C for 15 s each) was also performed to test primer specificity. The fold enrichment method was finally used for analysis the immunoprecipitated DNA, with each ChIP signal normalized to its respective DNA concentration, input sample and background signal obtained for the IgG negative control. Primer pair sequences for ChIP analyses are
Rictor-F: AAACAAACCCATCTCCACTGC;
Rictor-R: TTTCCCTTCTGCTTCCCTTT;
p27Kip1-F: CCGTTTGGCTAGTTTGTTTGTCT;
p27kip1-R: TGACTGCTGGAGGGGTACTG;
RAD51-F: ATGCGAGTAGGAGGCTCAGA;
RAD51-R: AGCGCTCTTGTGGTTTGTTT;
4.10. Sulphorhodamine B (SRB) Colourimetric Assay
Drug sensitivity to Lapatinib was tested using the SRB colorimetric assay. Drug sensitive and resistant BT474 cells were seeded 24-h post-transfection in 96-well plates, 3000 and 1000 cells per well, respectively. Cells were allowed to attach overnight and then treated with the Lapatinib concentration indicated (0.1, 1.0 or 1.5 µM). Cells were then fixed 24, 48 or 72 h following treatment using ice cold 20% (w/v) trichloroacetic acid (TCA) for 1 h. Cells were then washed, allowed to dry overnight and stained with 0.4% SRB in 0.1% (v/v) acetic acid for 30 min. Excess dye was washed off with 1% (v/v) acetic acid and 10 mM Tris was then used to dissolve SRB by shaking for 30 min at room temperature. Absorbance was finally measured at 492 nm using a Sunrise microplate reader (Tecan, Männedorf, Switzerland).
4.11. Clonogenic Assay
Long term sensitivity to Lapatinib was tested by the clonogenic assay. One day post-transfection, the cells were counted and plated into 6-well plates at a density of 1000 and 500 cells per well for drug sensitive and resistant BT474 cells, respectively. Colony formation was allowed for 28 and 14 days in sensitive and resistant BT474 cells, respectively. At the end of this period, cells were fixed with 1% Formaldehyde in PBS, washed three times with PBS and stained with 0.5% (w/v) crystal violet. Pictures were taken and the stain was solubilized with 33% acetic acid by shaking at room temperature until completely dissolved. Absorbance was finally measured at 592 nm using a Sunrise microplate reader (Tecan, Männedorf, Switzerland).
4.12. Patients and Tissue Specimens
The patient tissue samples came from a study consisting of 688 breast cancer patients who underwent surgery for primary breast cancer between 1993 and 1998, at the Department of Pathology, Asan Medical Center, Seoul, Korea. Formalin-fixed, paraffin-embedded tissue samples from these preoperatively chemo- and radiotherapy naive patients were available for analysis as previously described [
35]. The expression levels of standard biomarkers, including estrogen receptor (ER), progesterone receptor (PR) and HER2, were reviewed by immunohistochemical staining at the time of diagnosis, independently by two pathologists. HER2-overexpressing tumors were defined as those that score of 3+ by immunohistochemistry (IHC) or by gene amplification using either fluorescence in situ hybridization or silver in situ hybridization [
36]. Exemption from informed consent after the de-identification of information was approved by the Institutional Review Board of Asan Medical Center (2013-0641). Tissue microarray sections were evaluated with an automated immunohistochemical staining device (Benchmark XT; Ventana Medical Systems, Tucson, AZ, USA). Antibodies to FOXO3 (1:400 dilution; EMD Millipore, Billerica, MA, USA), SIRT1 (1:50 dilution; Abcam, Cambridge, MA, USA), SIRT6 (1:50 dilution; Cell Signaling Technology, Danvers, MA, USA) were used. Each case was evaluated by estimating the percentage and staining intensity (negative, 0; weak, 1; moderate, 2; and strong, 3) of tumor cells showing a cytoplasmic or nuclear staining pattern for FOXO3. We then classified the expression levels as high or low based on the mean staining intensity value obtained by the multiplication of each protein.
4.13. Statistical Analysis
Chi-square statistical analysis were used to test the correlations between protein expression of breast cancer patients, respectively using SPSS 16.0 (Imperial College London, Software Shop, UK); * p ≤ 0.05 and ** p ≤ 0.01 were considered as statistically significant and very significant, respectively. Other results shown represent the average of 3 independent experiments, which were each performed in triplicate (presented as the mean ± SEM). GraphPad Prism was used for statistical analysis (version 5, San Diego, CA, USA) and two-tailed Student’s t-test was used to compare the means. Differences between the two groups were considered statistically significant at p < 0.05. For comparisons between groups of more than two unpaired values, one-way analysis of variance (ANOVA) was used. Two-way ANOVA was used between groups of 2 variables and p < 0.05 was considered as statistically significant.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}