Diminished Condensin Gene Expression Drives Chromosome Instability That May Contribute to Colorectal Cancer Pathogenesis


Abstract
1. Introduction
2. Results
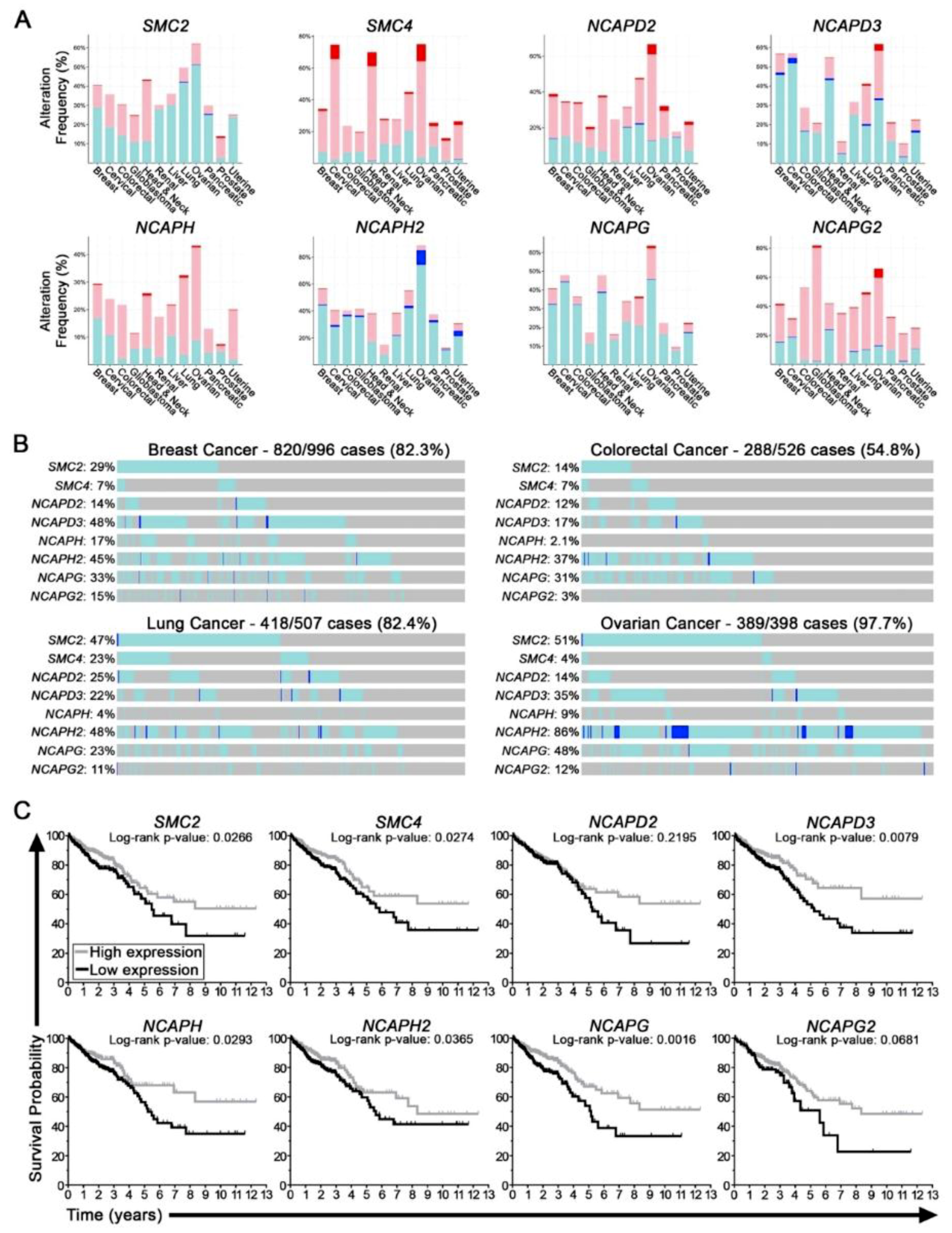
2.1. Condensin Genes Are Frequently Altered in Cancer and Correlate with Worse Patient Outcomes in Colorectal Cancer
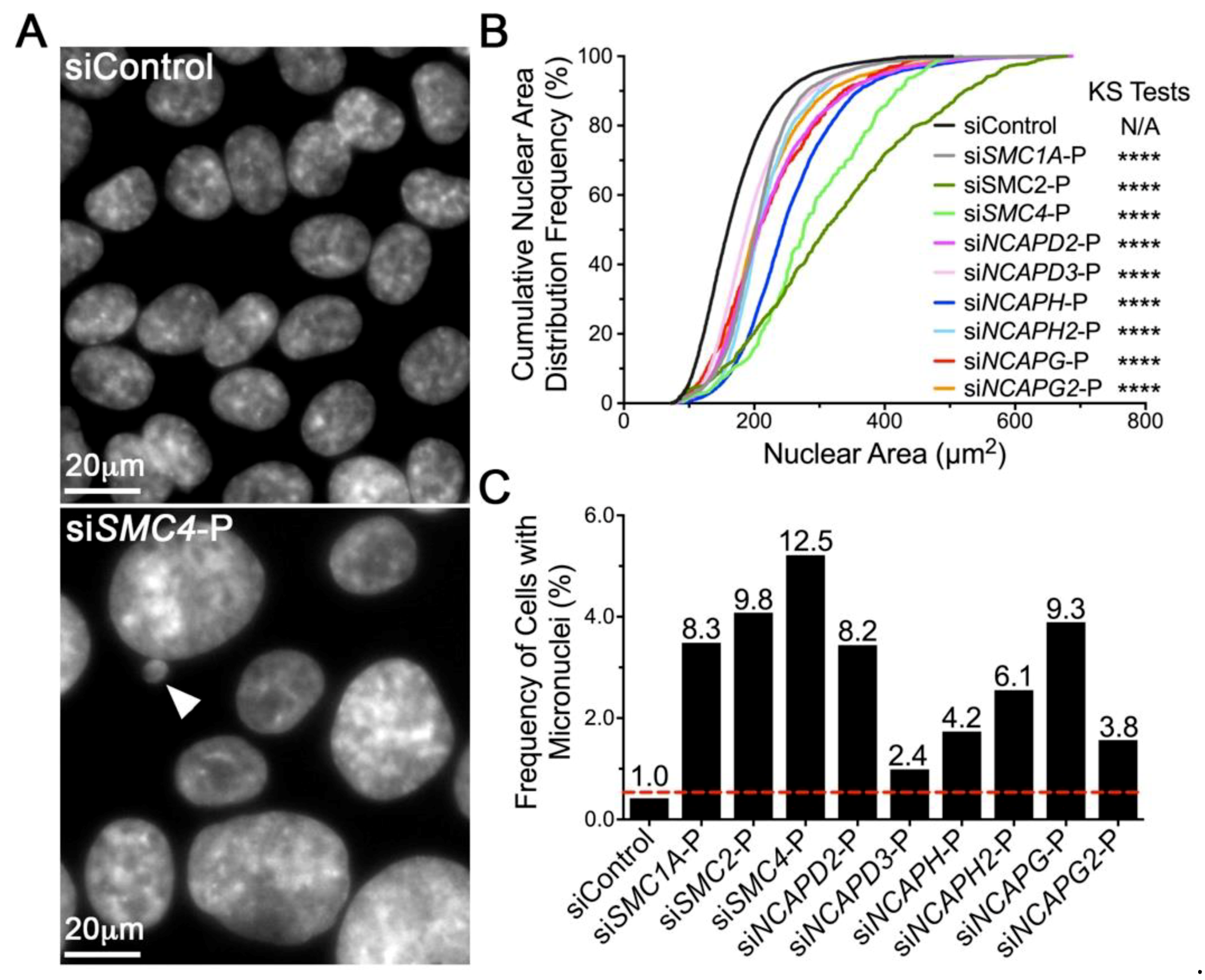
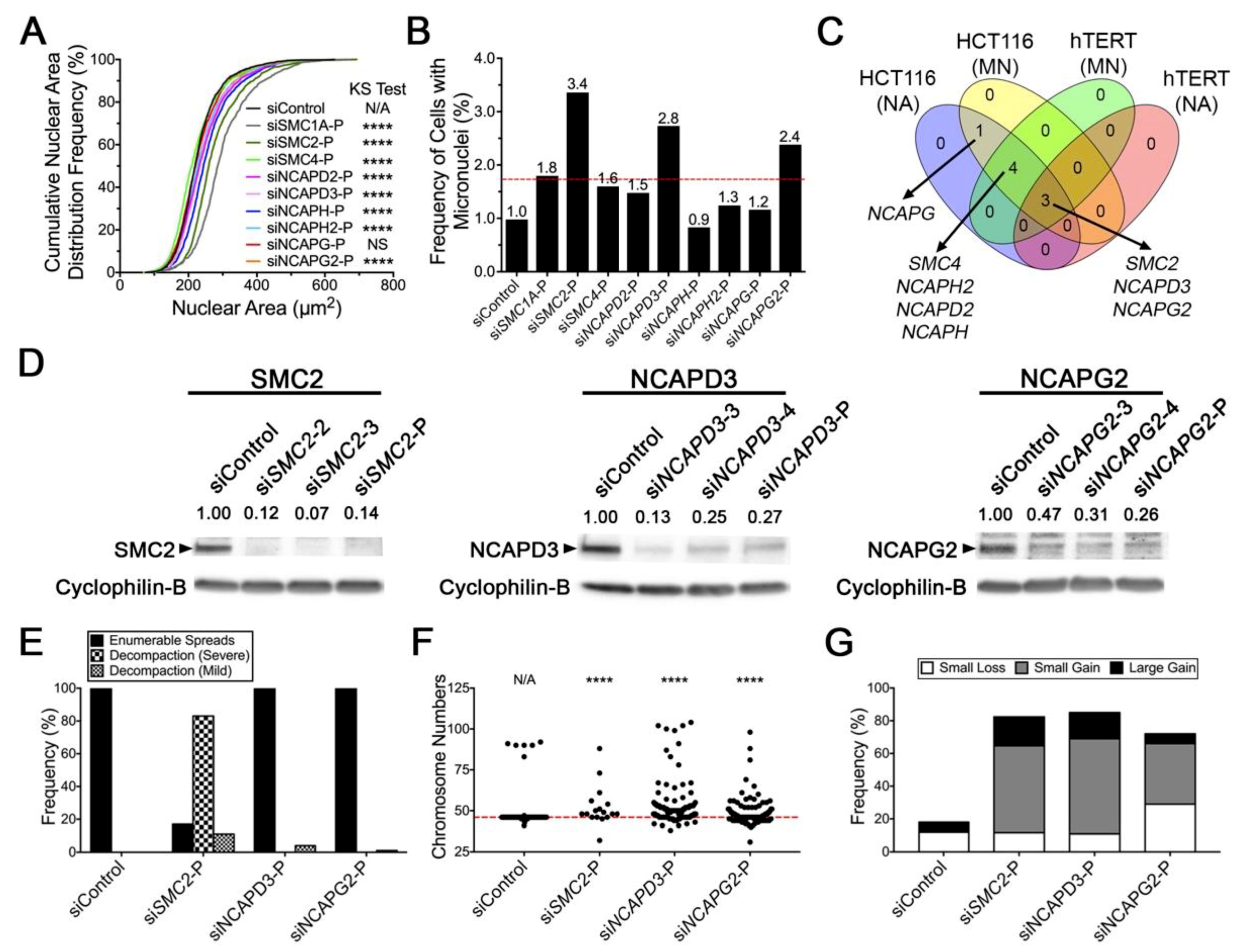
2.2. Diminished Condensin Gene Expression Induces Increases in Nuclear Areas (NAs) and Micronucleus (MN) Formation in HCT116 Cells
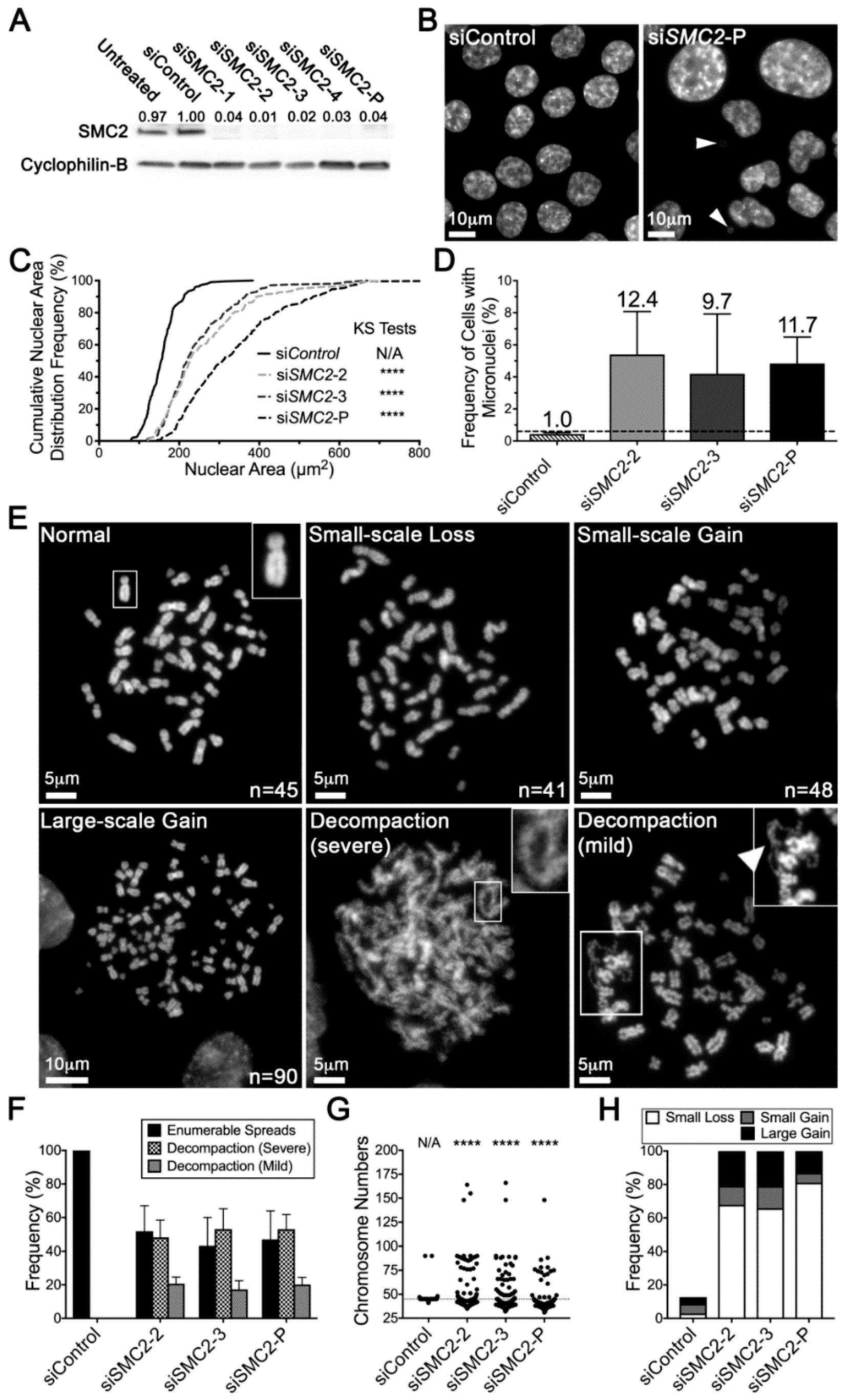
2.3. Reduced SMC2 Expression Is Associated with CIN and Chromosome Decompaction
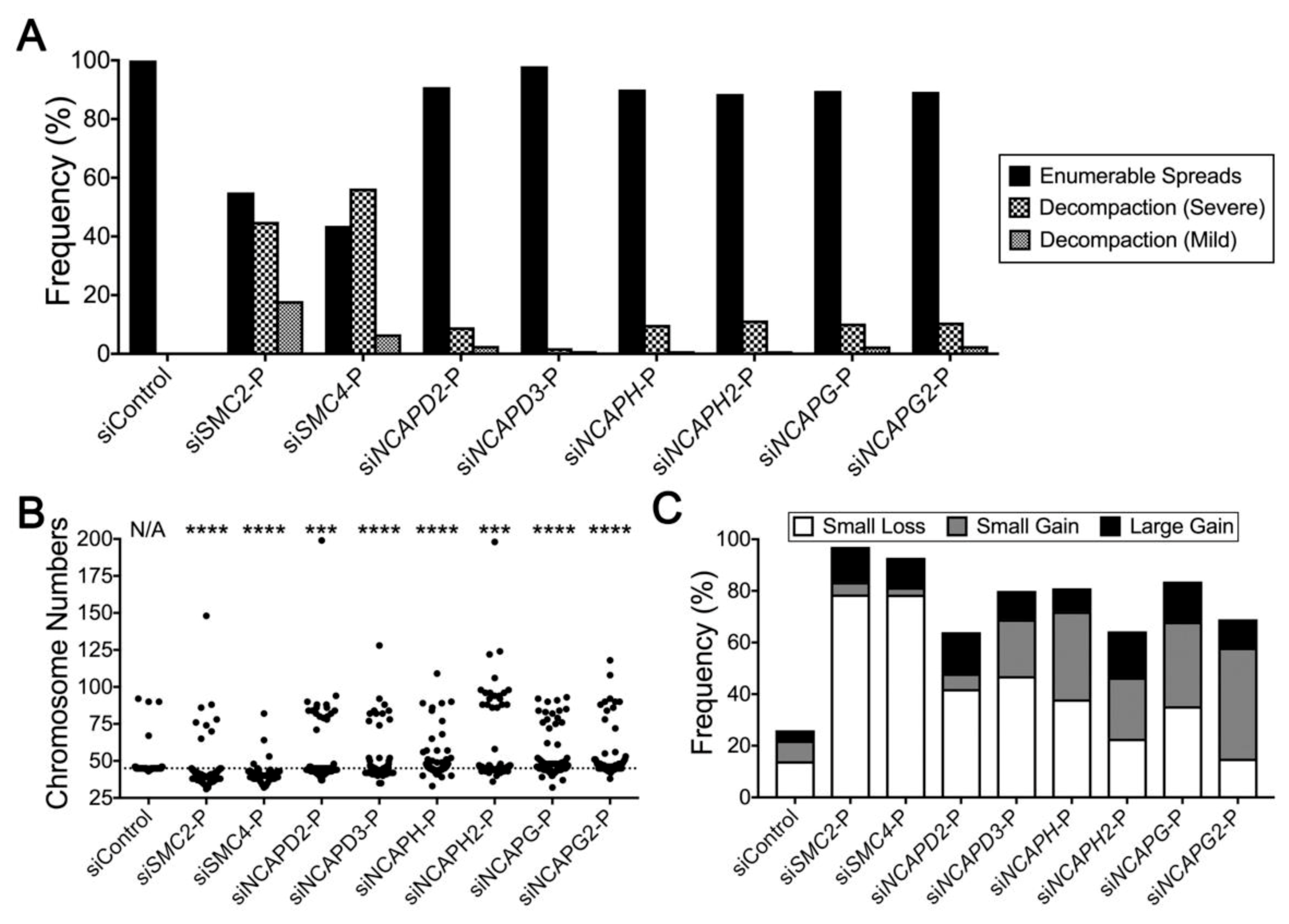
2.4. Condensin Gene Silencing Induces Numerical CIN and Structural Defects in HCT116 Cells
2.5. Reduced Condensin Gene Expression Induces CIN Phenotypes That Are Independent of Cell Type
3. Discussion
4. Materials and Methods
4.1. The Cancer Genome Atlas—Gene Alteration and Outcome Analyses
4.2. Cell Culture
4.3. Gene Silencing and Western Blot Analysis
4.4. Single Cell Quantitative Imaging Microscopy: Nuclear Areas and Micronucleus Formation
4.5. Mitotic Chromosome Spreads and Chromosome Enumeration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Facts Figures 2019; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Canadian Cancer Society’s Advisory Committee on Cancer Statistics. Canadian Cancer Statistics 2017; Canadian Cancer Society: Toronto, ON, Canada, 2017. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.C.; Morden, C.R.; Palmer, M.C.L.; Nachtigal, M.W.; McManus, K.J. Detecting Chromosome Instability in Cancer: Approaches to Resolve Cell-to-Cell Heterogeneity. Cancers (Basel) 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Tijhuis, A.E.; Johnson, S.C.; McClelland, S.E. The emerging links between chromosomal instability (CIN), metastasis, inflammation and tumour immunity. Mol. Cytogenet. 2019, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Komarova, N.L.; Sengupta, A.; Jallepalli, P.V.; Shih Ie, M.; Vogelstein, B.; Lengauer, C. The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. USA 2002, 99, 16226–16231. [Google Scholar] [CrossRef] [PubMed]
- Losi, L.; Baisse, B.; Bouzourene, H.; Benhattar, J. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis 2005, 26, 916–922. [Google Scholar] [CrossRef]
- Gao, C.; Su, Y.; Koeman, J.; Haak, E.; Dykema, K.; Essenberg, C.; Hudson, E.; Petillo, D.; Khoo, S.K.; Vande Woude, G.F. Chromosome instability drives phenotypic switching to metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 14793–14798. [Google Scholar] [CrossRef]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Stirling, P.C.; Bloom, M.S.; Solanki-Patil, T.; Smith, S.; Sipahimalani, P.; Li, Z.; Kofoed, M.; Ben-Aroya, S.; Myung, K.; Hieter, P. The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet. 2011, 7, e1002057. [Google Scholar] [CrossRef]
- de la Barre, A.E.; Gerson, V.; Gout, S.; Creaven, M.; Allis, C.D.; Dimitrov, S. Core histone N-termini play an essential role in mitotic chromosome condensation. EMBO J. 2000, 19, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Piskadlo, E.; Oliveira, R.A. Novel insights into mitotic chromosome condensation. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yu, L.; Bowen, J.; Gorovsky, M.A.; Allis, C.D. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 1999, 97, 99–109. [Google Scholar] [CrossRef]
- Hirano, T. Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 2016, 164, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F. SMC complexes: from DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 2016, 17, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Losada, A.; Hirano, M.; Myers, M.P.; Neuwald, A.F.; Hirano, T. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 2003, 115, 109–121. [Google Scholar] [CrossRef]
- Hirano, T.; Kobayashi, R.; Hirano, M. Condensins chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell 1997, 89, 511–521. [Google Scholar] [CrossRef]
- Houlard, M.; Godwin, J.; Metson, J.; Lee, J.; Hirano, T.; Nasmyth, K. Condensin confers the longitudinal rigidity of chromosomes. Nat. Cell Biol. 2015, 17, 771–781. [Google Scholar] [CrossRef]
- Martin, C.A.; Murray, J.E.; Carroll, P.; Leitch, A.; Mackenzie, K.J.; Halachev, M.; Fetit, A.E.; Keith, C.; Bicknell, L.S.; Fluteau, A.; et al. Mutations in genes encoding condensin complex proteins cause microcephaly through decatenation failure at mitosis. Genes. Dev. 2016, 30, 2158–2172. [Google Scholar] [CrossRef]
- Palou, R.; Dhanaraman, T.; Marrakchi, R.; Pascariu, M.; Tyers, M.; D’Amours, D. Condensin ATPase motifs contribute differentially to the maintenance of chromosome morphology and genome stability. PLoS Biol. 2018, 16, e2003980. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; McManus, K.J. A novel multiplexed, image-based approach to detect phenotypes that underlie chromosome instability in human cells. PLoS ONE 2015, 10, e0123200. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Liu, G.; Stevens, J.B.; Abdallah, B.Y.; Horne, S.D.; Ye, K.J.; Bremer, S.W.; Chowdhury, S.K.; Ye, C.J. Karyotype heterogeneity and unclassified chromosomal abnormalities. Cytogenet. Genome Res. 2013, 139, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Kumar, Y. Cancer cell micronucleus: an update on clinical and diagnostic applications. APMIS 2013, 121, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Stopper, H.; Muller, S.O. Micronuclei as a biological endpoint for genotoxicity: A minireview. Toxicol. In Vitro 1997, 11, 661–667. [Google Scholar] [CrossRef]
- Barber, T.D.; McManus, K.; Yuen, K.W.; Reis, M.; Parmigiani, G.; Shen, D.; Barrett, I.; Nouhi, Y.; Spencer, F.; Markowitz, S.; et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 3443–3448. [Google Scholar] [CrossRef] [PubMed]
- Sajesh, B.V.; Bailey, M.; Lichtensztejn, Z.; Hieter, P.; McManus, K.J. Synthetic lethal targeting of superoxide dismutase 1 selectively kills RAD54B-deficient colorectal cancer cells. Genetics 2013, 195, 757–767. [Google Scholar] [CrossRef]
- Asbaghi, Y.; Thompson, L.L.; Lichtensztejn, Z.; McManus, K.J. KIF11 silencing and inhibition induces chromosome instability that may contribute to cancer. Genes Chromosomes Cancer 2017, 56, 668–680. [Google Scholar] [CrossRef]
- Guppy, B.J.; McManus, K.J. Mitotic accumulation of dimethylated lysine 79 of histone H3 is important for maintaining genome integrity during mitosis in human cells. Genetics 2015, 199, 423–433. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Cogan, N.; Baird, D.M.; Phillips, R.; Crompton, L.A.; Caldwell, M.A.; Rubio, M.A.; Newson, R.; Lyng, F.; Case, C.P. DNA damaging bystander signalling from stem cells, cancer cells and fibroblasts after Cr(VI) exposure and its dependence on telomerase. Mutat. Res. 2010, 683, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; Gupta, A.; Wang, H.; Scherthan, H.; Dhar, S.; Gandhi, V.; Iliakis, G.; Shay, J.W.; Young, C.S.; Pandita, T.K. hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene 2003, 22, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Nishide, K.; Hirano, T. Overlapping and non-overlapping functions of condensins I and II in neural stem cell divisions. PLoS Genet. 2014, 10, e1004847. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.P.; Beesley, J.; Amin Al Olama, A.; Michailidou, K.; Tyrer, J.; Kote-Jarai, Z.; Lawrenson, K.; Lindstrom, S.; Ramus, S.J.; Thompson, D.J.; et al. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016, 6, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yu, H. The Smc complexes in DNA damage response. Cell Biosci. 2012, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Heale, J.T.; Ball, A.R., Jr.; Schmiesing, J.A.; Kim, J.S.; Kong, X.; Zhou, S.; Hudson, D.F.; Earnshaw, W.C.; Yokomori, K. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol. Cell 2006, 21, 837–848. [Google Scholar] [CrossRef]
- Kong, X.; Stephens, J.; Ball, A.R., Jr.; Heale, J.T.; Newkirk, D.A.; Berns, M.W.; Yokomori, K. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS ONE 2011, 6, e23548. [Google Scholar] [CrossRef]
- Wood, J.L.; Liang, Y.; Li, K.; Chen, J. Microcephalin/MCPH1 associates with the Condensin II complex to function in homologous recombination repair. J. Biol. Chem. 2008, 283, 29586–29592. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Kaushal, S.; Freudenreich, C.H. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 2019, 58, 270–283. [Google Scholar] [CrossRef]
- Stephens, A.D.; Haase, J.; Vicci, L.; Taylor, R.M.; Bloom, K. Cohesin, condensin, and the intramolecular centromere loop together generate the mitotic chromatin spring. J. Cell Biol. 2011, 193, 1167–1180. [Google Scholar] [CrossRef]
- Moore, L.L.; Stanvitch, G.; Roth, M.B.; Rosen, D. HCP-4/CENP-C promotes the prophase timing of centromere resolution by enabling the centromere association of HCP-6 in Caenorhabditis elegans. Mol. Cell Biol. 2005, 25, 2583–2592. [Google Scholar] [CrossRef]
- Thompson, L.L.; Jeusset, L.M.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers (Basel) 2017, 9, 151. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef]
- Cunningham, C.E.; MacAuley, M.J.; Yadav, G.; Vizeacoumar, F.S.; Freywald, A.; Vizeacoumar, F.J. Targeting the CINful genome: Strategies to overcome tumor heterogeneity. Prog. Biophys. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Target | Catalogue No. | Source | Host Species | Working Dilution |
|---|---|---|---|---|
| SMC2 | ab10399 | Abcam | Rabbit | 1:5000 |
| SMC4 | ab179831 | Abcam | Rabbit | 1:1000 |
| NCAPD2 | HPA037363 | Sigma-Aldrich | Rabbit | 1:1000 |
| NCAPD3 | ab70349 | Abcam | Rabbit | 1:1000 |
| NCAPG | SAB1405247 | Sigma-Aldrich | Rabbit | 1:1000 |
| NCAPG2 | ab70350 | Abcam | Rabbit | 1:2000 |
| NCAPH | HPA002647 | Sigma-Aldrich | Rabbit | 1:1000 |
| NCAPH2 | ab71459 | Abcam | Rabbit | 1:500 |
| Cyclophilin B | ab16045 | Abcam | Rabbit | 1:30,000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baergen, A.K.; Jeusset, L.M.; Lichtensztejn, Z.; McManus, K.J. Diminished Condensin Gene Expression Drives Chromosome Instability That May Contribute to Colorectal Cancer Pathogenesis. Cancers 2019, 11, 1066. https://doi.org/10.3390/cancers11081066
Baergen AK, Jeusset LM, Lichtensztejn Z, McManus KJ. Diminished Condensin Gene Expression Drives Chromosome Instability That May Contribute to Colorectal Cancer Pathogenesis. Cancers. 2019; 11(8):1066. https://doi.org/10.3390/cancers11081066
Chicago/Turabian StyleBaergen, Allison K., Lucile M. Jeusset, Zelda Lichtensztejn, and Kirk J. McManus. 2019. "Diminished Condensin Gene Expression Drives Chromosome Instability That May Contribute to Colorectal Cancer Pathogenesis" Cancers 11, no. 8: 1066. https://doi.org/10.3390/cancers11081066
APA StyleBaergen, A. K., Jeusset, L. M., Lichtensztejn, Z., & McManus, K. J. (2019). Diminished Condensin Gene Expression Drives Chromosome Instability That May Contribute to Colorectal Cancer Pathogenesis. Cancers, 11(8), 1066. https://doi.org/10.3390/cancers11081066