The Role of Forkhead Box Proteins in Acute Myeloid Leukemia

 , and
, and 
Abstract
1. Introduction
2. Current Classification of Acute Myeloid Leukemia
3. Forkhead Box Proteins in RUNX1-RUNX1T1 Acute Myeloid Leukemia
4. Forkhead Box Proteins in PML/RARA (Promyelocytic Leukemia/Retinoic Acid Receptor-Alpha) Acute Promyelocytic Leukemia
5. Role of Forkhead Box Proteins According to Specific Gene Mutations Frequently Identified in Acute Myeloid Leukemia
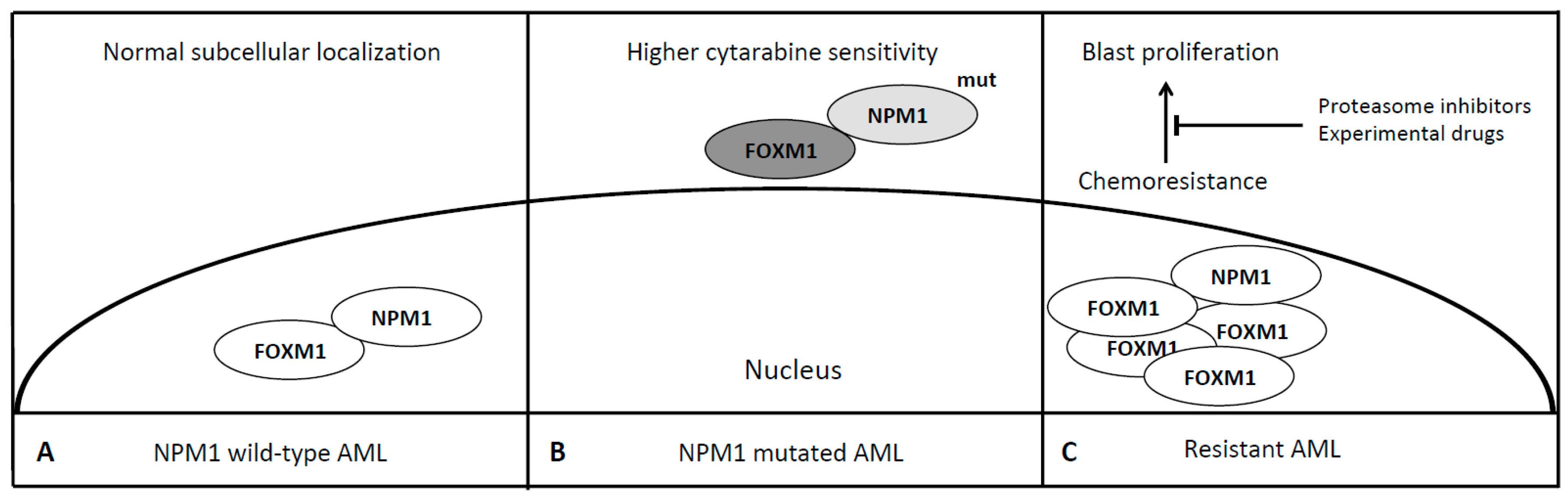
6. Forkhead Box Proteins in Nucleophosmin (NPM1) Mutated Acute Myeloid Leukemia
7. Forkhead Box Proteins in FLT3-ITD Mutated Acute Myeloid Leukemia
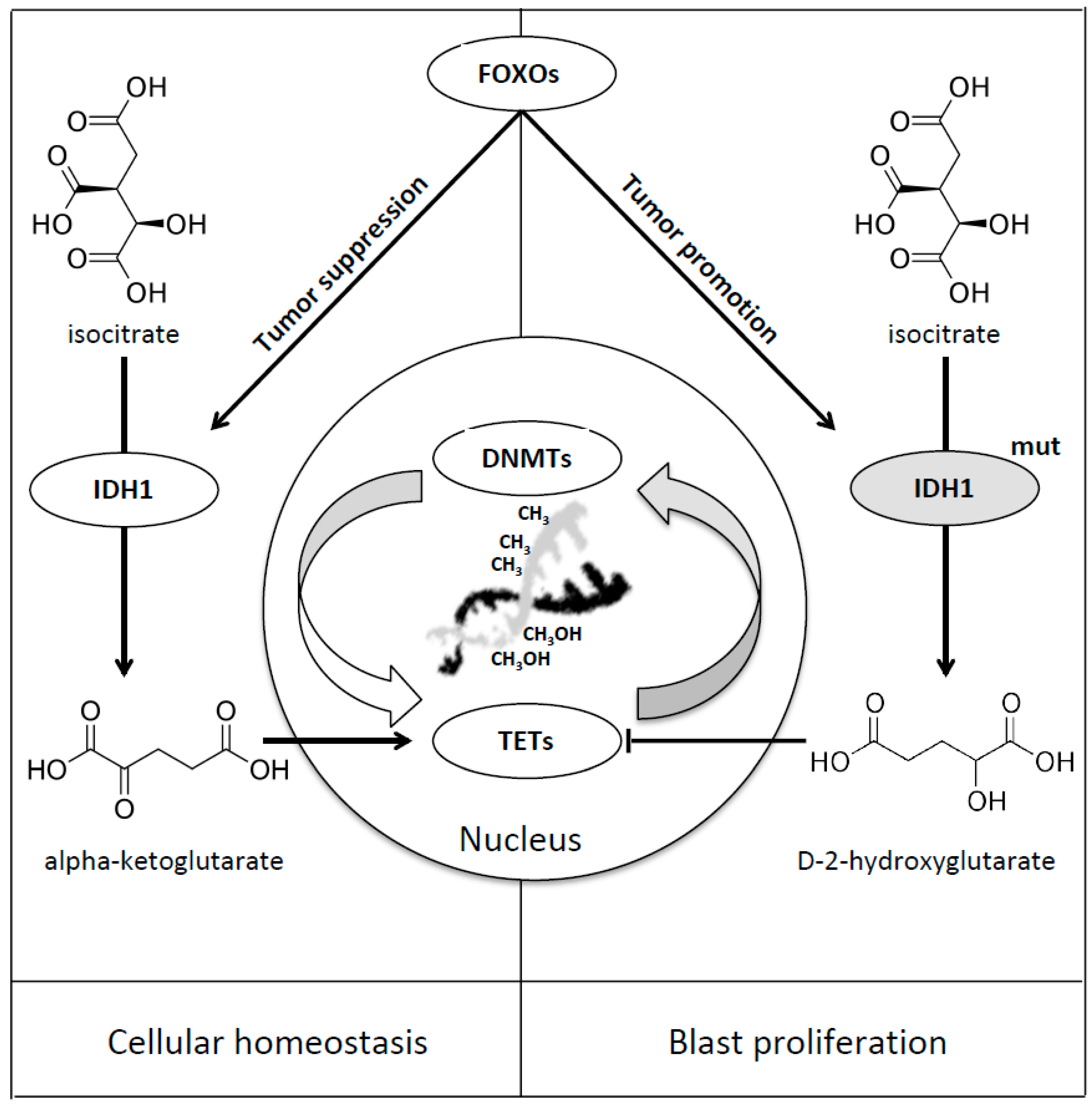
8. Forkhead Box Proteins in IDHs Mutated Acute Myeloid Leukaemia
9. Forkhead Box Proteins in Refractory Acute Myeloid Leukemia: Mechanism of Chemoresistance and Treatment Failure
10. Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Weigel, D.; Jürgens, G.; Küttner, F.; Seifert, E.; Jäckle, H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 1989, 57, 645–658. [Google Scholar] [CrossRef]
- Hannenhalli, S.; Kaestner, K.H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 2009, 10, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.C.; Carpenter, C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse forkhead box (FOX) gene families. Hum. Genom. 2010, 4, 345–352. [Google Scholar] [PubMed]
- Wang, J.; Li, W.; Zhao, K.; Fu, W.; Zheng, X.; Pang, X.; Du, G. Members of FOX family could be drug targets of cancers. Pharmacol. Ther. 2018, 181, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Gomes, A.P.; Wang, X.; Yoon, S.O.; Lee, G.; Nagiec, M.J.; Cho, S.; Chavez, A.; Islam, T.; Yu, Y.; et al. mTORC1 Promotes Metabolic Reprogramming by the Suppression of GSK3-Dependent Foxk1 Phosphorylation. Mol. Cell 2018, 70, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Maachani, U.B.; Shankavaram, U.; Kramp, T.; Tofilon, P.; Camphausen, K.; Tandle, A.T. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget 2016, 7, 77365–77377. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Fernández, M.; Medema, R.H. Novel functions of FoxM1: From molecular mechanisms to cancer therapy. Front. Oncol. 2013, 3, 30. [Google Scholar] [CrossRef]
- Ackermann, S.; Kocak, H.; Hero, B.; Ehemann, V.; Kahlert, Y.; Oberthuer, A.; Roels, F.; Theißen, J.; Odenthal, M.; Berthold, F.; et al. FOXP1 inhibits cell growth and attenuates tumorigenicity of neuroblastoma. BMC Cancer 2014, 14, 840. [Google Scholar] [CrossRef]
- Wang, P.; Lv, C.; Zhang, T.; Liu, J.; Yang, J.; Guan, F.; Hong, T. FOXQ1 regulates senescence-associated inflammation via activation of SIRT1 expression. Cell Death Dis. 2017, 8, e2946. [Google Scholar] [CrossRef]
- Macedo, J.C.; Vaz, S.; Bakker, B.; Ribeiro, R.; Bakker, P.L.; Escandell, J.M.; Ferreira, M.G.; Medema, R.; Foijer, F.; Logarinho, E. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat. Commun. 2018, 9, 2834. [Google Scholar] [CrossRef] [PubMed]
- Song, B.N.; Chu, I.S. A gene expression signature of FOXM1 predicts the prognosis of hepatocellular carcinoma. Exp. Mol. Med. 2018, 50, e418. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.F.; Zhu, T.; Mao, X.Y.; Mao, C.X.; Li, L.; Yin, J.Y.; Zhou, H.H.; Liu, Z.Q. Silencing of Forkhead box D1 inhibits proliferation and migration in glioma cells. Oncol. Rep. 2017, 37, 1196–1202. [Google Scholar] [CrossRef]
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Karadedou, C.T.; Gomes, A.R.; Chen, J.; Petkovic, M.; Ho, K.K.; Zwolinska, A.K.; Feltes, A.; Wong, S.Y.; Chan, K.Y.; Cheung, Y.N.; et al. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene 2012, 31, 1845–1858. [Google Scholar] [CrossRef] [PubMed]
- Rocca, D.L.; Wilkinson, K.A.; Henley, J.M. SUMOylation of FOXP1 regulates transcriptional repression via CtBP1 to drive dendritic morphogenesis. Sci. Rep. 2017, 7, 877. [Google Scholar] [CrossRef] [PubMed]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Lin, S.; Ptasinska, A.; Chen, X.; Shrestha, M.; Assi, S.A.; Chin, P.S.; Imperato, M.R.; Aronow, B.J.; Zhang, J.; Weirauch, M.T.; et al. A FOXO1-induced oncogenic network defines the AML1-ETO preleukemic program. Blood 2017, 130, 1213–1222. [Google Scholar] [CrossRef]
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E. Current Management of Childhood Acute Myeloid Leukemia. Paediatr. Drugs 2017, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.; Wetzler, M.; Lowenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.M.; Duque, J.; Shizuru, J.A.; Lübbert, M. Complementing mutations in core binding factor leukemias: From mouse models to clinical applications. Oncogene 2008, 27, 5759–5773. [Google Scholar] [CrossRef] [PubMed]
- Tonks, A.; Pearn, L.; Musson, M.; Gilkes, A.; Mills, K.I.; Burnett, A.K.; Darley, R.L. Transcriptional dysregulation mediated by RUNX1-RUNX1T1 in normal human progenitor cells and in acute myeloid leukaemia. Leukemia 2007, 21, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Singh, A.; Khosravi-Far, R. FoxO tumor suppressors and BCR-ABL-induced leukemia: A matter of evasion of apoptosis. Biochim. Biophys. Acta 2008, 1785, 63–84. [Google Scholar] [CrossRef] [PubMed]
- Kornblau, S.M.; Singh, N.; Qiu, Y.; Chen, W.; Zhang, N.; Coombes, K.R. Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin. Cancer Res. 2010, 16, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Obrador-Hevia, A.; Serra-Sitjar, M.; Rodríguez, J.; Villalonga, P.; Fernández de Mattos, S. The tumour suppressor FOXO3 is a key regulator of mantle cell lymphoma proliferation and survival. Br. J. Haematol. 2012, 156, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Ushmorov, A.; Leithäuser, F.; Guan, H.; Steidl, C.; Färbinger, J.; Pelzer, C.; Vogel, M.J.; Maier, H.J.; Gascoyne, R.D.; et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood 2012, 119, 3503–3511. [Google Scholar] [CrossRef]
- Yao, S.; Mahmud, Z.; Sachini, N.; Aimjongjun, S.; Saavedra-García, P.; Lam, E.W. Characterization of FOXO Acetylation. Methods Mol. Biol. 2019, 1890, 77–90. [Google Scholar] [PubMed]
- Rehman, A.; Kim, Y.; Kim, H.; Sim, J.; Ahn, H.; Chung, M.S.; Shin, S.J.; Jang, K. FOXO3a expression is associated with lymph node metastasis and poor disease-free survival in triple-negative breast cancer. J. Clin. Pathol. 2018, 71, 806–813. [Google Scholar] [CrossRef]
- Hou, T.; Li, Z.; Zhao, Y.; Zhu, W.G. Mechanisms controlling the anti-neoplastic functions of FoxO proteins. Semin. Cancer Biol. 2018, 50, 101–114. [Google Scholar] [CrossRef]
- Pan, C.W.; Jin, X.; Zhao, Y.; Pan, Y.; Yang, J.; Karnes, R.J.; Zhang, J.; Wang, L.; Huang, H. AKT-phosphorylated FOXO1 suppresses ERK activation and chemoresistance by disrupting IQGAP1-MAPK interaction. EMBO J. 2017, 36, 995–1010. [Google Scholar] [CrossRef]
- Zhu, H. Targeting forkhead box transcription factors FOXM1 and FOXO in leukemia. Oncol. Rep. 2014, 32, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.C.; Francis, R.E.; Guest, S.K.; Costa, J.R.; Gomes, A.R.; Myatt, S.S.; Brosens, J.J.; Lam, E.W. Doxorubicin activates FOXO3a to induce the expression of multidrug resistance gene ABCB1 (MDR1) in K562 leukemic cells. Mol. Cancer Ther. 2008, 7, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Cho, K.B.; Choi, H.S.; Han, H.K.; Kang, K.W. Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis 2008, 29, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Sakoe, Y.; Sakoe, K.; Kirito, K.; Ozawa, K.; Komatsu, N. FOXO3A as a key molecule for all-trans retinoic acid–induced granulocytic differentiation and apoptosis in acute promyelocytic leukemia. Blood 2010, 115, 3787–3795. [Google Scholar] [CrossRef] [PubMed]
- Somerville, T.D.; Wiseman, D.H.; Spencer, G.J.; Huang, X.; Lynch, J.T.; Leong, H.S.; Williams, E.L.; Cheesman, E.; Somervaille, T.C. Frequent Derepression of the Mesenchymal Transcription Factor Gene FOXC1 in Acute Myeloid Leukemia. Cancer Cell 2015, 28, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, E.; Falconi, G.; Noguera, N.I.; Saulle, E.; Cicconi, L.; Divona, M.; Banella, C.; Picardi, A.; Cerio, AM.; Boe, L.; et al. The forkhead box C1 (FOXC1) transcription factor is downregulated in acute promyelocytic leukemia. Oncotarget 2017, 8, 84074–84085. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laoukili, J.; Stahl, M.; Medema, R.H. FoxM1: At the crossroads of ageing and cancer. Biochim. Biophys. Acta 2007, 1775, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Hirano, I.; Okinaka, K.; Takemura, T.; Yokota, D.; Ono, T.; Shigeno, K.; Shibata, K.; Fujisawa, S.; Ohnishi, K. The FOXM1 transcriptional factor promotes the proliferation of leukemia cells through modulation of cell cycle progression in acute myeloid leukemia. Carcinogenesis 2010, 31, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Halasi, M.; Patel, A.; Schultz, R.; Kalakota, N.; Chen, Y.H.; Aardsma, N.; Liu, L.; Crispino, J.D.; Mahmud, N.; et al. FOXM1 contributes to treatment failure in acute myeloid leukemia. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, B.; Ngo, H.T.; Kang, H.; Griffin, J.D. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene 2004, 23, 3338–3349. [Google Scholar] [CrossRef] [PubMed]
- Seedhouse, C.H.; Mills, K.I.; Ahluwalia, S.; Grundy, M.; Shang, S.; Burnett, A.K.; Russell, N.H.; Pallis, M. Distinct poor prognostic subgroups of acute myeloid leukaemia, FLT3-ITD and P-glycoprotein-positive, have contrasting levels of FOXO1. Leuk. Res. 2014, 38, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Zhang, D.H.; Mao, X.; Zhang, X.H.; Zhang, B. Over-expression of FoxM1 is associated with adverse prognosis and FLT3-ITD in acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2014, 446, 280–285. [Google Scholar] [CrossRef]
- Charitou, P.; Rodriguez-Colman, M.; Gerrits, J.; van Triest, M.; Groot Koerkamp, M.; Hornsveld, M.; Holstege, F.; Verhoeven-Duif, N.M.; Burgering, B.M. FOXOs support the metabolic requirements of normal and tumor cells by promoting IDH1 expression. EMBO Rep. 2015, 16, 456–466. [Google Scholar] [CrossRef]
- Grignani, F.; Ferrucci, P.F.; Testa, U.; Talamo, G.; Fagioli, M.; Alcalay, M.; Mencarelli, A.; Grignani, F.; Peschle, C.; Nicoletti, I.; et al. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell 1993, 74, 423–431. [Google Scholar] [CrossRef]
- Altucci, L.; Rossin, A.; Raffelsberger, W.; Reitmair, A.; Chomienne, C.; Gronemeyer, H. Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat. Med. 2001, 7, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Birkenkamp, K.U.; Coffer, P.J. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem. Soc. Trans. 2003, 31, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Watanabe, T.; Uchida, M.; Mori, M.; Kirito, K.; Kikuchi, S.; Liu, Q.; Tauchi, T.; Miyazawa, K.; Endo, H.; et al. A member of Forkhead transcription factor FKHRL1 is a downstream effector of STI571-induced cell cycle arrest in BCR-ABL-expressing cells. J. Biol. Chem. 2003, 278, 6411–6419. [Google Scholar] [CrossRef] [PubMed]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, L.; Zhan, S.; Chen, L.; Wang, Y.; Zhang, Y.; Du, J.; Wu, Y.; Gu, L. Arsenic Trioxide Suppressed Migration and Angiogenesis by Targeting FOXO3a in Gastric Cancer Cells. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, V.; Iaccarino, L.; Ottone, T.; Cicconi, L.; Lavorgna, S.; Divona, M.; Cairoli, R.; Cristiano, A.; Ciardi, C.; Travaglini, S.; et al. Early and sensitive detection of PML-A216V mutation by droplet digital PCR in ATO-resistant acute promyelocytic leukemia. Leukemia 2019. [Google Scholar] [CrossRef] [PubMed]
- Soncini, M.; Santoro, F.; Gutierrez, A.; Frigè, G.; Romanenghi, M.; Botrugno, O.A.; Pallavicini, I.; Pelicci, P.; Di Croce, L.; Minucci, S. The DNA demethylating agent decitabine activates the TRAIL pathway and induces apoptosis in acute myeloid leukemia. Biochim. Biophys. Acta 2013, 1832, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 740. [Google Scholar] [CrossRef]
- Schnittger, S.; Schoch, C.; Kern, W.; Mecucci, C.; Tschulik, C.; Martelli, M.F.; Haferlach, T.; Hiddemann, W.; Falini, B. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 2005, 106, 3733–3739. [Google Scholar] [CrossRef]
- Bhat, U.G.; Jagadeeswaran, R.; Halasi, M.; Gartel, A.L. Nucleophosmin interacts with FOXM1 and modulates the level and localization of FOXM1 in human cancer cells. J. Biol. Chem. 2011, 286, 41425–41433. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Roose, J.; Clevers, H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997, 25, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Massa, P.E.; Olivotto, E.; Bianchi, F.; Borzi, R.M.; Hanidu, A.; Li, X.; Li, J.; Marcu, K.B. Sustained NF-kappaB activation produces a short-term cell proliferation block in conjunction with repressing effectors of cell cycle progression controlled by E2F or FoxM1. J. Cell. Physiol. 2009, 218, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 2019, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Quentmeier, H.; Martelli, M.O.; Dirks, W.G.; Bolli, N.; Liso, A.; Macleod, R.A.; Nicoletti, I.; Mannucci, R.; Pucciarini, A.; Bigerna, B.; et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia 2005, 19, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Halasi, M.; Zia, M.F.; Gann, P.; Gaitonde, S.; Mahmud, N.; Gartel, A.L. Nuclear FOXM1 drives chemoresistance in AML. Leukemia 2017, 31, 251–255. [Google Scholar] [CrossRef]
- Halasi, M.; Váraljai, R.; Benevolenskaya, E.; Gartel, A.L. A Novel Function of Molecular Chaperone HSP70: Suppression of oncogenic FOXM1 after proteotoxic stress. J. Biol. Chem. 2016, 291, 142–148. [Google Scholar] [CrossRef]
- Attar, E.C.; Johnson, J.L.; Amrein, P.C.; Lozanski, G.; Wadleigh, M.; DeAngelo, D.J.; Kolitz, J.E.; Powell, B.L.; Voorhees, P.; Wang, E.S.; et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J. Clin. Oncol. 2013, 31, 923–929. [Google Scholar] [CrossRef]
- Bertaina, A.; Vinti, L.; Strocchio, L.; Gaspari, S.; Caruso, R.; Algeri, M.; Coletti, V.; Gurnari, C.; Romano, M.; Cefalo, M.G.; et al. The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br. J. Haematol. 2017, 176, 629–636. [Google Scholar] [CrossRef]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, C.; Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, M.; Jensen, T.W.; Ray, T.; Andruska, N.D.; Shah, A.; Egner, J.R.; Ray, P.S. FOXC1 Expression in Acute Myeloid Leukemia: Potential Predictor of Disease Relapse and/or Refractory Disease. Blood 2016, 128, 5260. [Google Scholar]
- Assi, S.A.; Imperato, M.R.; Coleman, D.J.L.; Pickin, A.; Potluri, S.; Ptasinska, A.; Chin, P.S.; Blair, H.; Cauchy, P.; James, S.R.; et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat. Genet. 2019, 51, 151–162. [Google Scholar] [CrossRef]
- Cauchy, P.; James, S.R.; Zacarias-Cabeza, J.; Ptasinska, A.; Imperato, M.R.; Assi, S.A.; Piper, J.; Canestraro, M.; Hoogenkamp, M.; Raghavan, M.; et al. Chronic FLT3-ITD Signaling in Acute Myeloid Leukemia Is Connected to a Specific Chromatin Signature. Cell Rep. 2015, 12, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Han, M.; Jia, L.; Lv, W.; Wang, L.; Cui, W. Epigenetic Enzyme Mutations: Role in Tumorigenesis and Molecular Inhibitors. Front. Oncol. 2019, 9, 194. [Google Scholar] [CrossRef]
- Hou, H.A.; Tien, H.F. Mutations in epigenetic modifiers in acute myeloid leukemia and their clinical utility. Expert Rev. Hematol. 2016, 9, 447–469. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro. Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Falconi, G.; Fabiani, E.; Piciocchi, A.; Criscuolo, M.; Fianchi, L.; Lindfors Rossi, E.L.; Finelli, C.; Cerqui, E.; Ottone, T.; Molteni, A.; et al. Somatic mutations as markers of outcome after azacitidine and allogeneic stem cell transplantation in higher-risk myelodysplastic syndromes. Leukemia 2019, 33, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.E.; Bittinger, M.A.; Su, S.M.; Fantin, V.R. Cancer-associated IDH mutations: Biomarker and therapeutic opportunities. Oncogene 2010, 29, 6409–6417. [Google Scholar] [CrossRef]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chinnaiyan, A.M. Metabolism unhinged: IDH mutations in cancer. Nat. Med. 2011, 17, 291–293. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef]
- Buege, M.J.; Di Pippo, A.; DiNardo, C.D. Evolving Treatment Strategies for Elderly Leukemia Patients with IDH Mutations. Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Nassereddine, S.; Lap, C.J.; Tabbara, I.A. Evaluating ivosidenib for the treatment of relapsed/refractory AML: Design, development, and place in therapy. OncoTargets Ther. 2019, 12, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Abou Dalle, I.; DiNardo, C.D. The role of enasidenib in the treatment of mutant IDH2 acute myeloid leukemia. Ther. Adv. Hematol. 2018, 9, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; AG221-C-001 Study Investigators. Differentiation Syndrome Associated with Enasidenib, a Selective Inhibitor of Mutant Isocitrate Dehydrogenase 2: Analysis of a Phase 1/2 Study. JAMA Oncol. 2018, 4, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Cerrano, M.; Itzykson, R. New Treatment Options for Acute Myeloid Leukemia in 2019. Curr. Oncol. Rep. 2019, 21, 16. [Google Scholar] [CrossRef] [PubMed]
- Boddu, P.; Borthakur, G. Therapeutic targeting of isocitrate dehydrogenase mutant AML. Expert Opin. Investig. Drugs 2017, 26, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, J.; Zhou, M.; Li, B.; Zhang, Y.; Huang, T.; Wang, L.; Jia, J.; Chen, C. The tumor suppressive role of miRNA-370 by targeting FoxM1 in acute myeloid leukemia. Mol. Cancer 2012, 11, 56. [Google Scholar] [CrossRef]
- Bhat, U.G.; Halasi, M.; Gartel, A.L. FoxM1 is a general target for proteasome inhibitors. PLoS ONE 2009, 4, e6593. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, N.; Tamburini, J.; Green, A.S.; Vignon, C.; Bardet, V.; Neyret, A.; Pannetier, M.; Willems, L.; Park, S.; Macone, A.; et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin. Cancer Res. 2010, 16, 5424–5435. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Lane, S.W.; Bullinger, L.; Kalaitzidis, D.; Yusuf, R.; Saez, B.; Ferraro, F.; Mercier, F.; Singh, H.; Brumme, K.M.; et al. AKT/FOXO Signaling Enforces Reversible Differentiation Blockade in Myeloid Leukemias. Cell 2011, 146, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, C.M.; Chillón, M.C.; García-Sanz, R.; Pérez, C.; Caballero, M.D.; Ramos, F.; de Coca, A.G.; Alonso, J.M.; Giraldo, P.; Bernal, T.; et al. High FOXO3a expression is associated with a poorer prognosis in AML with normal cytogenetics. Leuk. Res. 2009, 33, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Thépot, S.; Lainey, E.; Cluzeau, T.; Sébert, M.; Leroy, C.; Adès, L.; Eclache, V. Hypomethylating agents reactivate FOXO3A in acute myeloid leukemia. Cell Cycle 2011, 10, 2323–2330. [Google Scholar]
- Naudin, C.; Hattabi, A.; Michelet, F.; Miri-Nezhad, A.; Benyoucef, A.; Pflumio, F.; Guillonneau, F.; Fichelson, S.; Vigon, I.; Dusanter-Fourt, I.; et al. PUMILIO/FOXP1 signaling drives expansion of hematopoietic stem/progenitor and leukemia cells. Blood 2017, 129, 2493–2506. [Google Scholar] [CrossRef]
- Piao, Z.; Kim, H.J.; Choi, J.Y.; Hong, C.R.; Lee, J.W.; Kang, H.J.; Park, K.D.; Shin, H. Y Effect of FOXP3 polymorphism on the clinical outcomes after allogeneic hematopoietic stem cell transplantation in pediatric acute leukemia patients. Int. Immunopharmacol. 2016, 31, 132–139. [Google Scholar] [CrossRef]
- Cabral, C.M.V.; Braga, W.M.T.; Arantes, A.M.; de Oliveira, J.S.R.; Colleoni, G.W.B.; Kerbauy, F.R. Expression of Foxp3 and Rorc Genes in Patients Undergoing Allogeneic Hematopoietic Cell Transplantation: Impact in the Development of Acute and Chronic Graft-Versus-Host Disease. Int. J. Blood Res. Disord. 2018, 5, 032. [Google Scholar]
- Wang, A.; Zhong, H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology 2018, 23, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Blau, O. Bone marrow stromal cells in the pathogenesis of acute myeloid leukemia. Front. Biosci. 2014, 19, 171–180. [Google Scholar] [CrossRef]
- Spees, J.L.; Lee, R.H.; Gregory, C.A. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res. Ther. 2016, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, Q.; He, H.R.; Zhai, Y.J.; Lu, J.; Hu, H.B.; Zhou, J.S.; Yang, Y.H.; Li, Y.J. The association between XRCC1 Arg399Gln polymorphism and risk of leukemia in different populations: A meta-analysis of case-control studies. Onco. Targets Ther. 2015, 8, 3277–3287. [Google Scholar] [PubMed]
- Voso, M.T.; Fabiani, E.; D’Alo’, F.; Guidi, F.; Di Ruscio, A.; Sica, S.; Pagano, L.; Greco, M.; Hohaus, S.; Leone, G. Increased risk of acute myeloid leukaemia due to polymorphisms in detoxification and DNA repair enzymes. Ann. Oncol. 2007, 18, 1523–1528. [Google Scholar] [CrossRef]
- Fabiani, E.; Fianchi, L.; Falconi, G.; Boncompagni, R.; Criscuolo, M.; Guidi, F.; La Brocca, A.; Hohaus, S.; Leone, G.; Voso, M.T. The BCL2L10 Leu21Arg variant and risk of therapy-related myeloid neoplasms and de novo myelodysplastic syndromes. Leuk. Lymphoma 2014, 55, 1538–1543. [Google Scholar] [CrossRef]
- Bargal, S.A.; Rafiee, R.; Crews, K.R.; Wu, H.; Cao, X.; Rubnitz, J.E.; Ribeiro, R.C.; Downing, J.R.; Pounds, S.B.; Lamba, J.K. Genome-wide association analysis identifies SNPs predictive of in vitro leukemic cell sensitivity to cytarabine in pediatric AML. Oncotarget 2018, 9, 34859–34875. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Yang, Z.; Dou, H.; Lu, L.; Bi, J.; Zou, L.; Yu, J.; Bao, L. Correlation of TET2 SNP rs2454206 with improved survival in children with acute myeloid leukemia featuring intermediate-risk cytogenetics. Genes Chromosomes Cancer 2018, 57, 379–386. [Google Scholar] [CrossRef]
- Kutny, M.A.; Alonzo, T.A.; Gamazon, E.R.; Gerbing, R.B.; Geraghty, D.; Lange, B.; Heerema, N.A.; Sung, L.; Aplenc, R.; Franklin, J.; et al. Ethnic variation of TET2 SNP rs2454206 and association with clinical outcome in childhood AML: A report from the Children’s Oncology Group. Leukemia 2015, 29, 2424–2426. [Google Scholar] [CrossRef][Green Version]
- Voso, M.T.; Hohaus, S.; Guidi, F.; Fabiani, E.; D’Alò, F.; Groner, S.; Späth, D.; Doehner, K.; Leone, G.; Doehner, H.; et al. Prognostic role of glutathione S-transferase polymorphisms in acute myeloid leukemia. Leukemia 2008, 22, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.; Shin, S.; Park, K.U.; Kim, I.; Yoon, S.S.; Kwon, T.K.; Song, E.Y. Association of FOXP3 Single Nucleotide Polymorphisms with Clinical Outcomes After Allogenic Hematopoietic Stem Cell Transplantation. Ann. Lab. Med. 2018, 38, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Haimovici, A.; Humbert, M.; Federzoni, E.A.; Shan-Krauer, D.; Brunner, T.; Frese, S.; Kaufmann, T.; Torbett, B.E.; Tschan, M.P.P.U. 1 supports TRAIL-induced cell death by inhibiting NF-κB-mediated cell survival and inducing DR5 expression. Cell Death Differ. 2017, 24, 866–877. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Recurrent Abnormalities in Acute Myeloid Leukemia | FOX Family Member | Biological Process | References |
|---|---|---|---|
| t(8;21); RUNX1-RUNX1T1 | FOXO1 | Self-renewal and Differentiation | Lin et al. [19] |
| PML-RARA | FOXO3A FOXC1 | Apoptosis and Granulocytic Differentiation Granulocytic Differentiation and Epigenetic Regulation | Sakoe et al. [40] Somerville et al. [41] and Fabiani et al. [42] |
| NPM1 | FOXM1 | Cell Proliferation, Division and Chemoresistance | Laoukili et al. [43] Nakamura et al. [44] Khan et al. [45] |
| FLT3 ITD | FOXO3A FOXO1 FOXM1 | Apoptosis, Survival and Proliferation Cell growth, Apoptosis and antioxidant defences Survival, Apoptosis and Chemoresistance | Scheijen et al. [46] Seedhouse et al. [47] Liu et al. [48] |
| IDH 1-2 | FOXOs | Cellular Differentiation and Tumor Suppression/Progression and Epigenetic Instability | Charitou et al. [49] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurnari, C.; Falconi, G.; De Bellis, E.; Voso, M.T.; Fabiani, E. The Role of Forkhead Box Proteins in Acute Myeloid Leukemia. Cancers 2019, 11, 865. https://doi.org/10.3390/cancers11060865
Gurnari C, Falconi G, De Bellis E, Voso MT, Fabiani E. The Role of Forkhead Box Proteins in Acute Myeloid Leukemia. Cancers. 2019; 11(6):865. https://doi.org/10.3390/cancers11060865
Chicago/Turabian StyleGurnari, Carmelo, Giulia Falconi, Eleonora De Bellis, Maria Teresa Voso, and Emiliano Fabiani. 2019. "The Role of Forkhead Box Proteins in Acute Myeloid Leukemia" Cancers 11, no. 6: 865. https://doi.org/10.3390/cancers11060865
APA StyleGurnari, C., Falconi, G., De Bellis, E., Voso, M. T., & Fabiani, E. (2019). The Role of Forkhead Box Proteins in Acute Myeloid Leukemia. Cancers, 11(6), 865. https://doi.org/10.3390/cancers11060865