The Role of Cancer Stem Cells and Mechanical Forces in Ovarian Cancer Metastasis

 ,
, 
Abstract
1. Introduction
2. Metastasis in Ovarian Cancers
3. The Roles of Ovarian Cancer Stem-Like Cells (CSCs) in Metastasis
4. Interactions between Ovarian Cancer Stem-Like Cells and Ovarian Tumor Microenvironment that Modulate Metastasis
5. Nexus between Cancer Stem Like Cell Phenotypes and Mechanotransduction
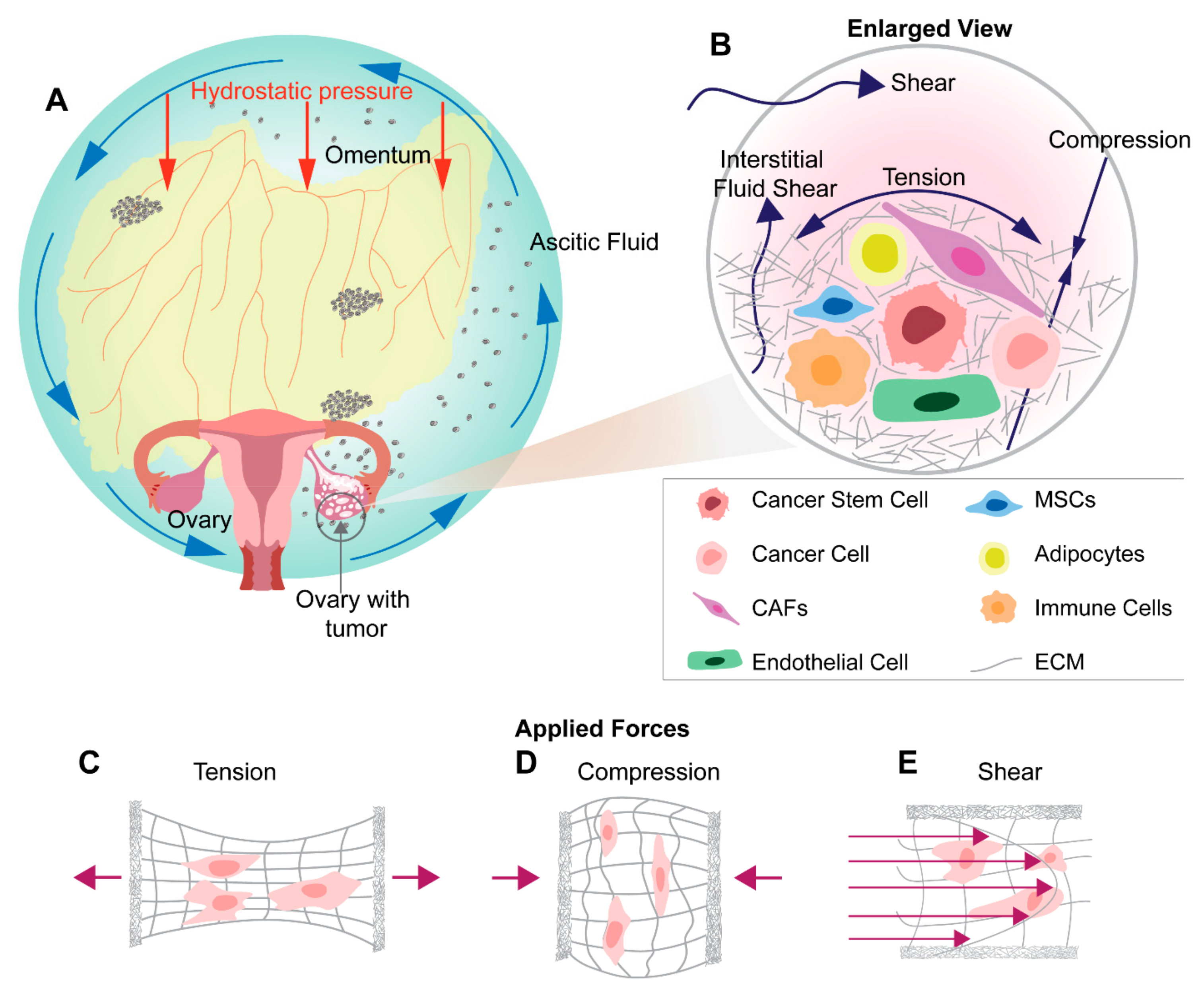
6. Mechanical Stimuli in Ovarian Tumor Microenvironment and Metastasis
7. Shear Stress in Ovarian Cancer Metastasis
8. Tensile Stress in Ovarian Cancer Metastasis
9. Compression Stress in Ovarian Cancer Metastasis
10. Matrix Stiffness Stress in Ovarian Cancer Metastasis
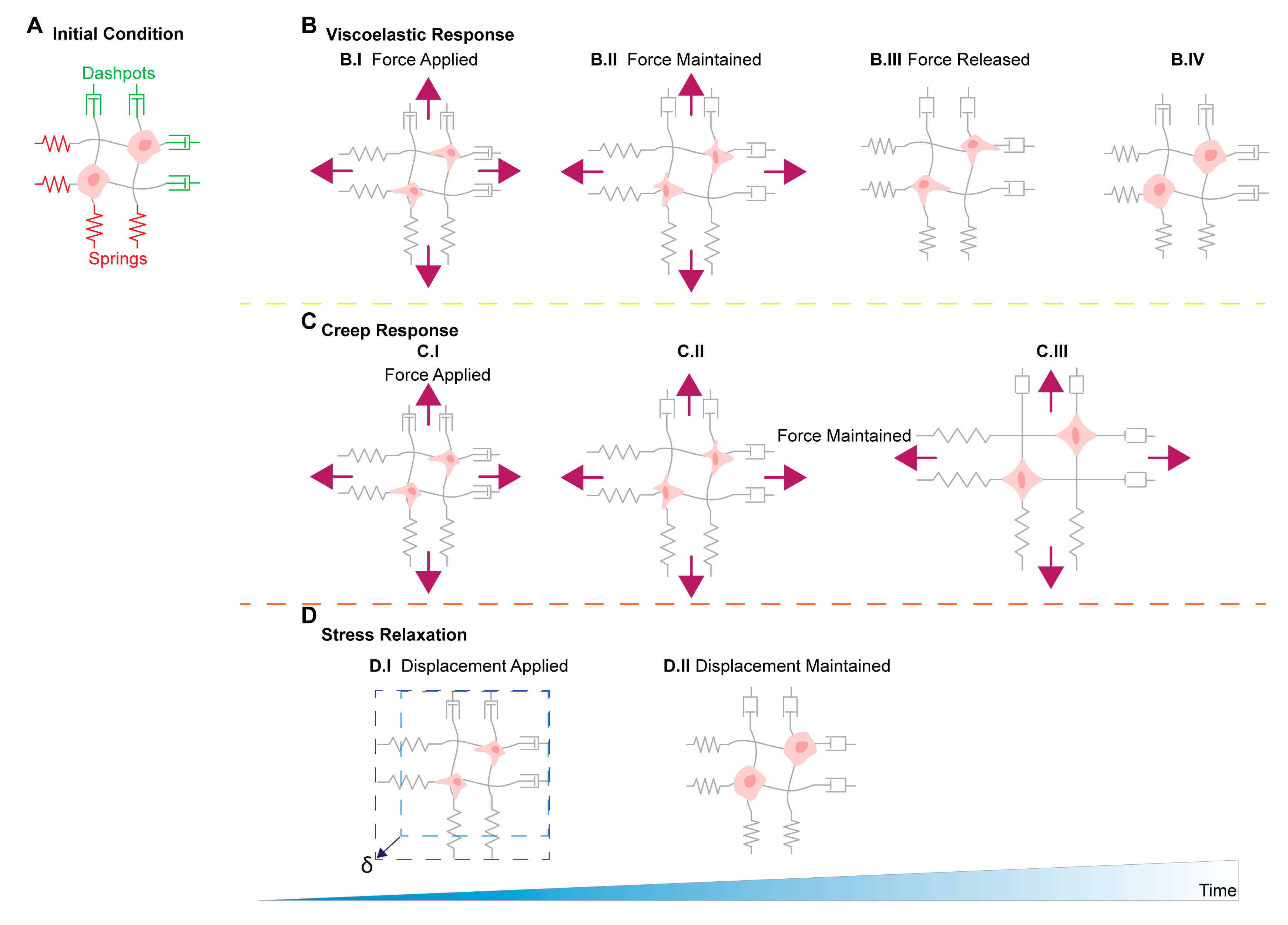
11. Stress Relaxation, Creep Compliance in Ovarian Cancer Metastasis
12. Challenges Confronting Future Metastasis Research in Ovarian Cancers
13. Challenges with Incorporating CSCs in Metastasis Research
14. Challenges with Physiologic In Vitro and In Vivo Models to Study Mechanical Stimuli in Ovarian Cancers
15. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Sajdak, S.; Huczyński, A.; Rehlis, S.; Markowska, J. Ovarian cancer stem cells: A target for oncological therapy. Adv. Clin. Exp. Med. 2018, 27, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Lupia, M.; Cavallaro, U. Ovarian cancer stem cells: Still an elusive entity? Mol. Cancer 2017, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; He, Y.; Li, Z.; Chen, X.; Liu, Q.; Zou, S.; Kong, C.; Liu, Y.; Gao, C.; Zhang, G.; et al. Constructing an ovarian cancer metastasis index by dissecting medical records. Oncotarget 2017, 8, 102212–102222. [Google Scholar] [CrossRef] [PubMed]
- Kossaï, M.; Leary, A.; Scoazec, J.-Y.; Genestie, C. Ovarian Cancer: A Heterogeneous Disease. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2018, 85, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, B.; Song, Y.S. Ascites modulates cancer cell behavior, contributing to tumor heterogeneity in ovarian cancer. Cancer Sci. 2016, 107, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- De Cristofaro, T.; Di Palma, T.; Soriano, A.A.; Monticelli, A.; Affinito, O.; Cocozza, S.; Zannini, M. Candidate genes and pathways downstream of PAX8 involved in ovarian high-grade serous carcinoma. Oncotarget 2016, 7, 41929–41947. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Sieh, W.; Köbel, M.; Longacre, T.A.; Bowtell, D.D.; deFazio, A.; Goodman, M.T.; Høgdall, E.; Deen, S.; Wentzensen, N.; Moysich, K.B.; et al. Hormone-receptor expression and ovarian cancer survival: An Ovarian Tumor Tissue Analysis consortium study. Lancet Oncol. 2013, 14, 853–862. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res. J. Lab. Clin. Med. 2016, 175, 92–102.e2. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.-J.; Choi, H.J.; Bischoff, F.Z.; et al. Hematogenous metastasis of ovarian cancer: Rethinking mode of spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, N.; Hirai, Y.; Umayahara, K.; Fujiwara, K.; Takizawa, K.; Hasumi, K. Lymph node metastasis in ovarian cancer: Difference between serous and non-serous primary tumors. Gynecol. Oncol. 2005, 99, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.; Bachmann, R.; Fend, F.; Wallwiener, D. Incidence and Impact of Lymph Node Metastases in Advanced Ovarian Cancer: Implications for Surgical Treatment. J. Cancer 2016, 7, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed]
- Judson, P.L.; Geller, M.A.; Bliss, R.L.; Boente, M.P.; Downs, L.S.; Argenta, P.A.; Carson, L.F. Preoperative detection of peripherally circulating cancer cells and its prognostic significance in ovarian cancer. Gynecol. Oncol. 2003, 91, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hijleh, M.F.; Habbal, O.A.; Moqattash, S.T. The role of the diaphragm in lymphatic absorption from the peritoneal cavity. J. Anat. 1995, 186, 453–467. [Google Scholar] [PubMed]
- Feki, A.; Berardi, P.; Bellingan, G.; Major, A.; Krause, K.-H.; Petignat, P.; Zehra, R.; Pervaiz, S.; Irminger-Finger, I. Dissemination of intraperitoneal ovarian cancer: Discussion of mechanisms and demonstration of lymphatic spreading in ovarian cancer model. Crit. Rev. Oncol. Hematol. 2009, 72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.-L.; Leung, C.S.; Yip, K.-P.; Au Yeung, C.L.; Wong, S.T.C.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed]
- Al Habyan, S.; Kalos, C.; Szymborski, J.; McCaffrey, L. Multicellular detachment generates metastatic spheroids during intra-abdominal dissemination in epithelial ovarian cancer. Oncogene 2018, 37, 5127–5135. [Google Scholar] [CrossRef] [PubMed]
- Harper, E.I.; Sheedy, E.F.; Stack, M.S. With Great Age Comes Great Metastatic Ability: Ovarian Cancer and the Appeal of the Aging Peritoneal Microenvironment. Cancers 2018, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, J.; Zhang, Z.; Li, H.; Cheng, W.; Liu, J. Cancer stem cells, epithelial-mesenchymal transition, and drug resistance in high-grade ovarian serous carcinoma. Hum. Pathol. 2013, 44, 2373–2384. [Google Scholar] [CrossRef]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef]
- Deng, J.; Wang, L.; Chen, H.; Hao, J.; Ni, J.; Chang, L.; Duan, W.; Graham, P.; Li, Y. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget 2016, 7, 55771–55788. [Google Scholar] [CrossRef]
- Burgos-Ojeda, D.; Wu, R.; McLean, K.; Chen, Y.-C.; Talpaz, M.; Yoon, E.; Cho, K.R.; Buckanovich, R.J. CD24+ Ovarian Cancer Cells Are Enriched for Cancer-Initiating Cells and Dependent on JAK2 Signaling for Growth and Metastasis. Mol. Cancer Ther. 2015, 14, 1717–1727. [Google Scholar] [CrossRef]
- Meng, E.; Long, B.; Sullivan, P.; McClellan, S.; Finan, M.A.; Reed, E.; Shevde, L.; Rocconi, R.P. CD44+/CD24− ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin. Exp. Metastasis 2012, 29, 939–948. [Google Scholar] [CrossRef]
- Gunjal, P.; Pedziwiatr, D.; Ismail, A.A.; Kakar, S.S.; Ratajczak, M.Z. An emerging question about putative cancer stem cells in established cell lines-are they true stem cells or a fluctuating cell phenotype? J. Cancer Stem Cell Res. 2015, 3, e1004. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Ward, M.R.; Bregenzer, M.E.; Fleck, E.M.A.; Tan, L.; McLean, K.; Buckanovich, R.J.; Mehta, G. Personalized Medicine-Based Approach to Model Patterns of Chemoresistance and Tumor Recurrence Using Ovarian Cancer Stem Cell Spheroids. Clin. Cancer Res. 2017, 23, 6934–6945. [Google Scholar] [CrossRef]
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Karaminejadranjbar, M.; Hu, Z.; et al. An evolving story of the metastatic voyage of ovarian cancer cells: Cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 2019, 38, 2885–2898. [Google Scholar] [CrossRef]
- Parte, S.C.; Batra, S.K.; Kakar, S.S. Characterization of stem cell and cancer stem cell populations in ovary and ovarian tumors. J. Ovarian Res. 2018, 11, 69. [Google Scholar] [CrossRef]
- Al-Alem, L.F.; Pandya, U.M.; Baker, A.T.; Bellio, C.; Zarrella, B.D.; Clark, J.; DiGloria, C.M.; Rueda, B.R. Ovarian cancer stem cells: What progress have we made? Int. J. Biochem. Cell Biol. 2019, 107, 92–103. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. MCR 2011, 9, 1608–1620. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell. Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal “Liaisons” during Cancer Progression. Crit. Rev. Oncog. 2016, 21, 155–168. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, Q.; Lau, W.B.; Lau, B.; Xu, L.; Zhao, L.; Yang, H.; Feng, M.; Xuan, Y.; Yang, Y.; et al. Tumor microenvironment: The culprit for ovarian cancer metastasis? Cancer Lett. 2016, 377, 174–182. [Google Scholar] [CrossRef]
- Guo, S.; Deng, C.-X. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef]
- Varas-Godoy, M.; Rice, G.; Illanes, S.E. The Crosstalk between Ovarian Cancer Stem Cell Niche and the Tumor Microenvironment. Stem Cells Int. 2017, 2017, 5263974. [Google Scholar] [CrossRef]
- Bregenzer, M.E.; Horst, E.N.; Mehta, P.; Novak, C.M.; Raghavan, S.; Snyder, C.S.; Mehta, G. Integrated cancer tissue engineering models for precision medicine. PLoS ONE 2019, 14, e0216564. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Castells, M.; Thibault, B.; Delord, J.-P.; Couderc, B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death. Int. J. Mol. Sci. 2012, 13, 9545–9571. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Sandoval, P.; Jiménez-Heffernan, J.A.; Rynne-Vidal, Á.; Pérez-Lozano, M.L.; Gilsanz, Á.; Ruiz-Carpio, V.; Reyes, R.; García-Bordas, J.; Stamatakis, K.; Dotor, J.; et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol. 2013, 231, 517–531. [Google Scholar] [CrossRef]
- Rynne-Vidal, A.; Au-Yeung, C.L.; Jiménez-Heffernan, J.A.; Pérez-Lozano, M.L.; Cremades-Jimeno, L.; Bárcena, C.; Cristóbal-García, I.; Fernández-Chacón, C.; Yeung, T.L.; Mok, S.C.; et al. Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J. Pathol. 2017, 242, 140–151. [Google Scholar] [CrossRef]
- Cai, J.; Tang, H.; Xu, L.; Wang, X.; Yang, C.; Ruan, S.; Guo, J.; Hu, S.; Wang, Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 2012, 33, 20–29. [Google Scholar] [CrossRef]
- Rynne-Vidal, A.; Jiménez-Heffernan, J.A.; Fernández-Chacón, C.; López-Cabrera, M.; Sandoval, P. The Mesothelial Origin of Carcinoma Associated-Fibroblasts in Peritoneal Metastasis. Cancers 2015, 7, 1994–2011. [Google Scholar] [CrossRef]
- Yeung, T.-L.; Leung, C.S.; Wong, K.-K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, H.; Cai, J.; Zhang, T.; Guo, J.; Feng, D.; Wang, Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011, 303, 47–55. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Lara-Pezzi, E.; Selgas, R.; Ramírez-Huesca, M.; Domínguez-Jiménez, C.; Jiménez-Heffernan, J.A.; Aguilera, A.; Sánchez-Tomero, J.A.; Bajo, M.A.; Alvarez, V.; et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N. Engl. J. Med. 2003, 348, 403–413. [Google Scholar] [CrossRef]
- Kwon, Y.; Smith, B.D.; Zhou, Y.; Kaufman, M.D.; Godwin, A.K. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene 2015, 34, 144–153. [Google Scholar] [CrossRef]
- Deying, W.; Feng, G.; Shumei, L.; Hui, Z.; Ming, L.; Hongqing, W. CAF-derived HGF promotes cell proliferation and drug resistance by up-regulating the c-Met/PI3K/Akt and GRP78 signalling in ovarian cancer cells. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Wei, R.; Lv, M.; Li, F.; Cheng, T.; Zhang, Z.; Jiang, G.; Zhou, Y.; Gao, R.; Wei, X.; Lou, J.; et al. Human CAFs promote lymphangiogenesis in ovarian cancer via the Hh-VEGF-C signaling axis. Oncotarget 2017, 8, 67315–67328. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Coffman, L.G.; Pearson, A.T.; Frisbie, L.G.; Freeman, Z.; Christie, E.; Bowtell, D.D.; Buckanovich, R.J. Ovarian Carcinoma-Associated Mesenchymal Stem Cells Arise from Tissue-Specific Normal Stroma. Stem Cells 2019, 37, 257–269. [Google Scholar] [CrossRef]
- McLean, K.; Gong, Y.; Choi, Y.; Deng, N.; Yang, K.; Bai, S.; Cabrera, L.; Keller, E.; McCauley, L.; Cho, K.R.; et al. Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Invest. 2011, 121, 3206–3219. [Google Scholar] [CrossRef]
- Pasquet, M.; Golzio, M.; Mery, E.; Rafii, A.; Benabbou, N.; Mirshahi, P.; Hennebelle, I.; Bourin, P.; Allal, B.; Teissie, J.; et al. Hospicells (ascites-derived stromal cells) promote tumorigenicity and angiogenesis. Int. J. Cancer 2010, 126, 2090–2101. [Google Scholar] [CrossRef]
- Liguori, M.; Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers 2011, 3, 3740–3761. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Carroll, M.J.; Fogg, K.C.; Patel, H.A.; Krause, H.B.; Mancha, A.-S.; Patankar, M.S.; Weisman, P.S.; Barroilhet, L.; Kreeger, P.K. Alternatively-Activated Macrophages Upregulate Mesothelial Expression of P-Selectin to Enhance Adhesion of Ovarian Cancer Cells. Cancer Res. 2018, 78, 3560–3573. [Google Scholar] [CrossRef]
- Fogg, K.C.; Olson, W.R.; Miller, J.N.; Khan, A.; Renner, C.; Hale, I.; Weisman, P.S.; Kreeger, P.K. Alternatively activated macrophage-derived secretome stimulates ovarian cancer spheroid spreading through a JAK2/STAT3 pathway. Cancer Lett. 2019, 458, 92–101. [Google Scholar] [CrossRef]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2015, 34, 165–176. [Google Scholar] [CrossRef]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Barnett, J.C.; Bean, S.M.; Whitaker, R.S.; Kondoh, E.; Baba, T.; Fujii, S.; Marks, J.R.; Dressman, H.K.; Murphy, S.K.; Berchuck, A. Ovarian cancer tumor infiltrating T-regulatory (Treg) cells are associated with a metastatic phenotype. Gynecol. Oncol. 2010, 116, 556–562. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. TNF-α/NF-κB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef]
- Masoumi Moghaddam, S.; Amini, A.; Morris, D.L.; Pourgholami, M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012, 31, 143–162. [Google Scholar] [CrossRef]
- Choi, H.-J.; Armaiz Pena, G.N.; Pradeep, S.; Cho, M.S.; Coleman, R.L.; Sood, A.K. Anti-vascular therapies in ovarian cancer: Moving beyond anti-VEGF approaches. Cancer Metastasis Rev. 2015, 34, 19–40. [Google Scholar] [CrossRef]
- Kryczek, I.; Lange, A.; Mottram, P.; Alvarez, X.; Cheng, P.; Hogan, M.; Moons, L.; Wei, S.; Zou, L.; Machelon, V.; et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res. 2005, 65, 465–472. [Google Scholar]
- Winiarski, B.K.; Wolanska, K.I.; Rai, S.; Ahmed, T.; Acheson, N.; Gutowski, N.J.; Whatmore, J.L. Epithelial ovarian cancer-induced angiogenic phenotype of human omental microvascular endothelial cells may occur independently of VEGF signaling. Transl. Oncol. 2013, 6, 703–714. [Google Scholar] [CrossRef]
- Schild, T.; Low, V.; Blenis, J.; Gomes, A.P. Unique Metabolic Adaptations Dictate Distal Organ-Specific Metastatic Colonization. Cancer Cell 2018, 33, 347–354. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, S.; Zhang, K.; Qing, Y.; Xu, T. A comprehensive overview of exosomes in ovarian cancer: Emerging biomarkers and therapeutic strategies. J. Ovarian Res. 2017, 10, 73. [Google Scholar] [CrossRef]
- Peng, P.; Yan, Y.; Keng, S. Exosomes in the ascites of ovarian cancer patients: Origin and effects on anti-tumor immunity. Oncol. Rep. 2011, 25, 749–762. [Google Scholar]
- Keller, S.; König, A.-K.; Marmé, F.; Runz, S.; Wolterink, S.; Koensgen, D.; Mustea, A.; Sehouli, J.; Altevogt, P. Systemic presence and tumor-growth promoting effect of ovarian carcinoma released exosomes. Cancer Lett. 2009, 278, 73–81. [Google Scholar] [CrossRef]
- Triantafillu, U.L.; Park, S.; Klaassen, N.L.; Raddatz, A.D.; Kim, Y. Fluid shear stress induces cancer stem cell-like phenotype in MCF7 breast cancer cell line without inducing epithelial to mesenchymal transition. Int. J. Oncol. 2017, 50, 993–1001. [Google Scholar] [CrossRef]
- Novak, C.; Horst, E.; Mehta, G. Review: Mechanotransduction in ovarian cancer: Shearing into the unknown. APL Bioeng. 2018, 2, 031701. [Google Scholar] [CrossRef]
- He, Q.; Luo, X.; Wang, K.; Zhou, Q.; Ao, H.; Yang, Y.; Li, S.; Li, Y.; Zhu, H.; Duan, T. Isolation and Characterization of Cancer Stem Cells from High-Grade Serous Ovarian Carcinomas. Cell. Physiol. Biochem. 2014, 33, 173–184. [Google Scholar] [CrossRef]
- Eyre, R.; Harvey, I.; Stemke-Hale, K.; Lennard, T.W.J.; Tyson-Capper, A.; Meeson, A.P. Reversing paclitaxel resistance in ovarian cancer cells via inhibition of the ABCB1 expressing side population. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 9879–9892. [Google Scholar] [CrossRef]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef]
- Roy Choudhury, A.; Gupta, S.; Chaturvedi, P.K.; Kumar, N.; Pandey, D. Mechanobiology of Cancer Stem Cells and Their Niche. Cancer Microenviron. 2019, 12, 17–27. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, Y.; Ye, R.; Zheng, Y.; Zhao, Z.; Li, J. Mechanotransduction in cancer stem cells. Cell Biol. Int. 2013, 37, 888–891. [Google Scholar] [CrossRef]
- Northey, J.J.; Przybyla, L.; Weaver, V.M. Tissue Force Programs Cell Fate and Tumor Aggression. Cancer Discov. 2017, 7, 1224–1237. [Google Scholar] [CrossRef]
- Ip, C.K.M.; Li, S.-S.; Tang, M.Y.H.; Sy, S.K.H.; Ren, Y.; Shum, H.C.; Wong, A.S.T. Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress. Sci. Rep. 2016, 6, 26788. [Google Scholar] [CrossRef]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Wang, X.; Li, X.; Liang, J.; Xing, H. Role of TWIST2, E-cadherin and Vimentin in epithelial ovarian carcinogenesis and prognosis and their interaction in cancer progression. Eur. J. Gynaecol. Oncol. 2016, 37, 100–108. [Google Scholar]
- Rizvi, I.; Gurkan, U.A.; Tasoglu, S.; Alagic, N.; Celli, J.P.; Mensah, L.B.; Mai, Z.; Demirci, U.; Hasan, T. Flow induces epithelial-mesenchymal transition, cellular heterogeneity and biomarker modulation in 3D ovarian cancer nodules. Proc. Natl. Acad. Sci. USA 2013, 110, E1974–E1983. [Google Scholar] [CrossRef]
- Avraham-Chakim, L.; Elad, D.; Zaretsky, U.; Kloog, Y.; Jaffa, A.; Grisaru, D. Fluid-flow induced wall shear stress and epithelial ovarian cancer peritoneal spreading. PLoS ONE 2013, 8, e60965. [Google Scholar] [CrossRef]
- Jia, P.; Wu, S.; Li, F.; Xu, Q.; Wu, M.; Chen, G.; Liao, G.; Wang, S.; Zhou, J.; Lu, Y.; et al. Breast cancer resistance protein-mediated topotecan resistance in ovarian cancer cells. Int. J. Gynecol. Cancer 2005, 15, 1042–1048. [Google Scholar] [CrossRef]
- Shukla, S.; Ohnuma, S.; Ambudkar, S.V. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr. Drug Targets 2011, 12, 621–630. [Google Scholar] [CrossRef]
- Samardzija, C.; Luwor, R.B.; Volchek, M.; Quinn, M.A.; Findlay, J.K.; Ahmed, N. A critical role of Oct4A in mediating metastasis and disease-free survival in a mouse model of ovarian cancer. Mol. Cancer 2015, 14, 152. [Google Scholar] [CrossRef]
- Chau, W.K.; Ip, C.K.; Mak, A.S.C.; Lai, H.-C.; Wong, A.S.T. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/β-catenin-ATP-binding cassette G2 signaling. Oncogene 2013, 32, 2767–2781. [Google Scholar] [CrossRef]
- Bryan, B.A.; Dennstedt, E.; Mitchell, D.C.; Walshe, T.E.; Noma, K.; Loureiro, R.; Saint-Geniez, M.; Campaigniac, J.-P.; Liao, J.K.; D’Amore, P.A. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010, 24, 3186–3195. [Google Scholar] [CrossRef]
- Ghosh, K.; Pan, Z.; Guan, E.; Ge, S.; Liu, Y.; Nakamura, T.; Ren, X.-D.; Rafailovich, M.; Clark, R.A.F. Cell adaptation to a physiologically relevant ECM mimic with different viscoelastic properties. Biomaterials 2007, 28, 671–679. [Google Scholar] [CrossRef]
- Hendricks, P.; Diaz, F.J.; Schmitt, S.; Sitta Sittampalam, G.; Nirmalanandhan, V.S. Effects of respiratory mechanical forces on the pharmacological response of lung cancer cells to chemotherapeutic agents. Fundam. Clin. Pharmacol. 2012, 26, 632–643. [Google Scholar] [CrossRef]
- Klymenko, Y.; Kim, O.; Stack, M.S. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers 2017, 9, 104. [Google Scholar] [CrossRef]
- Burkhalter, R. Microenvironmental regulation of ovarian cancer dissemination via activation of the Wnt signaling pathway. Ph.D. Thesis, University of Missouri, Columbia, MO, USA, May 2012. [Google Scholar]
- McGrail, D.J.; Kieu, Q.M.N.; Dawson, M.R. The malignancy of metastatic ovarian cancer cells is increased on soft matrices through a mechanosensitive Rho-ROCK pathway. J. Cell Sci. 2014, 127, 2621–2626. [Google Scholar] [CrossRef]
- Nam, S.; Lee, J.; Brownfield, D.G.; Chaudhuri, O. Viscoplasticity Enables Mechanical Remodeling of Matrix by Cells. Biophys. J. 2016, 111, 2296–2308. [Google Scholar] [CrossRef]
- Wisdom, K.M.; Adebowale, K.; Chang, J.; Lee, J.Y.; Nam, S.; Desai, R.; Rossen, N.S.; Rafat, M.; West, R.B.; Hodgson, L.; et al. Matrix mechanical plasticity regulates cancer cell migration through confining microenvironments. Nat. Commun. 2018, 9, 4144. [Google Scholar] [CrossRef]
- Wullkopf, L.; West, A.-K.V.; Leijnse, N.; Cox, T.R.; Madsen, C.D.; Oddershede, L.B.; Erler, J.T. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol. Biol. Cell 2018, 29, 2378–2385. [Google Scholar] [CrossRef]
- Kipps, E.; Tan, D.S.P.; Kaye, S.B. Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-Mesenchymal Transition in Breast Cancer Relates to the Basal-like Phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef]
- Kossatz, U.; Dietrich, N.; Zender, L.; Buer, J.; Manns, M.P.; Malek, N.P. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004, 18, 2602–2607. [Google Scholar] [CrossRef]
- Doetsch, F.; Verdugo, J.M.-G.; Caille, I.; Alvarez-Buylla, A.; Chao, M.V.; Casaccia-Bonnefil, P. Lack of the Cell-Cycle Inhibitor p27Kip1 Results in Selective Increase of Transit-Amplifying Cells for Adult Neurogenesis. J. Neurosci. 2002, 22, 2255–2264. [Google Scholar] [CrossRef]
- Chen, D.; Krasinski, K.; Sylvester, A.; Chen, J.; Nisen, P.D.; Andrés, V. Downregulation of cyclin-dependent kinase 2 activity and cyclin A promoter activity in vascular smooth muscle cells by p27(KIP1), an inhibitor of neointima formation in the rat carotid artery. J. Clin. Invest. 1997, 99, 2334–2341. [Google Scholar] [CrossRef]
- Novak, C.M.; Horst, E.N.; Taylor, C.; Liu, C.; Mehta, G. Breast cancer cells become chemoresistant, display invasive phenotypes, and upregulate Plau when exposed to fluid shear stress in an innovative 3D bioreactor. Biotechnol. Bioenginnering 2019. (accepted). [Google Scholar]
- Kumar, S.M.; Liu, S.; Lu, H.; Zhang, H.; Zhang, P.J.; Gimotty, P.A.; Guerra, M.; Guo, W.; Xu, X. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 2012, 31, 4898–4911. [Google Scholar] [CrossRef]
- Wu, D.; Huang, H.; He, C.; Wang, K. MicroRNA-199a-3p regulates endometrial cancer cell proliferation by targeting mammalian target of rapamycin (mTOR). Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2013, 23, 1191–1197. [Google Scholar] [CrossRef]
- Shen, L.; Sun, C.; Li, Y.; Li, X.; Sun, T.; Liu, C.; Zhou, Y.; Du, Z. MicroRNA-199a-3p suppresses glioma cell proliferation by regulating the AKT/mTOR signaling pathway. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2015, 36, 6929–6938. [Google Scholar] [CrossRef]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The Role of Mechanical Forces in Tumor Growth and Therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef]
- Ghosh, K.; Thodeti, C.K.; Dudley, A.C.; Mammoto, A.; Klagsbrun, M.; Ingber, D.E. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc. Natl. Acad. Sci. USA 2008, 105, 11305–11310. [Google Scholar] [CrossRef]
- Paszek, M.J.; Weaver, V.M. The Tension Mounts: Mechanics Meets Morphogenesis and Malignancy. J. Mammary Gland Biol. Neoplasia 2004, 9, 325–342. [Google Scholar] [CrossRef]
- Zhang, Y.; McNerny, E.G.; Terajima, M.; Raghavan, M.; Romanowicz, G.; Zhang, Z.; Zhang, H.; Kamiya, N.; Tantillo, M.; Zhu, P.; et al. Loss of BMP signaling through BMPR1A in osteoblasts leads to greater collagen cross-link maturation and material-level mechanical properties in mouse femoral trabecular compartments. Bone 2016, 88, 74–84. [Google Scholar] [CrossRef]
- Klymenko, Y.; Wates, R.B.; Weiss-Bilka, H.; Lombard, R.; Liu, Y.; Campbell, L.; Kim, O.; Wagner, D.; Ravosa, M.J.; Stack, M.S. Modeling the effect of ascites-induced compression on ovarian cancer multicellular aggregates. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Tse, J.M.; Cheng, G.; Tyrrell, J.A.; Wilcox-Adelman, S.A.; Boucher, Y.; Jain, R.K.; Munn, L.L. Mechanical compression drives cancer cells toward invasive phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 911–916. [Google Scholar] [CrossRef]
- Arend, R.C.; Londoño-Joshi, A.I.; Straughn, J.M.; Buchsbaum, D.J. The Wnt/β-catenin pathway in ovarian cancer: A review. Gynecol. Oncol. 2013, 131, 772–779. [Google Scholar] [CrossRef]
- Kalli, M.; Minia, A.; Pliaka, V.; Fotis, C.; Alexopoulos, L.G.; Stylianopoulos, T. Solid stress-induced migration is mediated by GDF15 through Akt pathway activation in pancreatic cancer cells. Sci. Rep. 2019, 9, 978. [Google Scholar] [CrossRef]
- Gkretsi, V.; Stylianopoulos, T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- McKenzie, A.J.; Hicks, S.R.; Svec, K.V.; Naughton, H.; Edmunds, Z.L.; Howe, A.K. The mechanical microenvironment regulates ovarian cancer cell morphology, migration, and spheroid disaggregation. Sci. Rep. 2018, 8, 7228. [Google Scholar] [CrossRef]
- Babahosseini, H.; Ketene, A.N.; Schmelz, E.M.; Roberts, P.C.; Agah, M. Biomechanical profile of cancer stem-like/tumor-initiating cells derived from a progressive ovarian cancer model. Nanomedicine Nanotechnol. Biol. Med. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Tan, F.; Huang, Y.; Pei, Q.; Liu, H.; Pei, H.; Zhu, H. Matrix stiffness mediates stemness characteristics via activating the Yes-associated protein in colorectal cancer cells. J. Cell. Biochem. 2018, 120, 2213–2225. [Google Scholar] [CrossRef]
- Jabbari, E.; Sarvestani, S.K.; Daneshian, L.; Moeinzadeh, S. Optimum 3D Matrix Stiffness for Maintenance of Cancer Stem Cells Is Dependent on Tissue Origin of Cancer Cells. PLoS ONE 2015, 10, e0132377. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Gu, L.; Klumpers, D.; Darnell, M.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Lee, H.-P.; Lippens, E.; Duda, G.N.; et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 2016, 15, 326–334. [Google Scholar] [CrossRef]
- Matellan, C.; del Río Hernández, A.E. Where No Hand Has Gone Before: Probing Mechanobiology at the Cellular Level. ACS Biomater. Sci. Eng. 2018. [Google Scholar] [CrossRef]
- Luo, Q.; Kuang, D.; Zhang, B.; Song, G. Cell stiffness determined by atomic force microscopy and its correlation with cell motility. Biochim. Biophys. Acta 2016, 1860, 1953–1960. [Google Scholar] [CrossRef]
- Sinkus, R.; Siegmann, K.; Xydeas, T.; Tanter, M.; Claussen, C.; Fink, M. MR elastography of breast lesions: Understanding the solid/liquid duality can improve the specificity of contrast-enhanced MR mammography. Magn. Reson. Med. 2007, 58, 1135–1144. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mechanical Stimuli | Physiological Sources of Mechanical Stimuli in Ovarian Cancer | Influences of Mechanical Stimuli on Cancer Progression and Metastasis |
|---|---|---|
| Shear |
| |
| Tension/Strain |
| Non-Ovarian Cancer Findings
|
| Compression |
| |
| Stiffness |
| |
| Others: Stress relaxation, Creep compliance | ECM restructuring from:
| Non-Ovarian Cancer Findings |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bregenzer, M.E.; Horst, E.N.; Mehta, P.; Novak, C.M.; Repetto, T.; Mehta, G. The Role of Cancer Stem Cells and Mechanical Forces in Ovarian Cancer Metastasis. Cancers 2019, 11, 1008. https://doi.org/10.3390/cancers11071008
Bregenzer ME, Horst EN, Mehta P, Novak CM, Repetto T, Mehta G. The Role of Cancer Stem Cells and Mechanical Forces in Ovarian Cancer Metastasis. Cancers. 2019; 11(7):1008. https://doi.org/10.3390/cancers11071008
Chicago/Turabian StyleBregenzer, Michael E., Eric N. Horst, Pooja Mehta, Caymen M. Novak, Taylor Repetto, and Geeta Mehta. 2019. "The Role of Cancer Stem Cells and Mechanical Forces in Ovarian Cancer Metastasis" Cancers 11, no. 7: 1008. https://doi.org/10.3390/cancers11071008
APA StyleBregenzer, M. E., Horst, E. N., Mehta, P., Novak, C. M., Repetto, T., & Mehta, G. (2019). The Role of Cancer Stem Cells and Mechanical Forces in Ovarian Cancer Metastasis. Cancers, 11(7), 1008. https://doi.org/10.3390/cancers11071008