Identification of Cancer Stem Cell Molecular Markers and Effects of hsa-miR-21-3p on Stemness in Esophageal Squamous Cell Carcinoma


Abstract
1. Introduction
2. Results
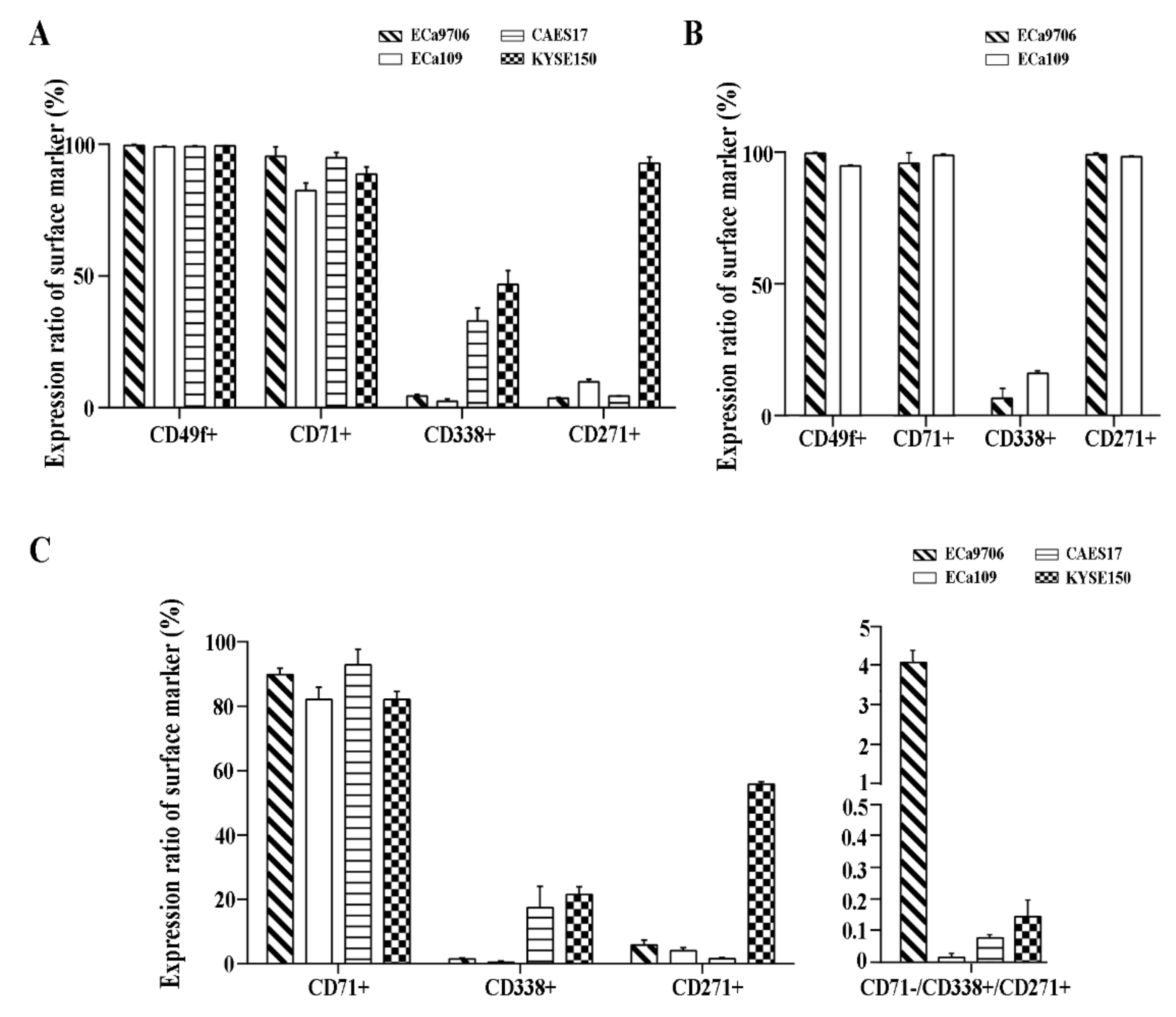
2.1. CD71, CD27,1 andCD338 Are Potential Biomarkers for Cancer Stem Cell (CSC) Sorting in Esophageal Squamous Cell Carcinoma (ESCC)
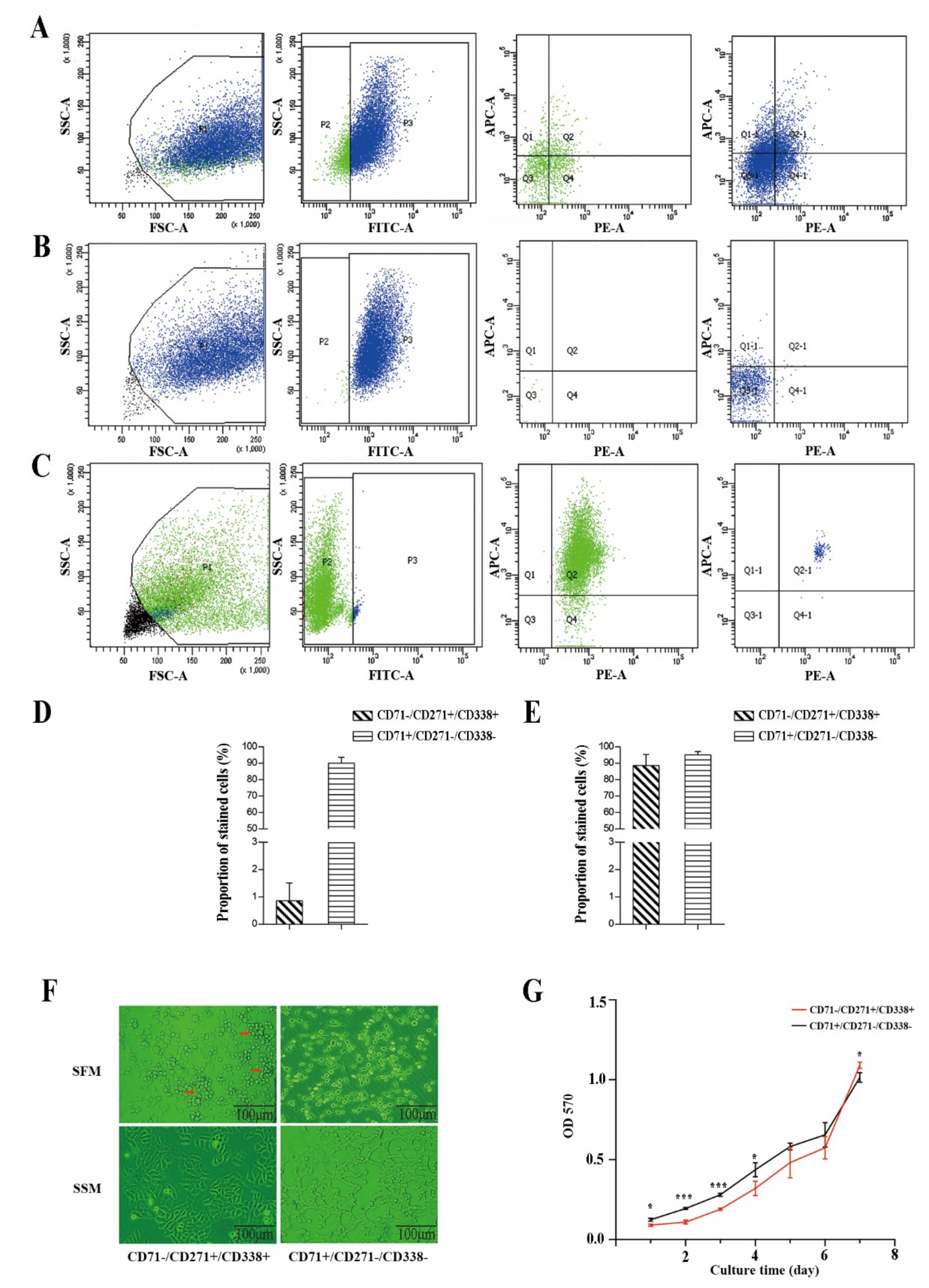
2.2. CD71−/CD271+/CD338+ Subpopulation Cells Possessed More Stem Cell Properties
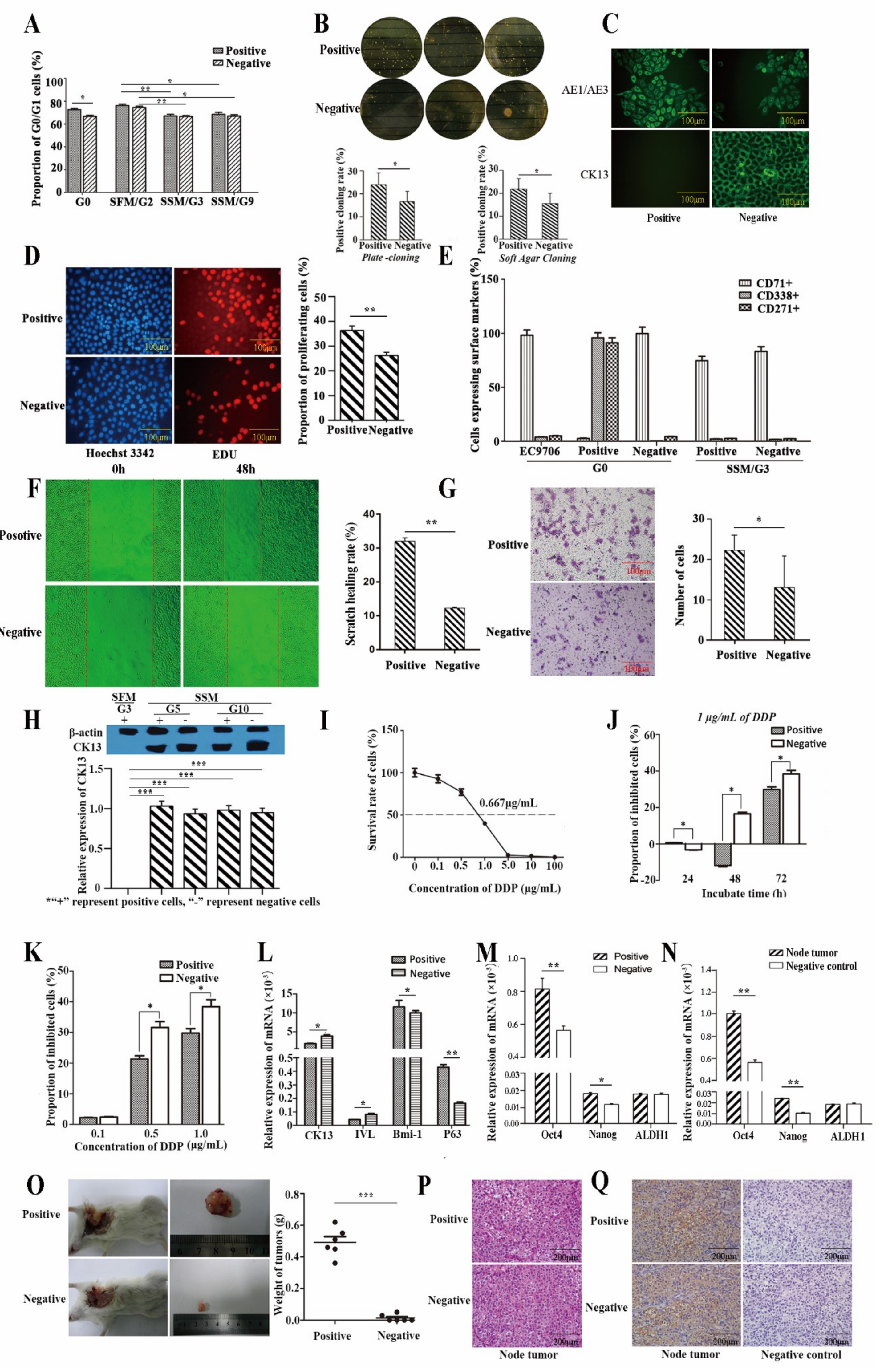
2.2.1. Proliferative Capacity
2.2.2. Self-Renewal Ability
2.2.3. Differentiative Capacity
2.2.4. Metastasis Ability
2.2.5. Drug Resistance
2.2.6. Gene Expression Related to Stemness
2.2.7. Tumor Xenograft in Nude Mice
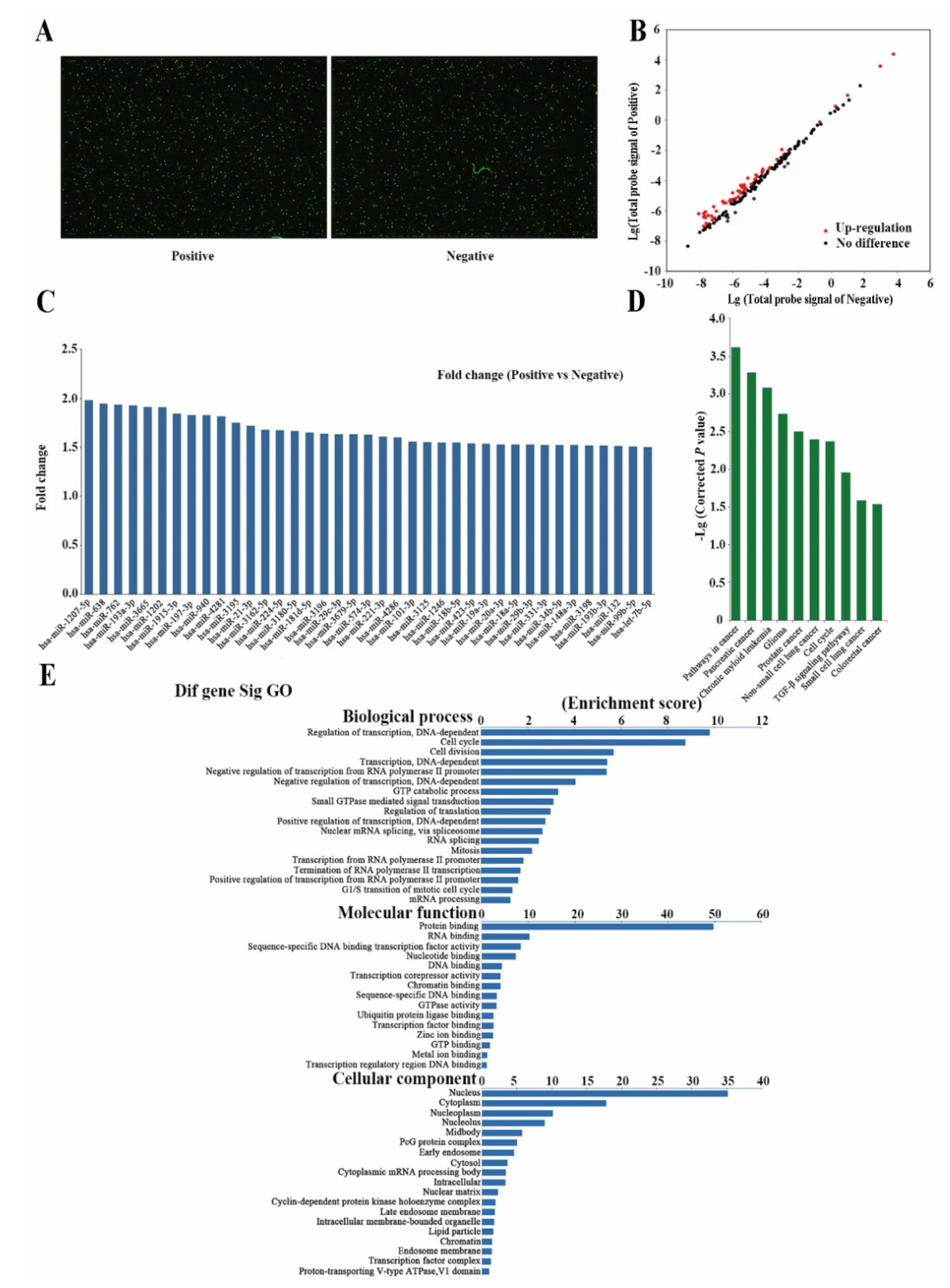
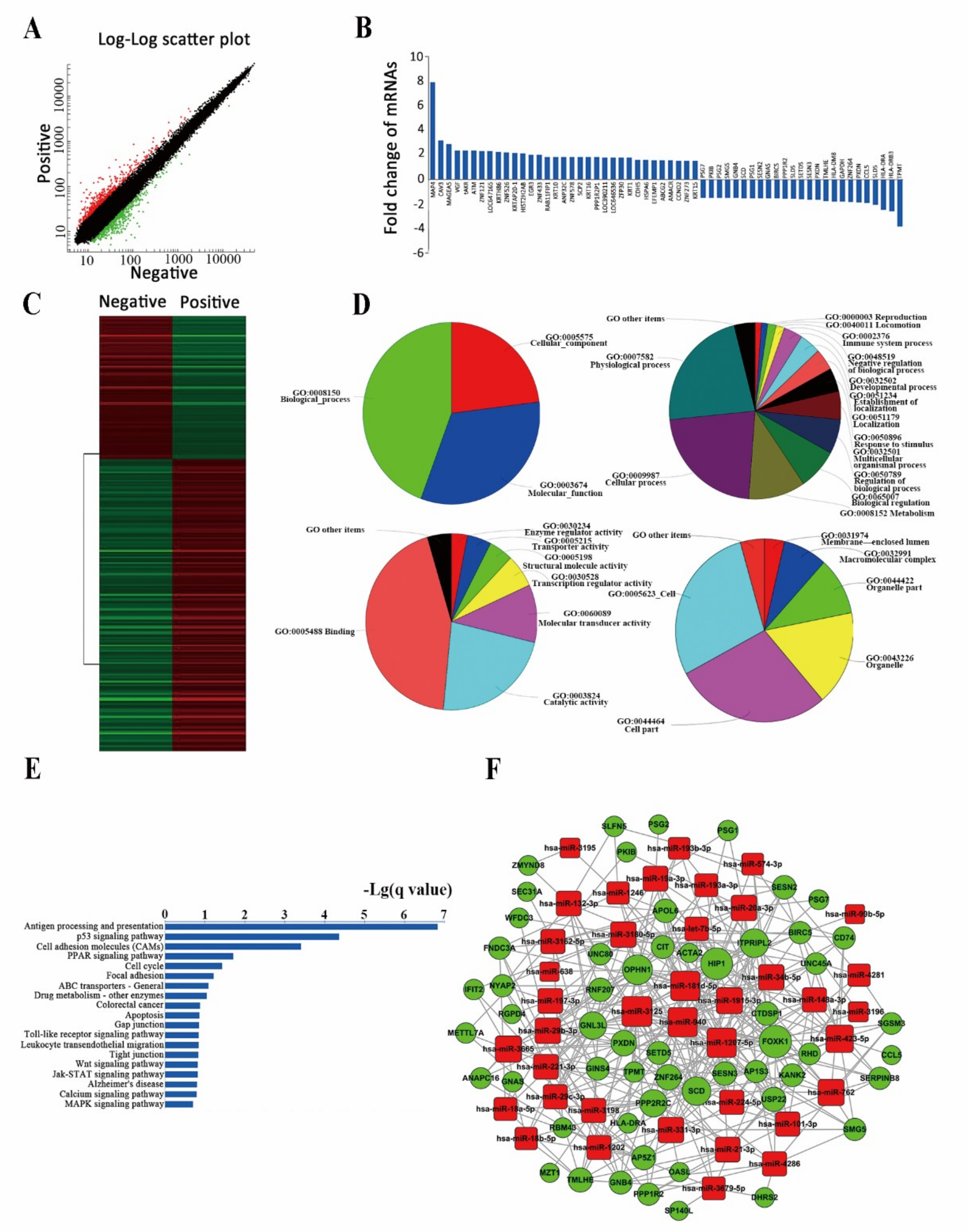
2.3. Differently Expressed miRNAs Are Vital for Cell Stemness
2.4. Analysis of Differently Expressed mRNAs between Two Subpopulations of Cells
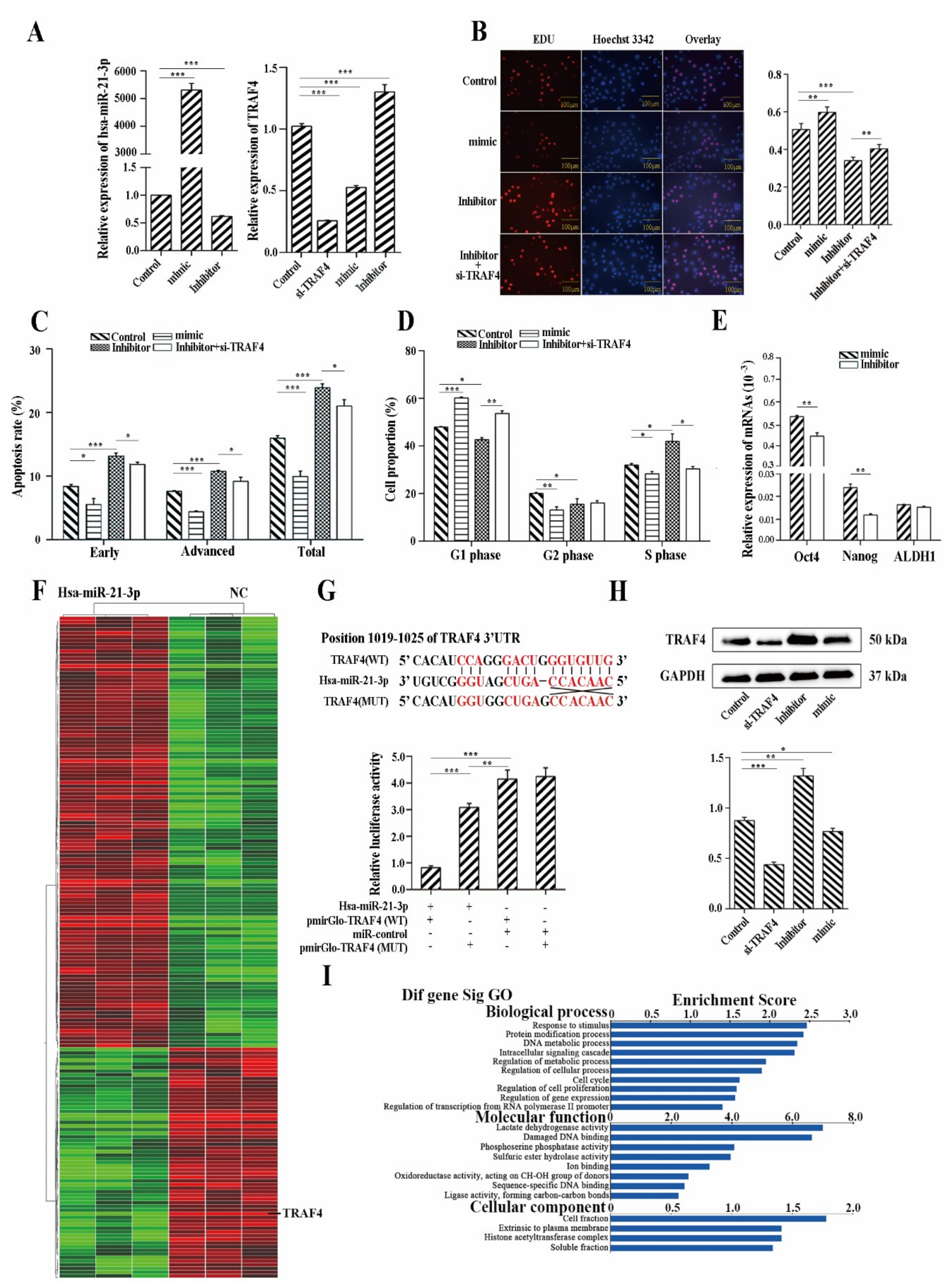
2.5. Hsa-miR-21-3p Is Critical for the Stemness Maintenance of CSCs
2.6. Hsa-miR-21-3p May Be a Potential Biomarker for Early Diagnosis of ESCC
3. Discussion
4. Materials and Methods
4.1. Population
4.2. Animal and Cell Lines
4.3. Antibodies and Reagents
4.4. Flow Cytometry and Cell Sorting
4.5. Apoptosis and Cell Cycle
4.6. Cell Proliferation Assay
4.7. Plate Cloning Assay
4.8. Soft Agar Cloning Assay
4.9. Scratch-Healing Experiment
4.10. Invasive Experiment
4.11. RNA Extraction and Genome-Wide mRNA Microarray
4.12. Bioinformatics Analysis of Microarray and Target Prediction
4.13. Western Blot Analysis
4.14. Luciferase Reporter Assay
4.15. RT-QPCR
4.16. Tumor Xenograft in Nude Mice
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CSCs | Cancer stem cells |
| ESCC | Esophageal squamous cell carcinoma |
| EdU | 5-ethynyl-2′-deoxyuridine |
| ALDH | Aldehyde dehydrogenase |
| MTT | Thiazolyl blue tetrazolium bromide |
| HE | Hematoxylin-eosin |
| SCID | Server Combined Immune-deficiency |
| p75NTR | p75 neurotmphin receptor |
| MMLV | Moloney Murine Leukemia Virus |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ANOVA | Analysis of variance |
| SSM | Serum-Supplied-DMEM-F12 Medium with 10% fetal bovine serum |
| SFM | Serum-Free-DMEM-F12 Medium |
| FACS | Fluorescence activated cell sorting |
References
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Wang, D.; Plukker, J.T.M.; Coppes, R.P. Cancer stem cells with increased metastatic potential as a therapeutic target for esophageal cancer. Semin. Cancer Biol. 2017, 44, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Napier, K.J.; Scheerer, M.; Misra, S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J. Gastrointest. Oncol. 2014, 6, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Fu, W.; Li, T.; Yuan, Q.; Wang, F.; Lv, G.; Lv, Y.; Fan, X.; Shen, Y.; Lin, F.; et al. Antagonism of EGFR and Notch limits resistance to EGFR inhibitors and radiation by decreasing tumor-initiating cell frequency. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.K.; Shao, C.; Wang, J.; Wei, Q.; Wang, X.; Collier, Z.; Tang, S.; Liu, H.; Zhang, F.; Huang, J.; et al. Wnt/beta-catenin signaling plays an ever-expanding role in stem cell self-renewal, tumorigenesis and cancer chemoresistance. Genes Dis. 2016, 3, 11–40. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Cai, J.; Chen, B.; Wu, S.; Li, R.; Xu, X.; Yang, Y.; Guan, H.; Zhu, X.; Zhang, L.; et al. Aberrantly expressed miR-582-3p maintains lung cancer stem cell-like traits by activating Wnt/beta-catenin signalling. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-S.; Li, W.-J.; Ge, D.; Zhang, P.-J.; Li, J.-J.; Lu, C.-L.; Ji, X.-D.; Guan, D.-X.; Gao, H.; Xu, L.-Y.; et al. Tumor Initiating Cells in Esophageal Squamous Cell Carcinomas Express High Levels of CD44. PLoS ONE 2011, 6, e21419. [Google Scholar] [CrossRef]
- Okumura, T.; Tsunoda, S.; Mori, Y.; Ito, T.; Kikuchi, K.; Wang, T.C.; Yasumoto, S.; Shimada, Y. The biological role of the low-affinity p75 neurotrophin receptor in esophageal squamous cell carcinoma. Clin. Cancer Res. 2006, 12, 5096–5103. [Google Scholar] [CrossRef]
- Tang, K.H.; Dai, Y.D.; Tong, M.; Chan, Y.P.; Kwan, P.S.; Fu, L.; Qin, Y.R.; Tsao, S.W.; Lung, H.L.; Lung, M.L.; et al. A CD90(+) Tumor-Initiating Cell Population with an Aggressive Signature and Metastatic Capacity in Esophageal Cancer. Cancer Res. 2013, 73, 2322–2332. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Zhu, H.; Tang, J.; Zhang, S.; Luo, J.; Sun, X. CD90 positive cells exhibit aggressive radioresistance in esophageal squamous cell carcinoma. J. Thorac. Dis. 2017, 9, 610–620. [Google Scholar] [CrossRef]
- Zhang, J.-X.; Chen, Z.-H.; Xu, Y.; Chen, J.-W.; Weng, H.-W.; Yun, M.; Zheng, Z.-S.; Chen, C.; Wu, B.-L.; Li, E.-M.; et al. Downregulation of MicroRNA-644a Promotes Esophageal Squamous Cell Carcinoma Aggressiveness and Stem Cell-like Phenotype via Dysregulation of PITX2. Clin. Cancer Res. 2017, 23, 298–310. [Google Scholar] [CrossRef]
- Chen, S.; Zheng, Y.; Zhang, S.; Jia, L.; Zhou, Y. Promotion Effects of miR-375 on the Osteogenic Differentiation of Human Adipose-Derived Mesenchymal Stem Cells. Stem Cell Rep. 2017, 8, 773–786. [Google Scholar] [CrossRef]
- Bitarte, N.; Bandres, E.; Boni, V.; Zarate, R.; Rodriguez, J.; Gonzalez-Huarriz, M.; Lopez, I.; Javier Sola, J.; Alonso, M.M.; Fortes, P.; et al. MicroRNA-451 Is Involved in the Self-renewal, Tumorigenicity, and Chemoresistance of Colorectal Cancer Stem Cells. Stem Cells 2011, 29, 1661–1671. [Google Scholar] [CrossRef]
- Peng, H.; Park, J.K.; Katsnelson, J.; Kaplan, N.; Yang, W.; Getsios, S.; Lavker, R.M. microRNA-103/107 Family Regulates Multiple Epithelial Stem Cell Characteristics. Stem Cells 2015, 33, 1642–1656. [Google Scholar] [CrossRef]
- Li, S.; Ge, S.; Yang, P. Expression of cytokeratins in enamel organ, junctional epithelium and epithelial cell rests of Malassez. J. Periodontal Res. 2015, 50, 846–854. [Google Scholar] [CrossRef]
- Shiraishi, O.; Yamasaki, M.; Makino, T.; Motoori, M.; Miyata, H.; Shinkai, M.; Kimura, Y.; Hirao, M.; Fujitani, K.; Tamura, S.; et al. Feasibility of Preoperative Chemotherapy with Docetaxel, Cisplatin, and 5-Fluorouracil versus Adriamycin, Cisplatin, and 5-Fluorouracil for Resectable Advanced Esophageal Cancer. Oncology 2017, 92, 101–108. [Google Scholar] [CrossRef]
- Takahashi, Y.; Noguchi, T.; Takeno, S.; Kimura, Y.; Okubo, M.; Kawahara, K. Reduced expression of p63 has prognostic implications for patients with esophageal squamous cell carcinoma. Oncol. Rep. 2006, 15, 323–328. [Google Scholar] [CrossRef]
- Hwang, C.-C.; Nieh, S.; Lai, C.-H.; Tsai, C.-S.; Chang, L.-C.; Hua, C.-C.; Chi, W.-Y.; Chien, H.-P.; Wang, C.-W.; Chan, S.-C.; et al. A Retrospective Review of the Prognostic Value of ALDH-1, Bmi-1 and Nanog Stem Cell Markers in Esophageal Squamous Cell Carcinoma. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Kim, B.E.; Leung, D.Y.M.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef]
- Chiou, S.H.; Wang, M.L.; Chou, Y.T.; Chen, C.J.; Hong, C.F.; Hsieh, W.J.; Chang, H.T.; Chen, Y.S.; Lin, T.W.; Hsu, H.S.; et al. Coexpression of Oct4 and Nanog Enhances Malignancy in Lung Adenocarcinoma by Inducing Cancer Stem Cell-Like Properties and Epithelial-Mesenchymal Transdifferentiation. Cancer Res. 2010, 70, 10433–10444. [Google Scholar] [CrossRef]
- Deng, S.; Yang, X.J.; Lassus, H.; Liang, S.; Kaur, S.; Ye, Q.R.; Li, C.S.; Wang, L.P.; Roby, K.F.; Orsulic, S.; et al. Distinct Expression Levels and Patterns of Stem Cell Marker, Aldehyde Dehydrogenase Isoform 1 (ALDH1), in Human Epithelial Cancers. PLoS ONE 2010, 5, 11. [Google Scholar] [CrossRef]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehres, H.; Shen, J.J.; Tang, D.G. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef]
- Kojima, H.; Okumura, T.; Yamaguchi, T.; Miwa, T.; Shimada, Y.; Nagata, T. Enhanced cancer stem cell properties of a mitotically quiescent subpopulation of p75NTR-positive cells in esophageal squamous cell carcinoma. Int. J. Oncol. 2017, 51, 49–62. [Google Scholar] [CrossRef]
- Okumura, T.; Yamaguchi, T.; Watanabe, T.; Nagata, T.; Shimada, Y. Clinical Relevance of a Candidate Stem Cell Marker, p75 Neurotrophin Receptor (p75NTR) Expression in Circulating Tumor Cells. Adv. Exp. Med. Biol. 2017, 994, 247–254. [Google Scholar] [CrossRef]
- Hang, D.; Dong, H.C.; Ning, T.; Dong, B.; Hou, D.L.; Xu, W.G. Prognostic value of the stem cell markers CD133 and ABCG2 expression in esophageal squamous cell carcinoma. Dis. Esophagus 2012, 25, 638–644. [Google Scholar] [CrossRef]
- Hemmesi, K.; Squadrito, M.L.; Mestdagh, P.; Conti, V.; Cominelli, M.; Piras, I.S.; Sergi, L.S.; Piccinin, S.; Maestro, R.; Poliani, P.L.; et al. miR-135a Inhibits Cancer Stem Cell-Driven Medulloblastoma Development by Directly Repressing Arhgef6 Expression. Stem Cells 2015, 33, 1377–1389. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, Y.; Wei, S.; Gao, W.; Chen, L.; Zhou, T.; Wang, Z.; Ying, M.; Zheng, Q. miRNA-148b suppresses hepatic cancer stem cell by targeting neuropilin-1. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Baez-Vega, P.M.; Vargas, I.M.E.; Valiyeva, F.; Rosado, J.E.; Roman, A.; Flores, J.; Marcos-Martinez, M.J.; Vivas-Mejia, P.E. Targeting miR-21-3p inhibits proliferation and invasion of ovarian cancer cells. Oncotarget 2016, 7, 36321–36337. [Google Scholar] [CrossRef]
- Pink, R.C.; Samuel, P.; Massa, D.; Caley, D.P.; Brooks, S.A.; Carter, D.R.F. The passenger strand, miR-21-3p, plays a role in mediating cisplatin resistance in ovarian cancer cells. Gynecol. Oncol. 2015, 137, 143–151. [Google Scholar] [CrossRef]
- Lo, T.F.; Tsai, W.C.; Chen, S.T. MicroRNA-21-3p, a Berberine-Induced miRNA, Directly Down-Regulates Human Methionine Adenosyltransferases 2A and 2B and Inhibits Hepatoma Cell Growth. PLoS ONE 2013, 8, 11. [Google Scholar] [CrossRef]
- Jiao, W.J.; Leng, X.Q.; Zhou, Q.; Wu, Y.Y.; Sun, L.N.; Tan, Y.; Ni, H.L.; Dong, X.Q.; Shen, T.; Liu, Y.; et al. Different miR-21-3p isoforms and their different features in colorectal cancer. Int. J. Cancer 2017, 141, 2103–2111. [Google Scholar] [CrossRef]
- Wang, B.; Yu, P.; Li, T.; Bian, Y.; Weng, X. MicroRNA expression in bone marrow mesenchymal stem cells from mice with steroid-induced osteonecrosis of the femoral head. Mol. Med. Rep. 2015, 12, 7447–7454. [Google Scholar] [CrossRef]
- Kedinger, V.; Rio, M.-C. TRAF4, the unique family member. Adv. Exp. Med. Biol. 2007, 597, 60–71. [Google Scholar]
- Sax, J.K.; El-Deiry, W.S. Identification and characterization of the cytoplasmic protein TRAF4 as a p53-regulated proapoptotic gene. J. Biol. Chem. 2003, 278, 36435–36444. [Google Scholar] [CrossRef]
- Rozan, L.M.; El-Deiry, W.S. Identification and characterization of proteins interacting with Traf4, an enigmatic p53 target. Cancer Biol. Ther. 2006, 5, 1228–1235. [Google Scholar] [CrossRef]
- Gu, X.; Coates, P.J.; MacCallum, S.F.; Boldrup, L.; Sjostrom, B.; Nylander, K. TRAR is potently induced by TAp63 isoforms and localised according to differentiation in SCCHN. Cancer Biol. Ther. 2007, 6, 1986–1990. [Google Scholar] [CrossRef]
- Kedinger, V.; Alpy, F.; Baguet, A.; Polette, M.; Stoll, I.; Chenard, M.-P.; Tomasetto, C.; Rio, M.-C. Tumor Necrosis Factor Receptor-Associated Factor 4 Is a Dynamic Tight Junction-Related Shuttle Protein Involved in Epithelium Homeostasis. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Fleckenstein, D.S.; Dirks, W.G.; Drexler, H.G.; Quentmeier, H. Tumor necrosis factor receptor-associated factor (TRAF) 4 is a new binding partner for the p70S6 serine/threonine kinase. Leuk. Res. 2003, 27, 687–694. [Google Scholar] [CrossRef]
- Ye, X.; Mehlen, P.; Rabizadeh, S.; VanArsdale, T.; Zhang, H.Y.; Shin, H.; Wang, J.J.L.; Leo, E.; Zapata, J.; Hauser, C.A.; et al. TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J. Biol. Chem. 1999, 274, 30202–30208. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | n | Mean ± SD | ΔΔCT * | 2−ΔΔCT | p Value | t Value |
|---|---|---|---|---|---|---|
| Tumor tissue | 137 | 10.201 ± 2.930 | −0.896 ± 3.865 | 1.863 | <0.01 | 2.737 |
| Para-tumor tissue | 173 | 11.097 ± 2.809 |
| Group | β | SE | χ2 | p Value | OR | 95% CI |
|---|---|---|---|---|---|---|
| Tumor tissue | −0.126 | 0.049 | 6.644 | 0.010 | 1.135 | 1.030–1.250 |
| Para-tumor tissue | 1.000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Z.; Liu, H.; Shi, Y.; Yin, L.; Zhu, Y.; Liu, R. Identification of Cancer Stem Cell Molecular Markers and Effects of hsa-miR-21-3p on Stemness in Esophageal Squamous Cell Carcinoma. Cancers 2019, 11, 518. https://doi.org/10.3390/cancers11040518
Gao Z, Liu H, Shi Y, Yin L, Zhu Y, Liu R. Identification of Cancer Stem Cell Molecular Markers and Effects of hsa-miR-21-3p on Stemness in Esophageal Squamous Cell Carcinoma. Cancers. 2019; 11(4):518. https://doi.org/10.3390/cancers11040518
Chicago/Turabian StyleGao, Zhikui, Hui Liu, Yajuan Shi, Lihong Yin, Yong Zhu, and Ran Liu. 2019. "Identification of Cancer Stem Cell Molecular Markers and Effects of hsa-miR-21-3p on Stemness in Esophageal Squamous Cell Carcinoma" Cancers 11, no. 4: 518. https://doi.org/10.3390/cancers11040518
APA StyleGao, Z., Liu, H., Shi, Y., Yin, L., Zhu, Y., & Liu, R. (2019). Identification of Cancer Stem Cell Molecular Markers and Effects of hsa-miR-21-3p on Stemness in Esophageal Squamous Cell Carcinoma. Cancers, 11(4), 518. https://doi.org/10.3390/cancers11040518