Endoplasmic Reticulum Stress Signaling as a Therapeutic Target in Malignant Pleural Mesothelioma


 , , ,
, , ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
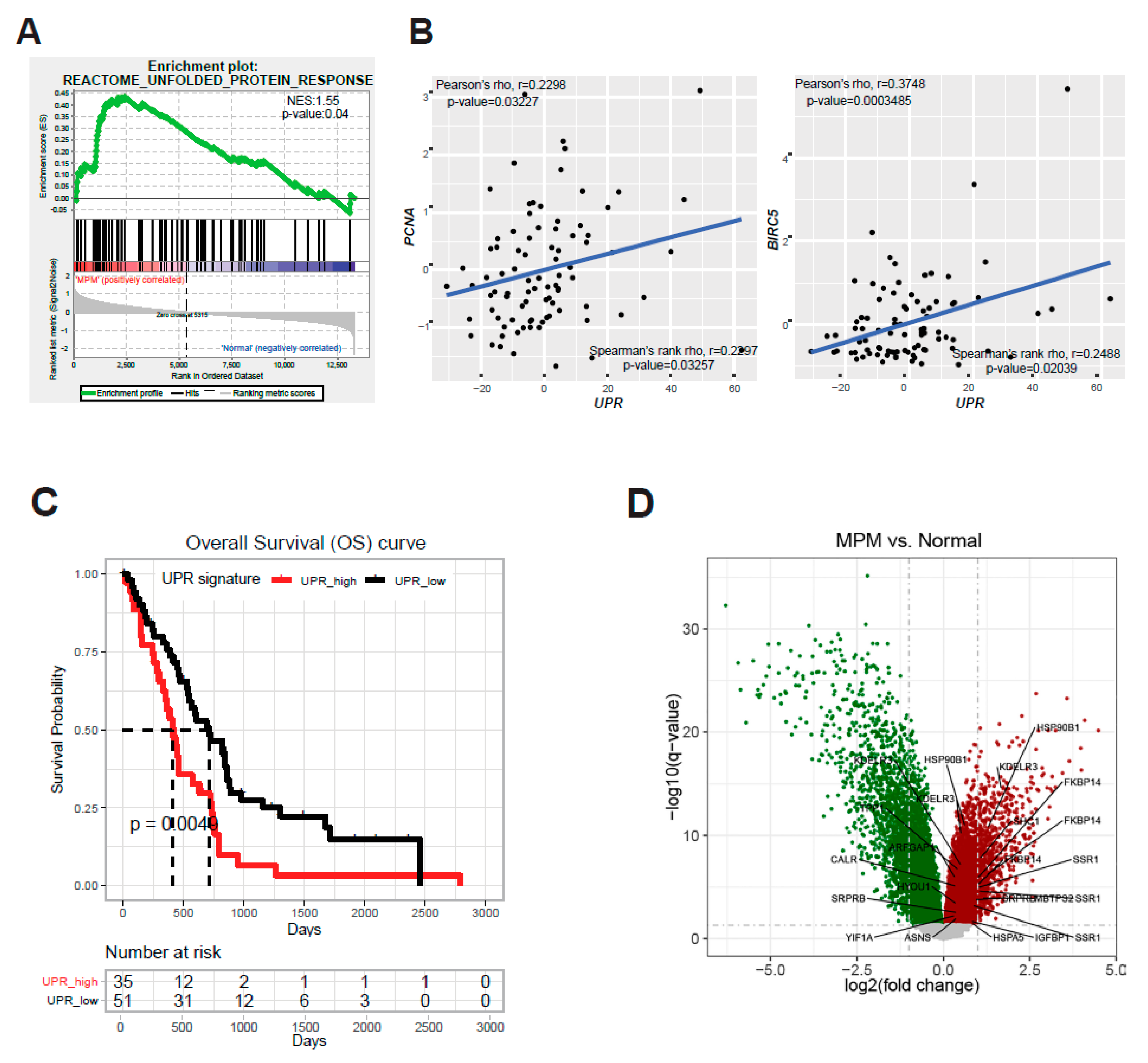
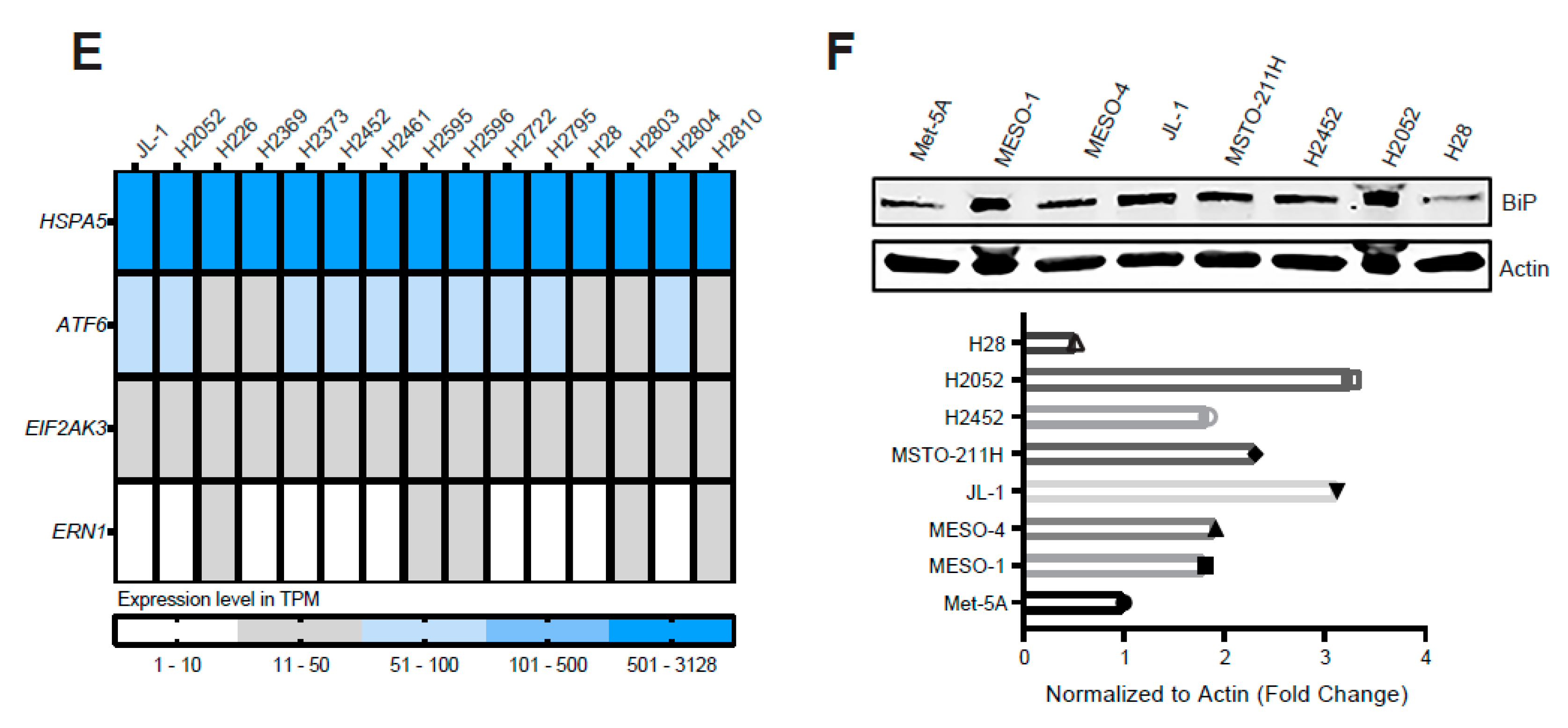
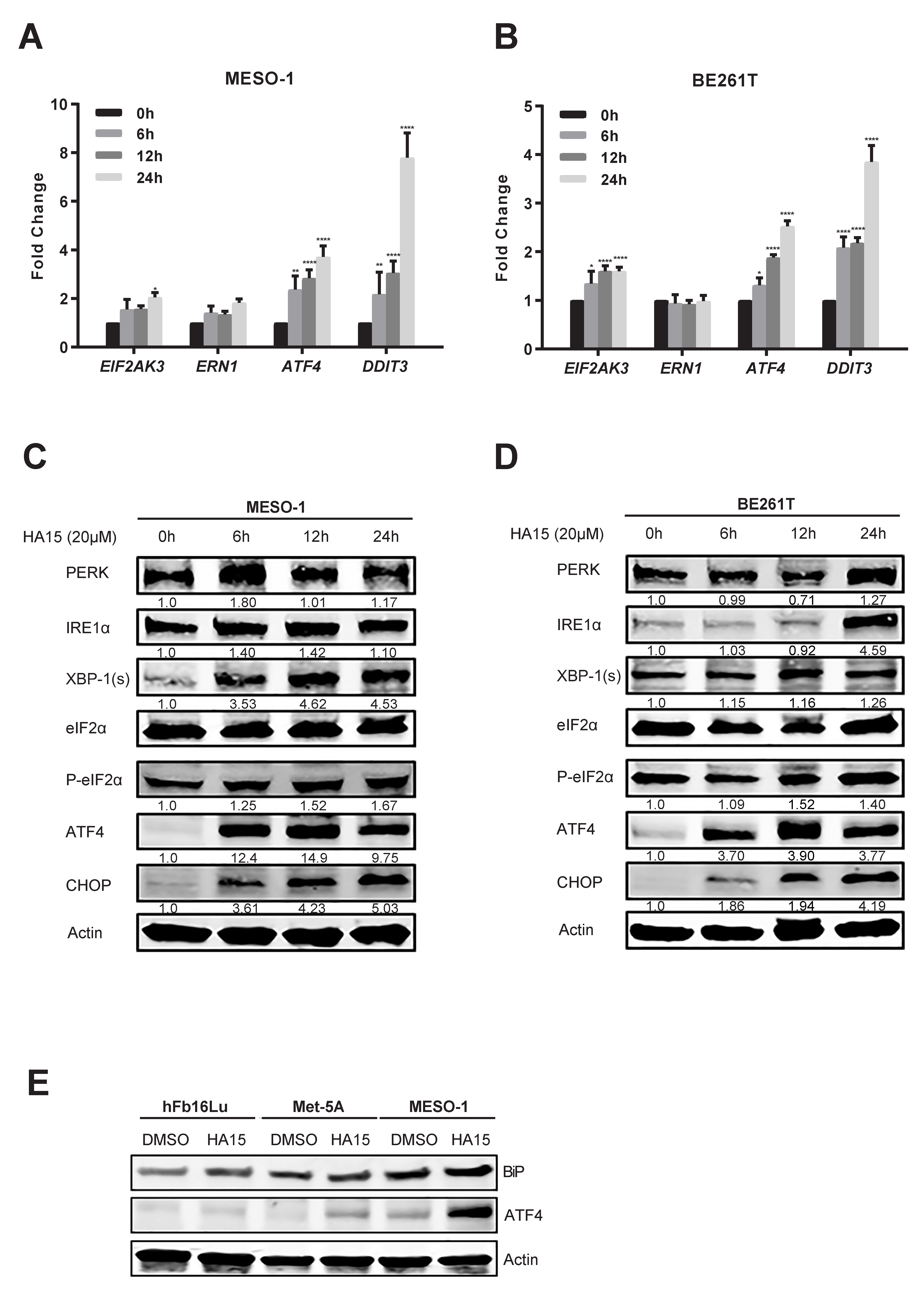
2.1. ER Stress and the Adaptive UPR Is Deregulated in Patients’ MPM
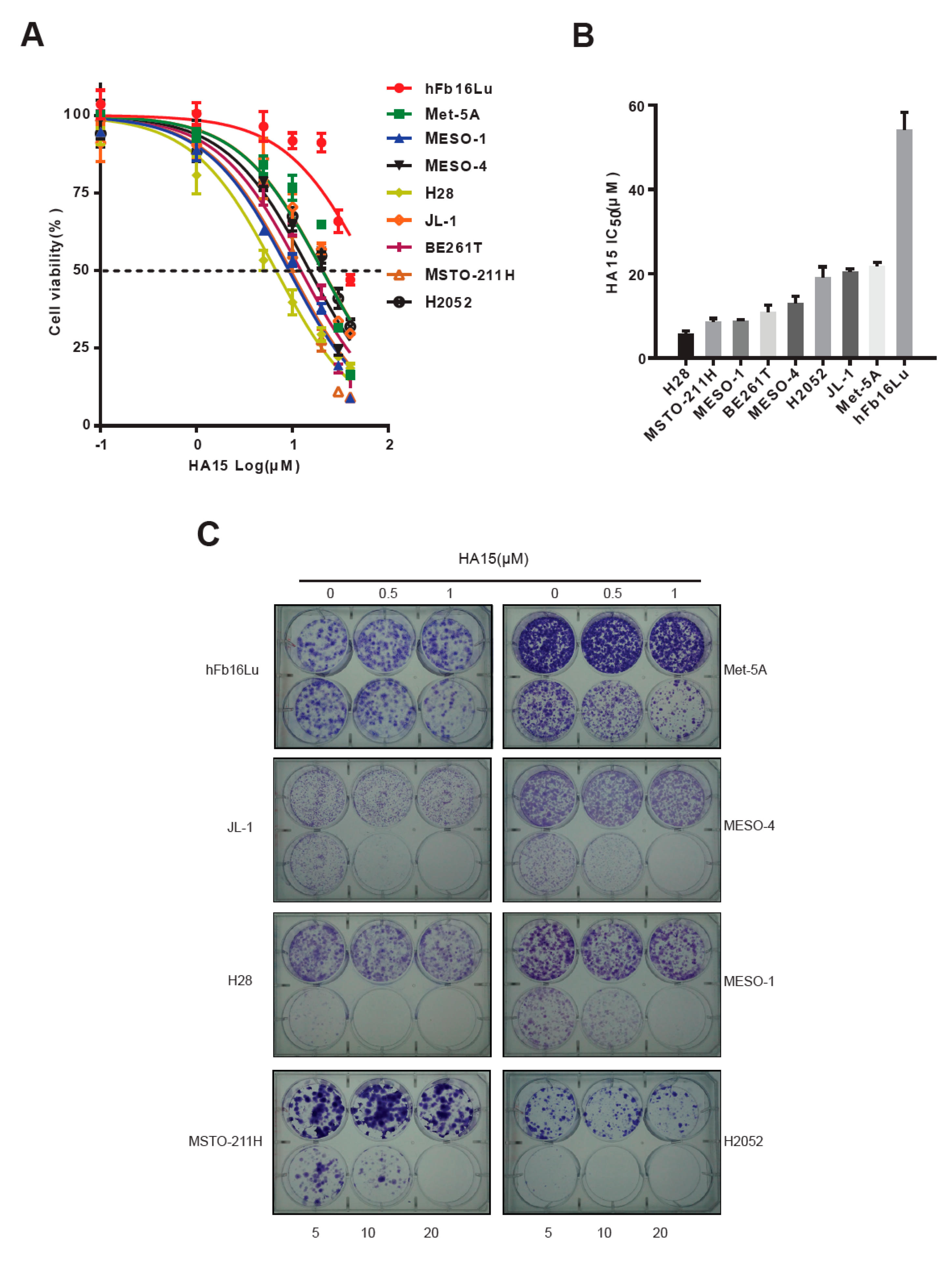
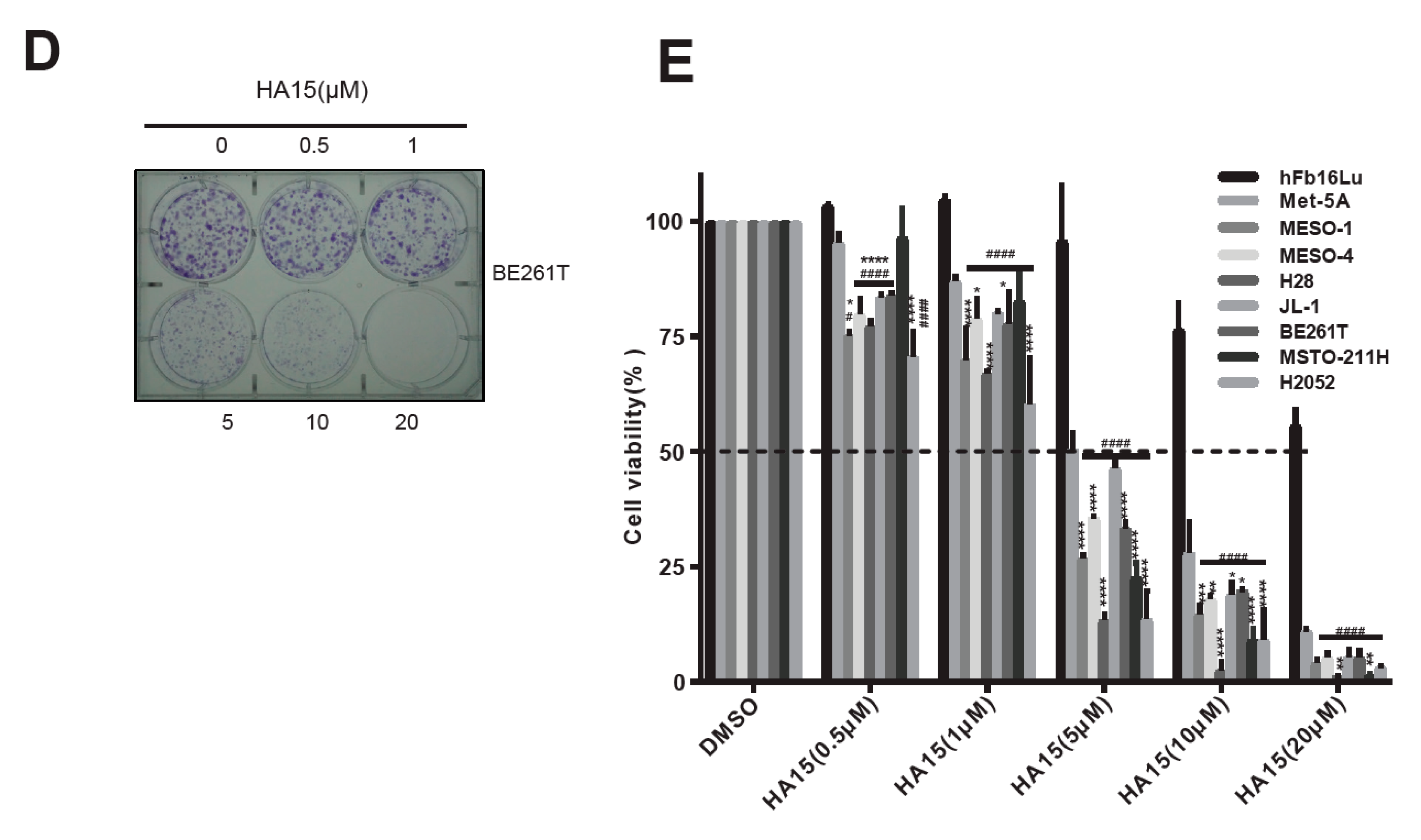
2.2. Excessive ER Stress Induced by HA15 Selectively Impairs MPM Cells
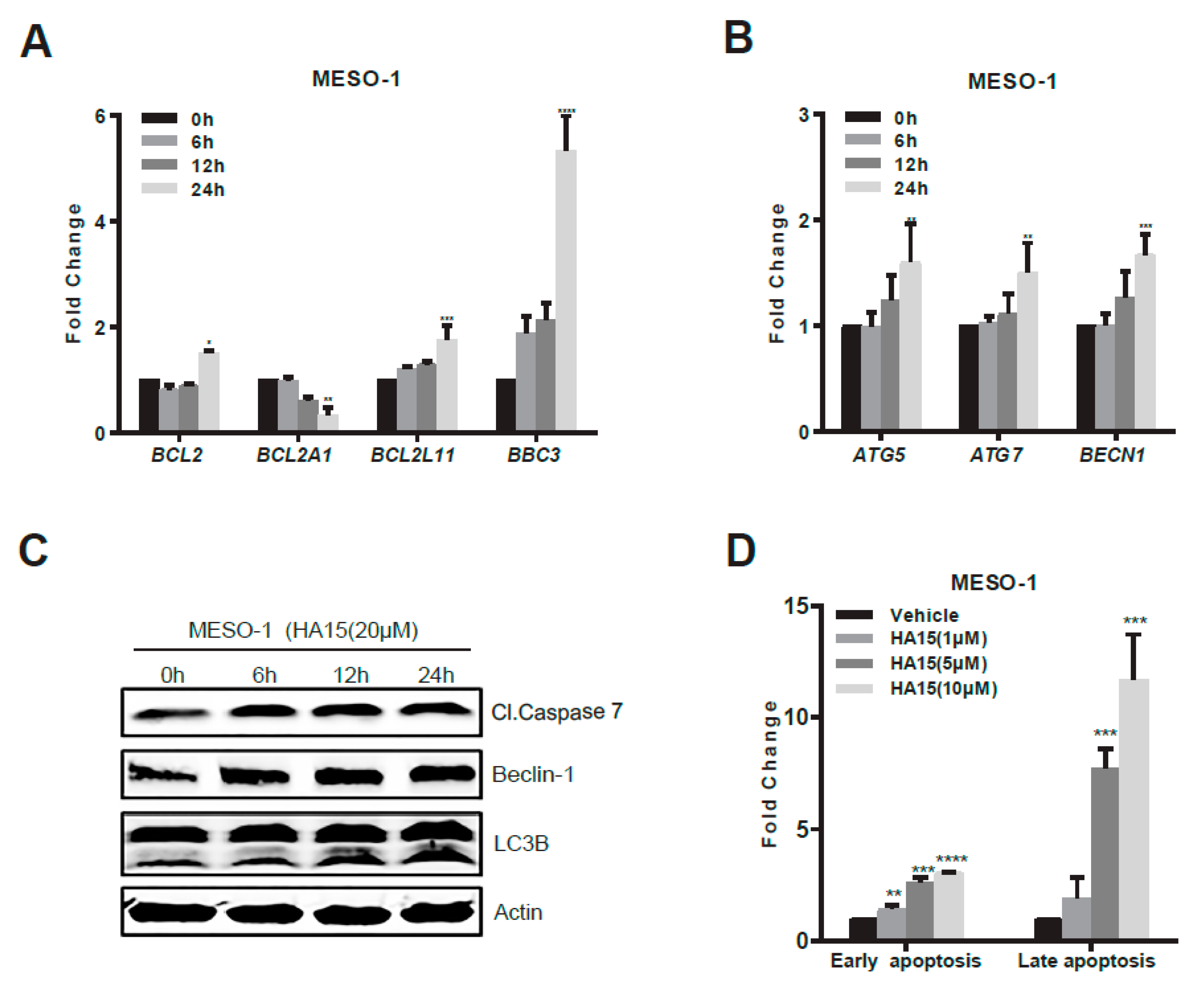
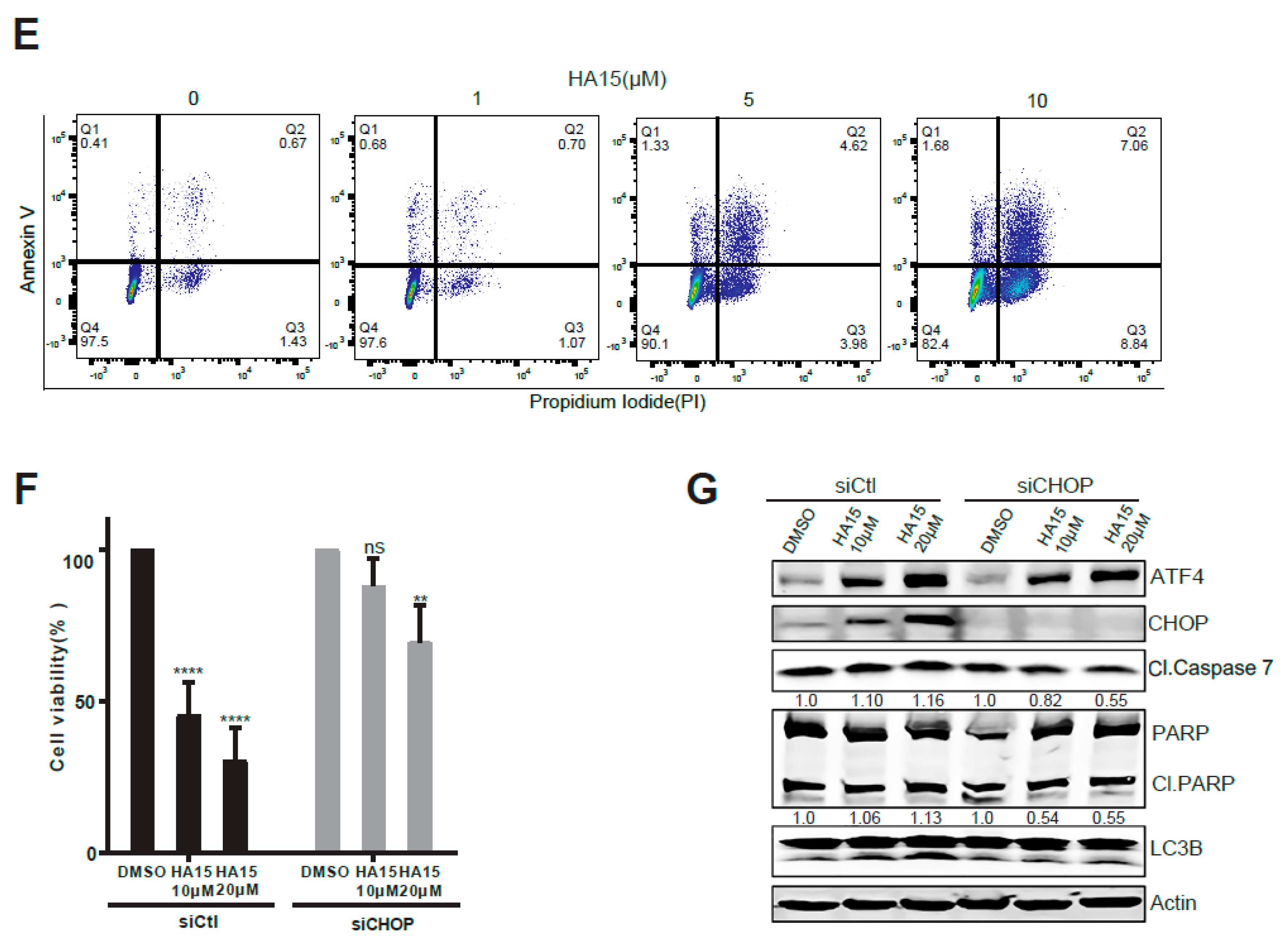
2.3. Activation of Malfunctional UPR and Autophagy Underpins HA15 Effectiveness in MPM Cells
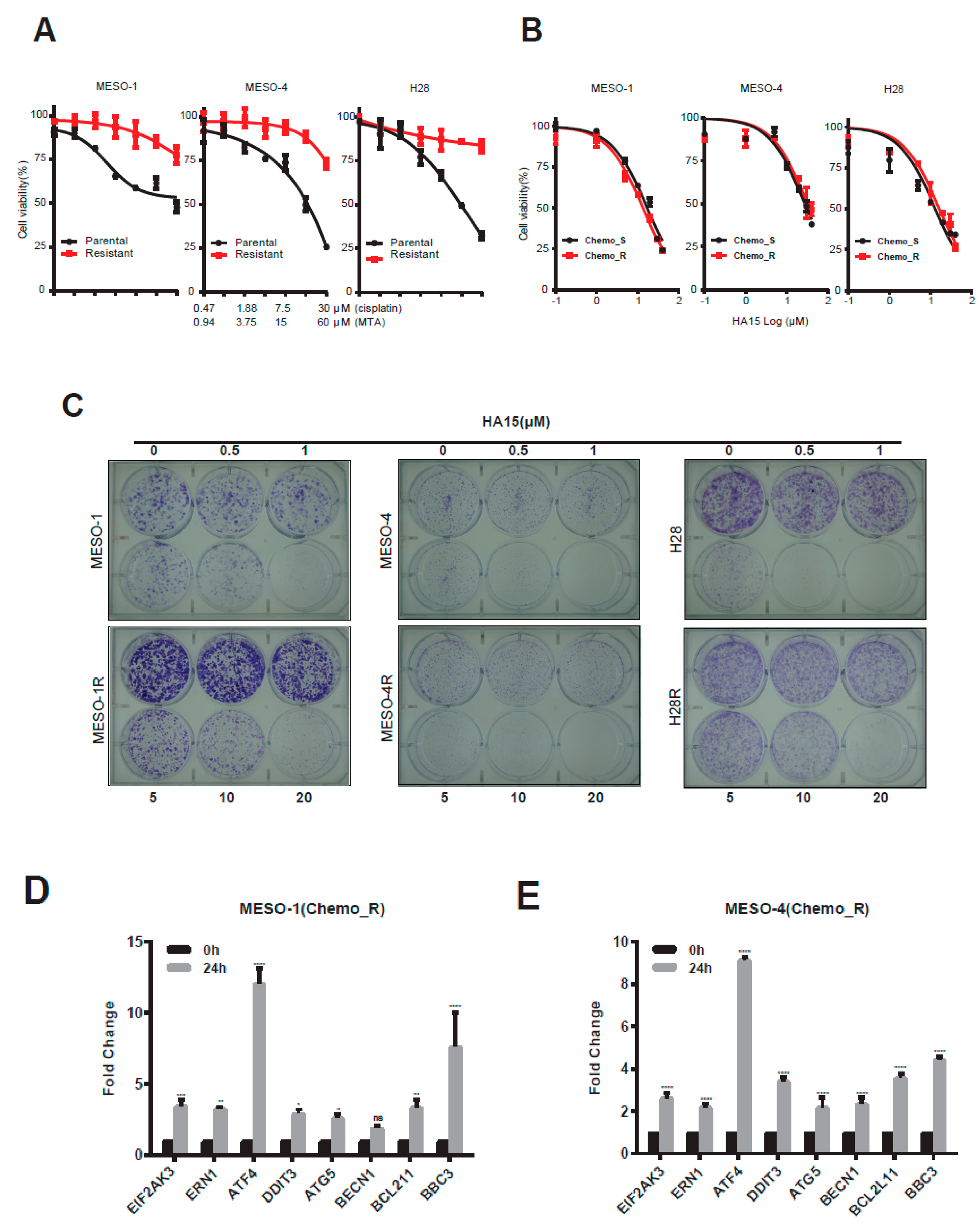
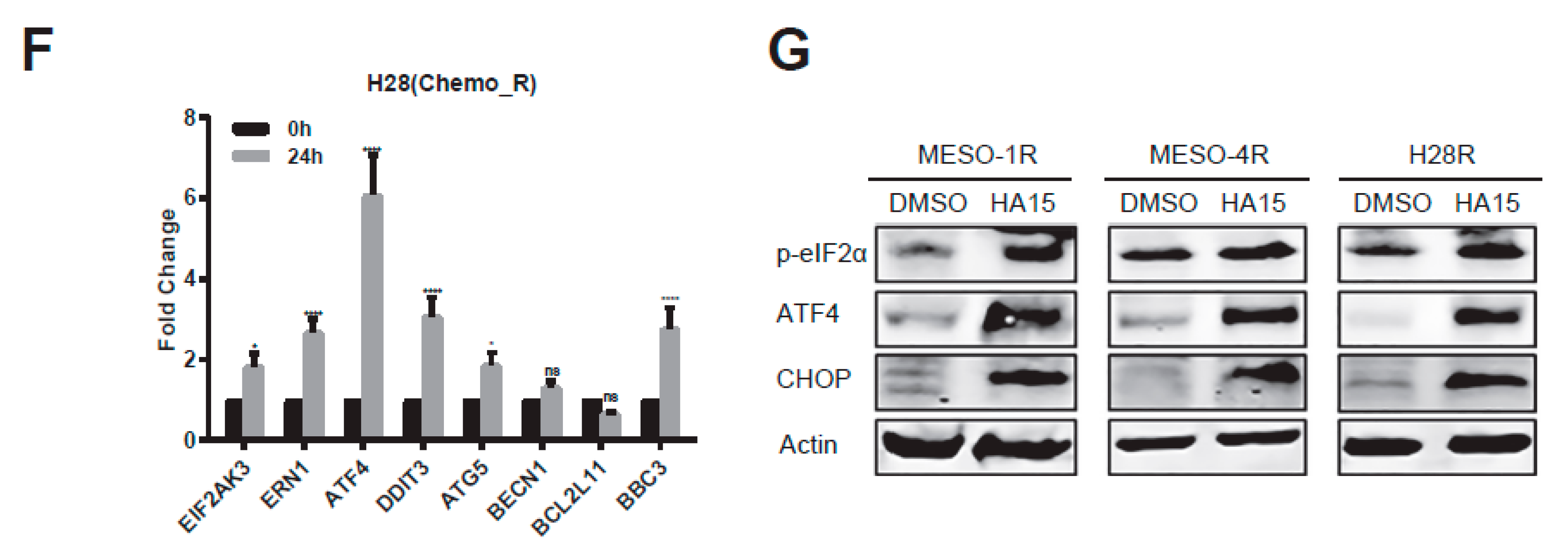
2.4. HA15 Potently Inhibits Chemo-Resistant MPM Cells
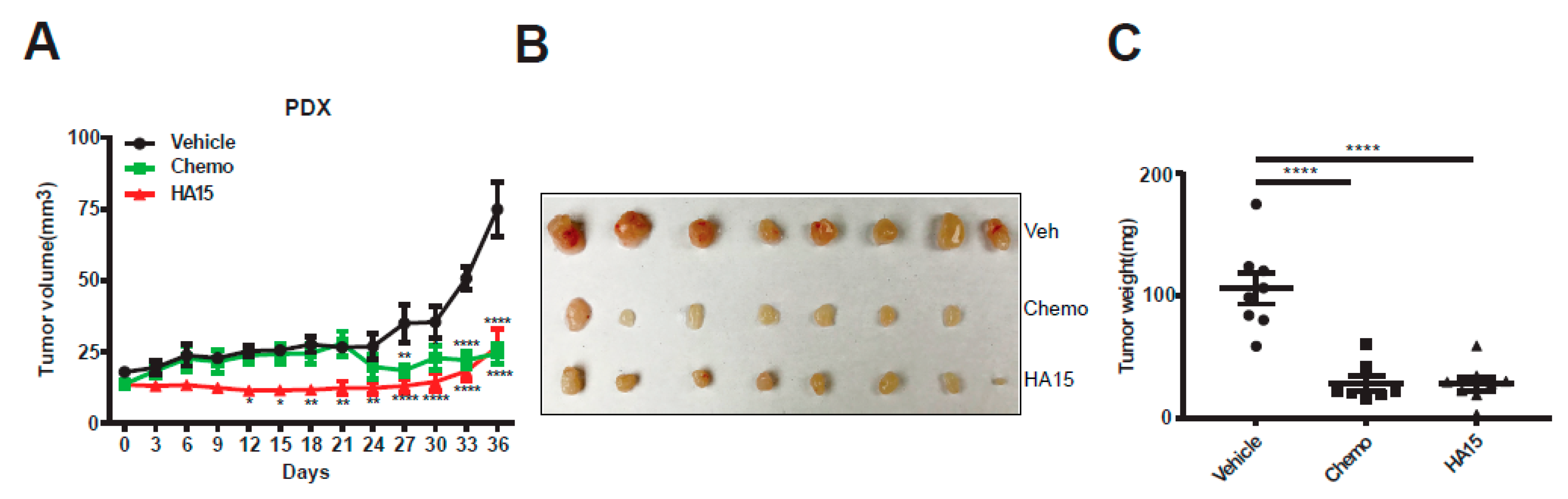
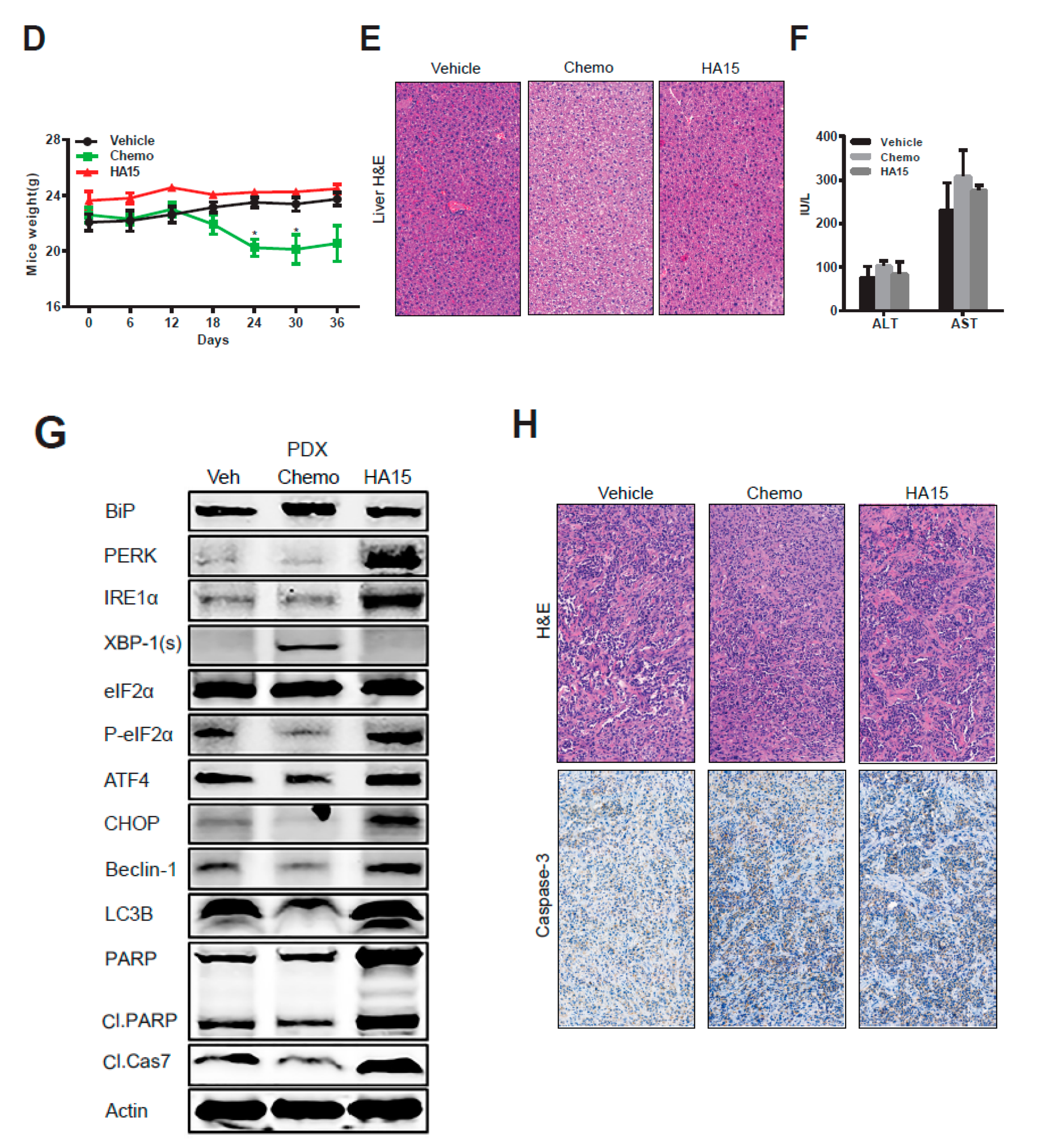
2.5. HA15 Suppresses MPM Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Viability and Clonogenic Survival Assay
4.3. Quantitative Real-Time PCR (qRT-PCR)
4.4. Western Blot and Immunohistochemistry
4.5. Apoptosis Assays
4.6. siRNA Knockdown
4.7. In Vivo Mouse Study
4.8. Public Databases (GEO, GSEA, TCGA, EMBL-EBI) and UPR Gene Signature
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McCambridge, A.J.; Napolitano, A.; Mansfield, A.S.; Fennell, D.A.; Sekido, Y.; Nowak, A.K.; Reungwetwattana, T.; Mao, W.; Pass, H.I.; Carbone, M.; et al. Progress in the Management of Malignant Pleural Mesothelioma in 2017. J. Thorac. Oncol. 2018, 13, 606–623. [Google Scholar] [CrossRef] [PubMed]
- Berzenji, L.; Van Schil, P. Multimodality treatment of malignant pleural mesothelioma. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. (Eds.) WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2015; Volume 7, p. 412. [Google Scholar]
- Zhang, W.; Wu, X.; Wu, L.; Zhang, W.; Zhao, X. Advances in the diagnosis, treatment and prognosis of malignant pleural mesothelioma. Ann. Transl. Med. 2015, 3, 182. [Google Scholar] [CrossRef] [PubMed]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.G.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef]
- Linton, A.; Cheng, Y.Y.; Griggs, K.; Kirschner, M.B.; Gattani, S.; Srikaran, S.; Chuan-Hao Kao, S.; McCaughan, B.C.; Klebe, S.; van Zandwijk, N.; et al. An RNAi-based screen reveals PLK1, CDK1 and NDC80 as potential therapeutic targets in malignant pleural mesothelioma. Br. J. Cancer 2014, 110, 510–519. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015, 75, 264–269. [Google Scholar] [CrossRef]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef]
- Wu, L.; Dell’Anno, I.; Lapidot, M.; Sekido, Y.; Chan, M.L.; Kohno, M.; Serre-Beinier, V.; Felley-Bosco, E.; de Perrot, M. Progress of malignant mesothelioma research in basic science: A review of the 14th international conference of the international mesothelioma interest group (iMig2018). Lung Cancer 2019, 127, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.E.A. Adaptive Stress Responses During Tumor Metastasis and Dormancy. Trends Cancer 2016, 2, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Villanueva, J.F.; Diaz-Molina, R.; Garcia-Gonzalez, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Sweep, F.C.; Span, P.N. The unfolded protein response as a target for cancer therapy. Biochim. Biophys. Acta 2014, 1846, 277–284. [Google Scholar] [CrossRef]
- Stengel, S.; Messner, B.; Falk-Paulsen, M.; Sommer, N.; Rosenstiel, P. Regulated proteolysis as an element of ER stress and autophagy: Implications for intestinal inflammation. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2183–2190. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Amodio, G.; Moltedo, O.; Faraonio, R.; Remondelli, P. Targeting the Endoplasmic Reticulum Unfolded Protein Response to Counteract the Oxidative Stress-Induced Endothelial Dysfunction. Oxid. Med. Cell Longev. 2018, 2018, 4946289. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Brown, L.; Ayyappan, P. Endoplasmic reticulum stress: A master regulator of metabolic syndrome. Eur. J. Pharmacol. 2019, 860, 172553. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; Gorman, A.M. Current Concepts in ER Stress-Induced Apoptosis. J. Carcinog. Mutagen. 2013, 6. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Xu, D.; Liang, S.Q.; Yang, H.; Luthi, U.; Riether, C.; Berezowska, S.; Marti, T.M.; Hall, S.R.R.; Bruggmann, R.; Kocher, G.J.; et al. Increased sensitivity to apoptosis upon endoplasmic reticulum stress-induced activation of the unfolded protein response in chemotherapy-resistant malignant pleural mesothelioma. Br. J. Cancer 2018, 119, 65–75. [Google Scholar] [CrossRef]
- Garg, A.D.; Maes, H.; van Vliet, A.R.; Agostinis, P. Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Mol. Cell Oncol. 2015, 2, e975089. [Google Scholar] [CrossRef]
- Xu, W.; Neckers, L. Gr(i)p the ER to Stress Out Melanoma. Cancer Cell 2016, 29, 769–771. [Google Scholar] [CrossRef][Green Version]
- Cerezo, M.; Lehraiki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H.; et al. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer Cell 2016, 29, 805–819. [Google Scholar] [CrossRef]
- Ruggiero, C.; Doghman-Bouguerra, M.; Ronco, C.; Benhida, R.; Rocchi, S.; Lalli, E. The GRP78/BiP inhibitor HA15 synergizes with mitotane action against adrenocortical carcinoma cells through convergent activation of ER stress pathways. Mol. Cell Endocrinol. 2018, 474, 57–64. [Google Scholar] [CrossRef]
- Gordon, G.J.; Rockwell, G.N.; Jensen, R.V.; Rheinwald, J.G.; Glickman, J.N.; Aronson, J.P.; Pottorf, B.J.; Nitz, M.D.; Richards, W.G.; Sugarbaker, D.J.; et al. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am. J. Pathol. 2005, 166, 1827–1840. [Google Scholar] [CrossRef]
- Suraokar, M.B.; Nunez, M.I.; Diao, L.; Chow, C.W.; Kim, D.; Behrens, C.; Lin, H.; Lee, S.; Raso, G.; Moran, C.; et al. Expression profiling stratifies mesothelioma tumors and signifies deregulation of spindle checkpoint pathway and microtubule network with therapeutic implications. Ann. Oncol. 2014, 25, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhao, B.; Gui, J.; Katlinski, K.V.; Brice, A.; Gao, Y.; Li, C.; Kushner, J.A.; Koumenis, C.; Diehl, J.A.; et al. Type I interferons mediate pancreatic toxicities of PERK inhibition. Proc. Natl. Acad. Sci. USA 2015, 112, 15420–15425. [Google Scholar] [CrossRef] [PubMed]
- Dufey, E.; Sepulveda, D.; Rojas-Rivera, D.; Hetz, C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Cell Physiol. 2014, 307, C582–C594. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [PubMed]
- Thevenot, P.T.; Sierra, R.A.; Raber, P.L.; Al-Khami, A.A.; Trillo-Tinoco, J.; Zarreii, P.; Ochoa, A.C.; Cui, Y.; Del Valle, L.; Rodriguez, P.C. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity 2014, 41, 389–401. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Usami, N.; Fukui, T.; Kondo, M.; Taniguchi, T.; Yokoyama, T.; Mori, S.; Yokoi, K.; Horio, Y.; Shimokata, K.; Sekido, Y.; et al. Establishment and characterization of four malignant pleural mesothelioma cell lines from Japanese patients. Cancer Sci. 2006, 97, 387–394. [Google Scholar] [CrossRef]
- Cortes-Dericks, L.; Yazd, E.F.; Mowla, S.J.; Schmid, R.A.; Karoubi, G. Suppression of OCT4B enhances sensitivity of lung adenocarcinoma A549 cells to cisplatin via increased apoptosis. Anticancer Res. 2013, 33, 5365–5373. [Google Scholar]
- Cortes-Dericks, L.; Froment, L.; Kocher, G.; Schmid, R.A. Human lung-derived mesenchymal stem cell-conditioned medium exerts in vitro antitumor effects in malignant pleural mesothelioma cell lines. Stem Cell Res. Ther. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.Q.; Marti, T.M.; Dorn, P.; Froment, L.; Hall, S.R.; Berezowska, S.; Kocher, G.; Schmid, R.A.; Peng, R.W. Blocking the epithelial-to-mesenchymal transition pathway abrogates resistance to anti-folate chemotherapy in lung cancer. Cell Death Dis. 2015, 6, e1824. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.Q.; Buhrer, E.D.; Berezowska, S.; Marti, T.M.; Xu, D.; Froment, L.; Yang, H.; Hall, S.R.R.; Vassella, E.; Yang, Z.; et al. mTOR mediates a mechanism of resistance to chemotherapy and defines a rational combination strategy to treat KRAS-mutant lung cancer. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, D.; Yang, H.; Yang, Z.; Berezowska, S.; Gao, Y.; Liang, S.-Q.; Marti, T.M.; Hall, S.R.R.; Dorn, P.; Kocher, G.J.; et al. Endoplasmic Reticulum Stress Signaling as a Therapeutic Target in Malignant Pleural Mesothelioma. Cancers 2019, 11, 1502. https://doi.org/10.3390/cancers11101502
Xu D, Yang H, Yang Z, Berezowska S, Gao Y, Liang S-Q, Marti TM, Hall SRR, Dorn P, Kocher GJ, et al. Endoplasmic Reticulum Stress Signaling as a Therapeutic Target in Malignant Pleural Mesothelioma. Cancers. 2019; 11(10):1502. https://doi.org/10.3390/cancers11101502
Chicago/Turabian StyleXu, Duo, Haitang Yang, Zhang Yang, Sabina Berezowska, Yanyun Gao, Shun-Qing Liang, Thomas M. Marti, Sean R. R. Hall, Patrick Dorn, Gregor J. Kocher, and et al. 2019. "Endoplasmic Reticulum Stress Signaling as a Therapeutic Target in Malignant Pleural Mesothelioma" Cancers 11, no. 10: 1502. https://doi.org/10.3390/cancers11101502
APA StyleXu, D., Yang, H., Yang, Z., Berezowska, S., Gao, Y., Liang, S.-Q., Marti, T. M., Hall, S. R. R., Dorn, P., Kocher, G. J., Schmid, R. A., & Peng, R.-W. (2019). Endoplasmic Reticulum Stress Signaling as a Therapeutic Target in Malignant Pleural Mesothelioma. Cancers, 11(10), 1502. https://doi.org/10.3390/cancers11101502