Potent Activity of Composite Cyclin Dependent Kinase Inhibition against Hepatocellular Carcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
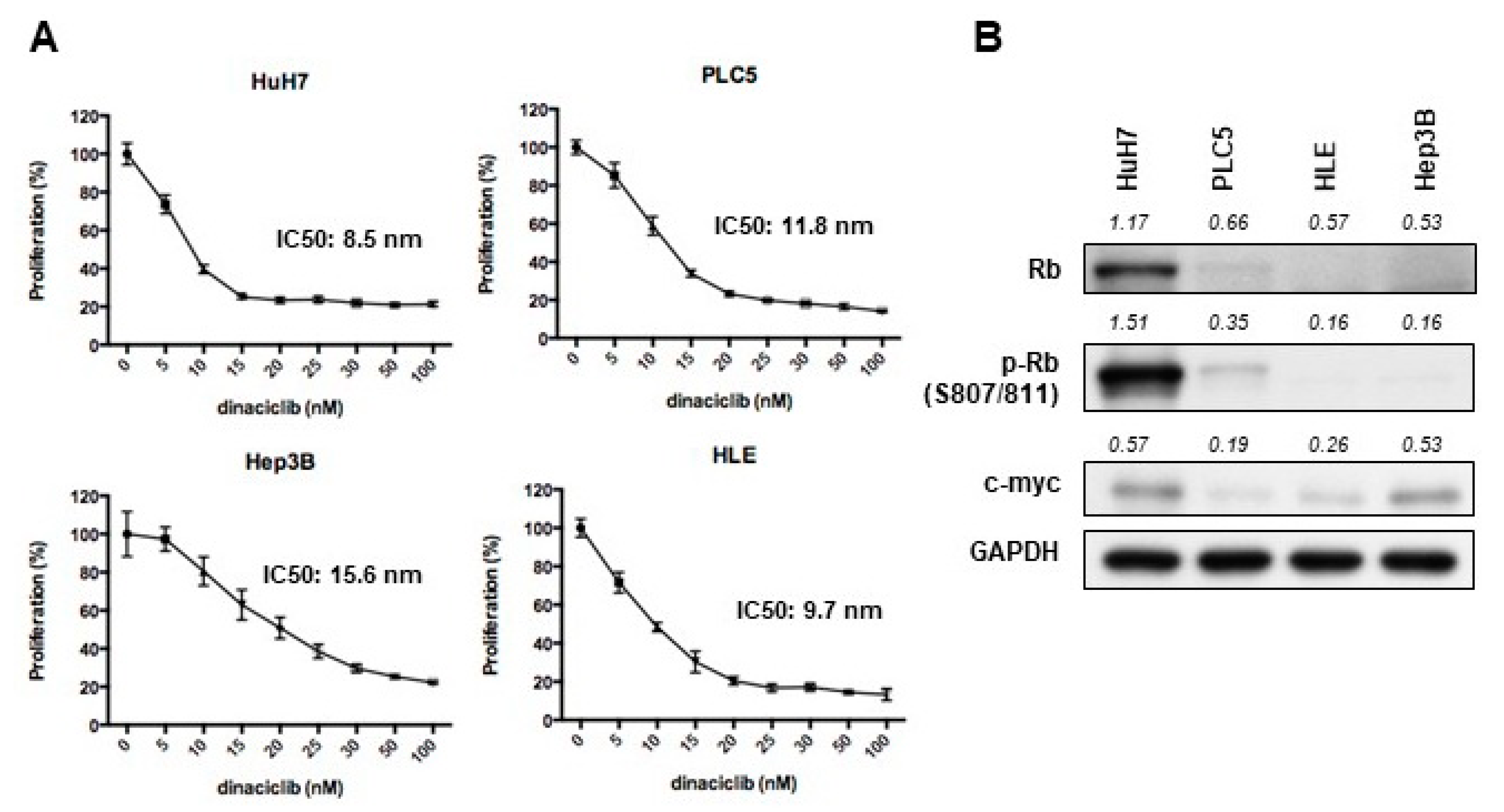
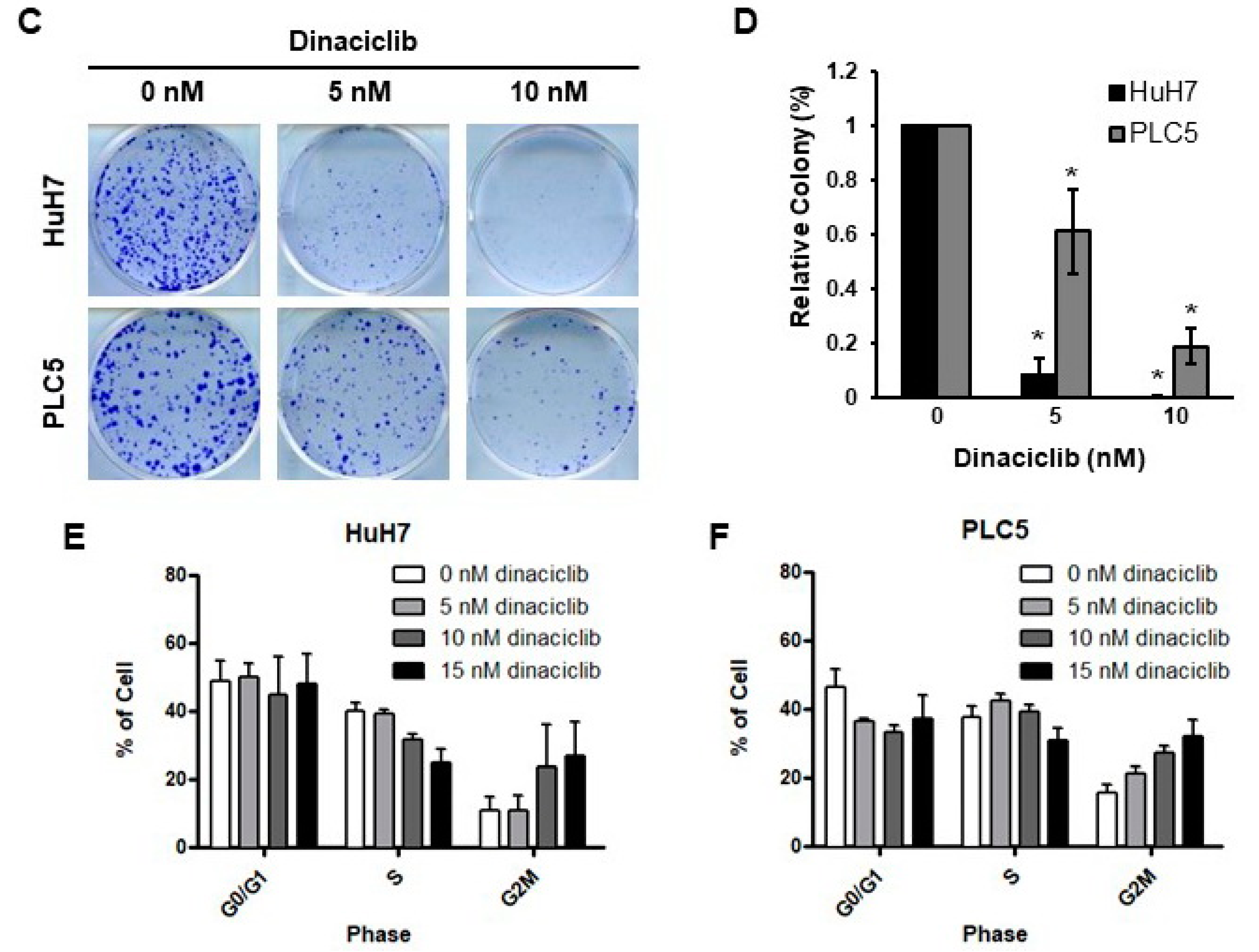
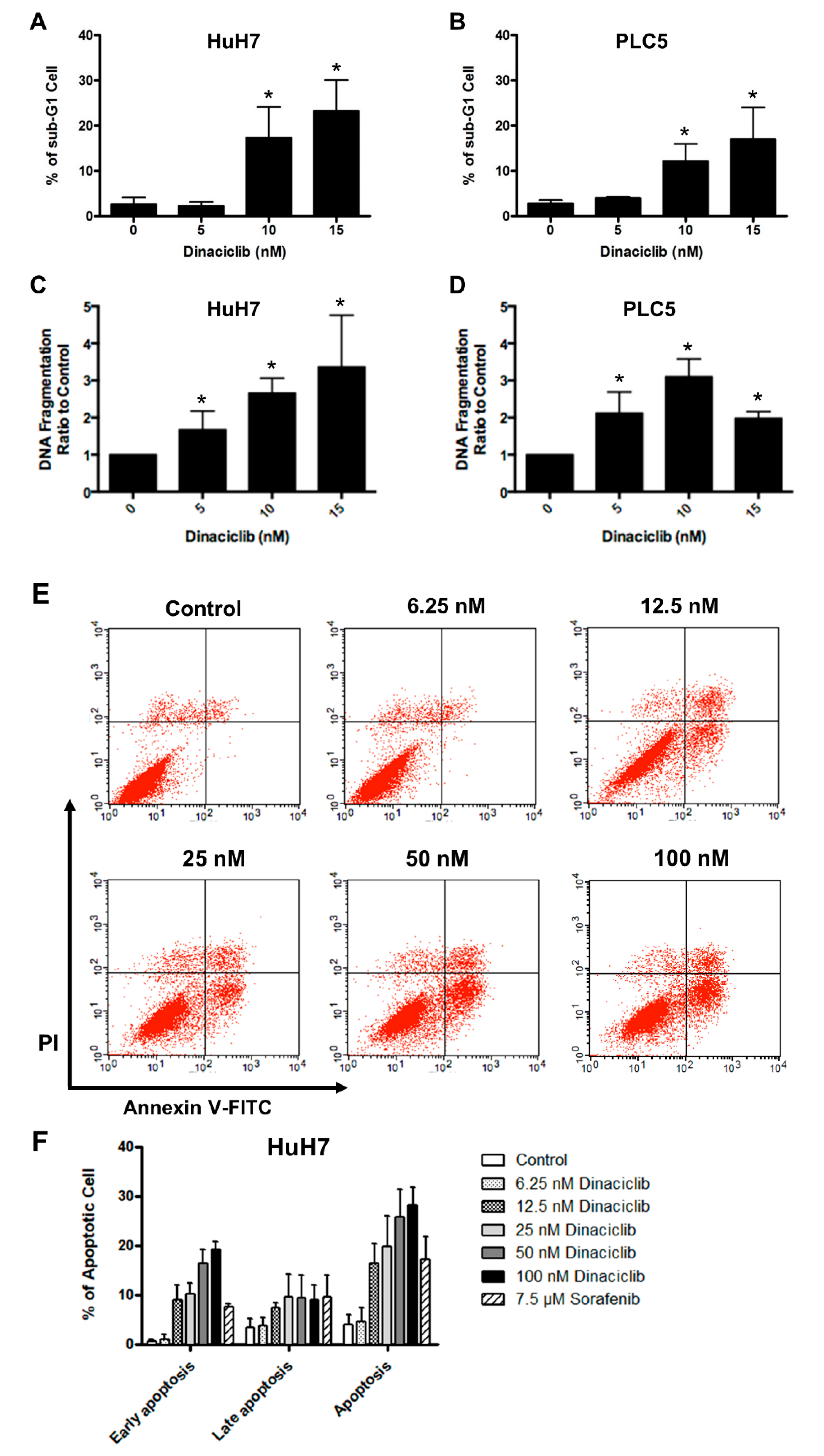
2.1. Dinaciclib Showed Potent In Vitro Activity in HCC Cells
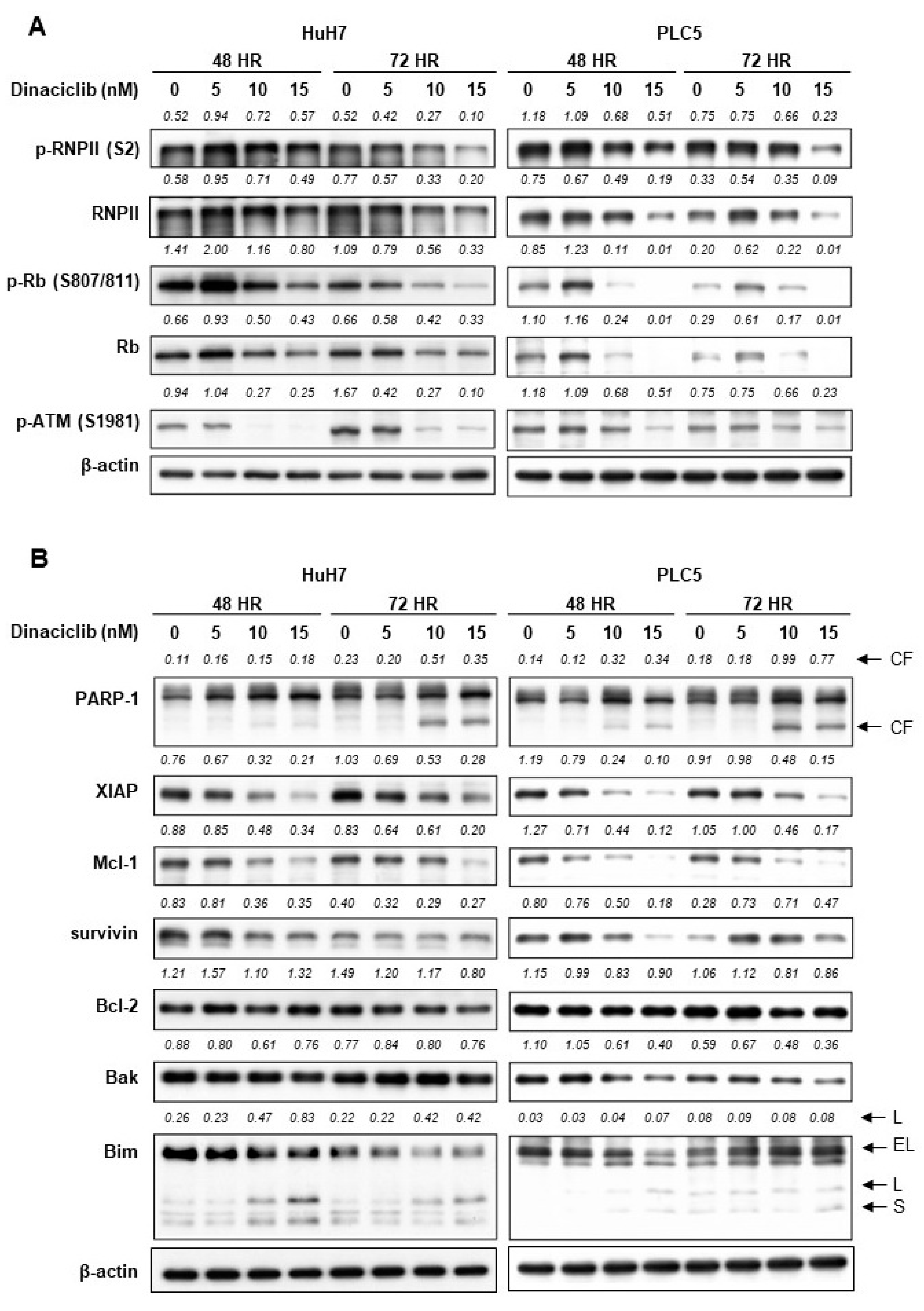
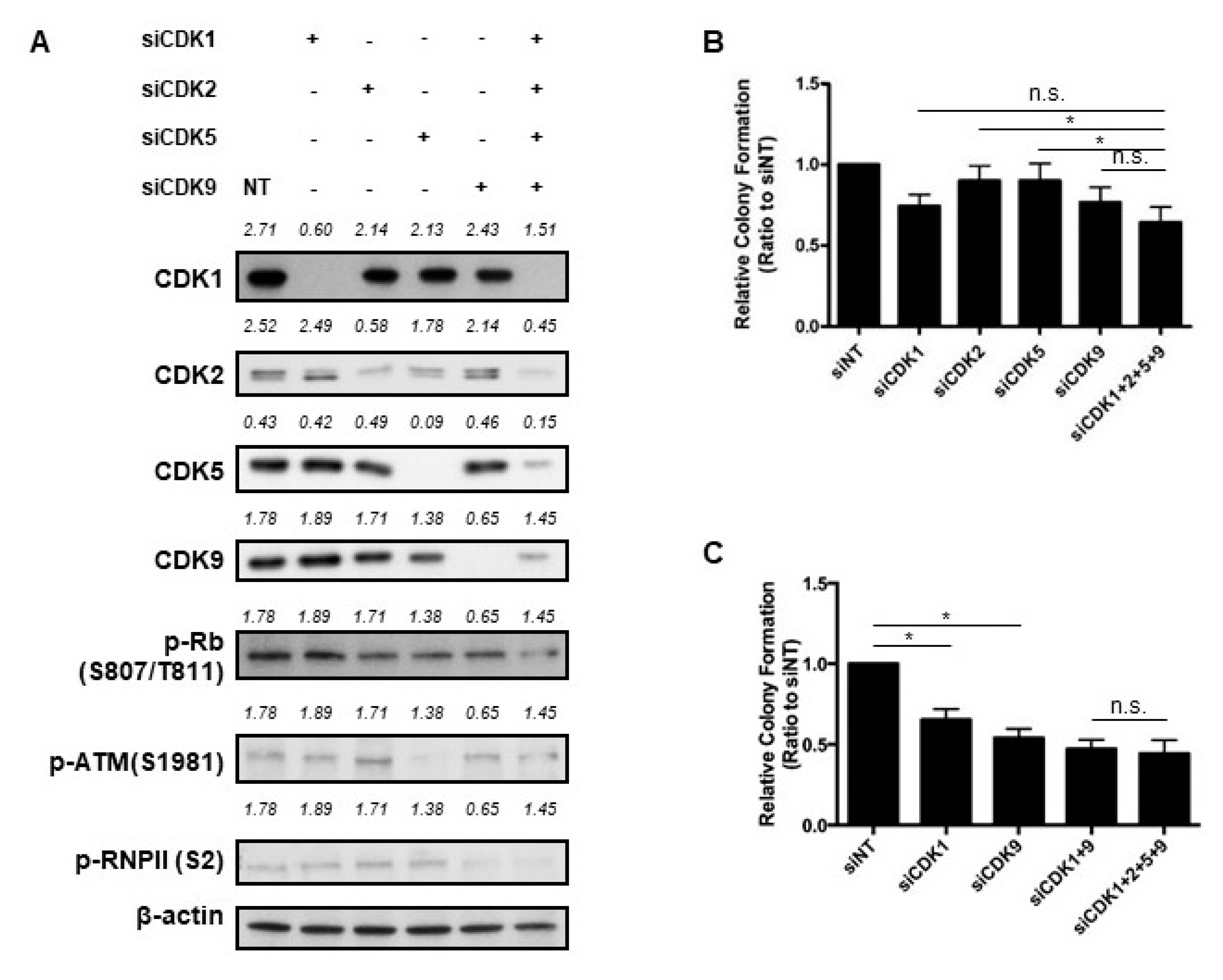
2.2. Dinaciclib Inhibited Function of CDKs 1, 2, 5, and 9 and Reduced Expression of Antiapoptotic Proteins
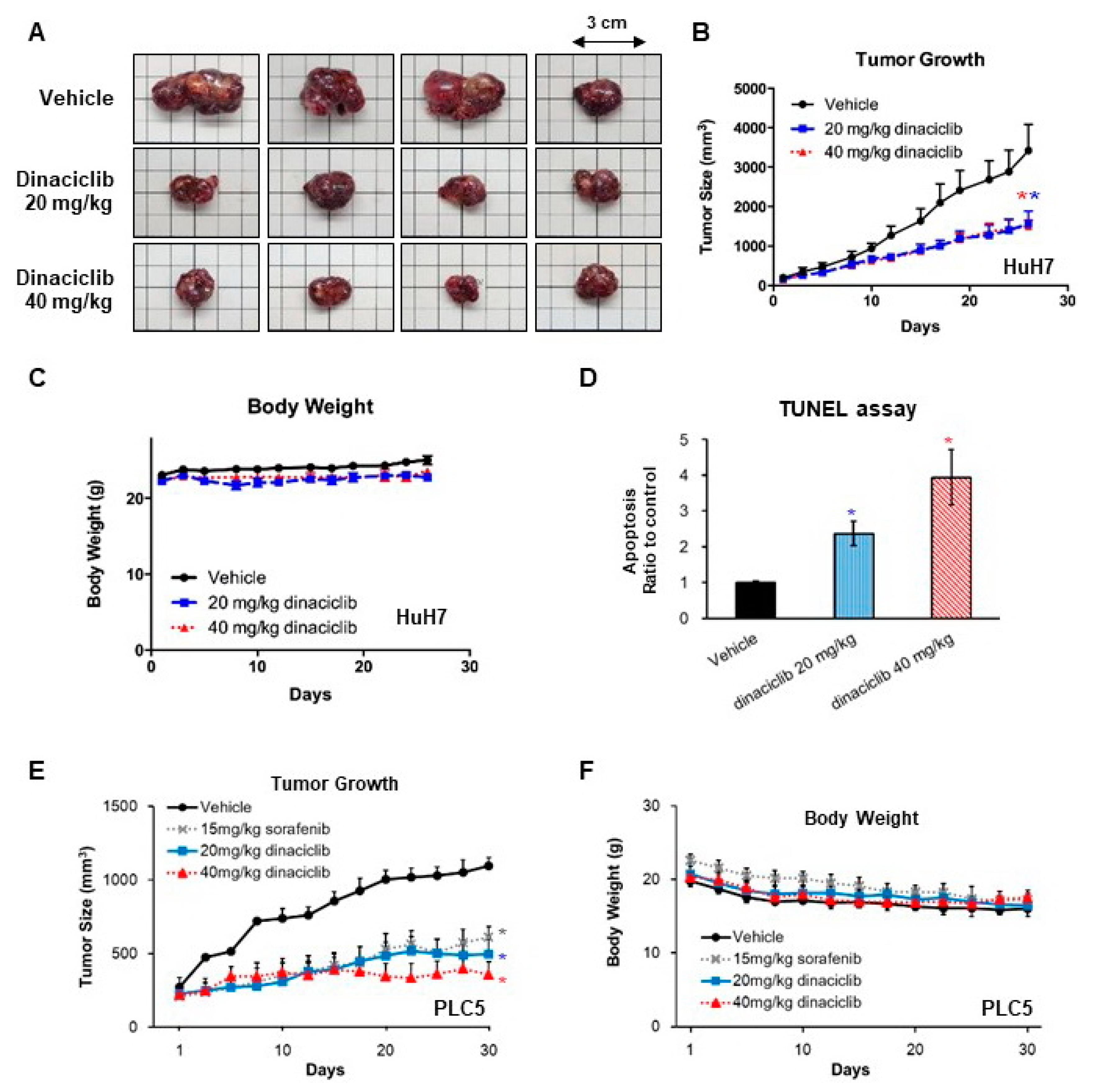
2.3. In Vivo Efficacy of Dinaciclib against HCC
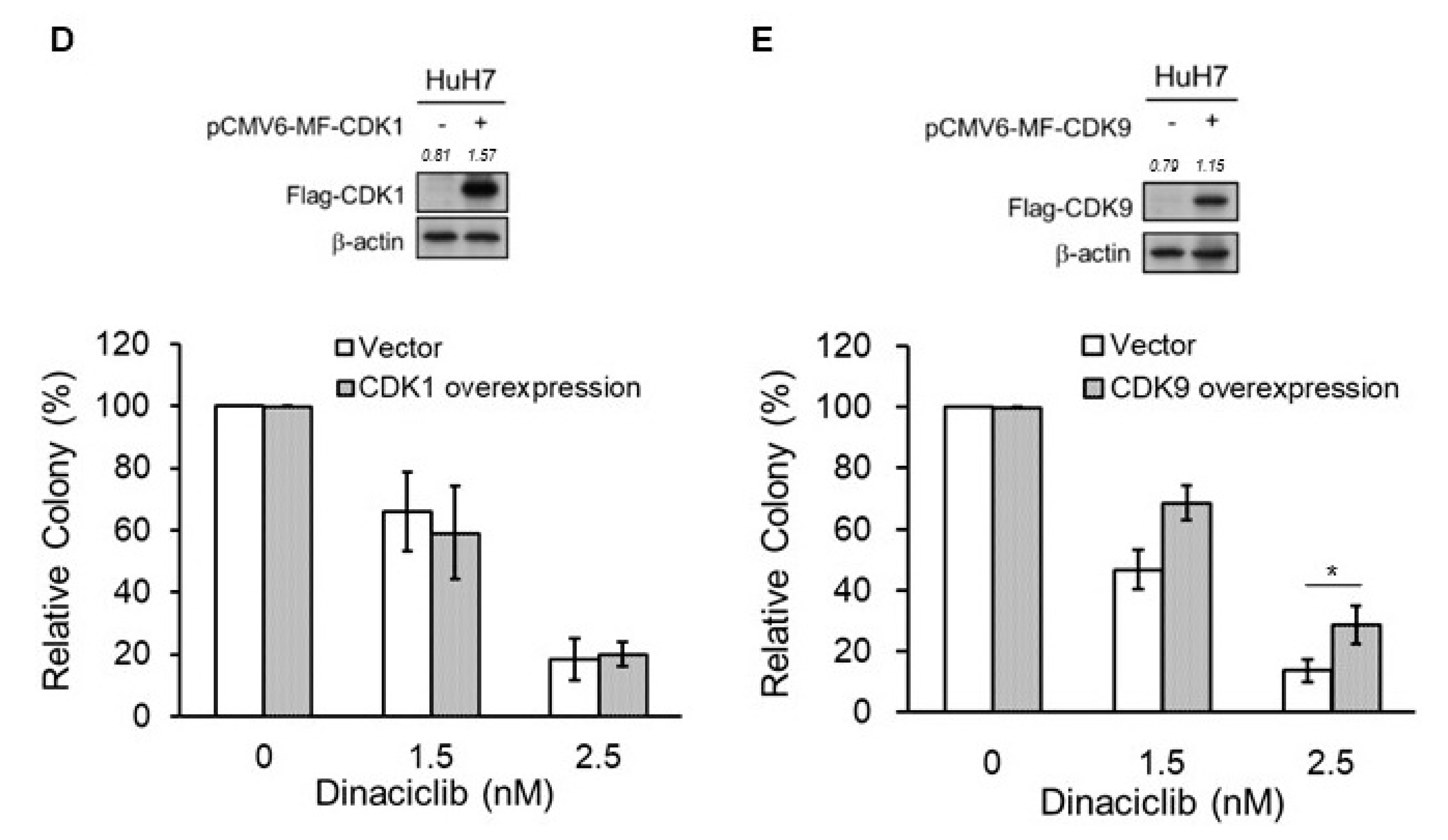
2.4. Key CDKs for the Efficacy of Dinaciclib
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Proliferation Assays
4.3. Western Blot Analysis
4.4. Cell Cycle Analysis
4.5. Apoptosis Assay
4.6. Xenograft Studies
4.7. RNA Knockdown and Overexpression
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Hampton, L.L.; Wirth, P.J.; Wang, N.J.; Xie, J.P.; Thorgeirsson, S.S. Alterations of tumor suppressor genes and allelic losses in human hepatocellular carcinomas in China. Cancer Res. 1994, 54, 281–285. [Google Scholar]
- Lin, Y.; Shi, C.Y.; Li, B.; Soo, B.H.; Mohammed-Ali, S.; Wee, A.; Oon, C.J.; Mack, P.O.; Chan, S.H. Tumour suppressor p53 and Rb genes in human hepatocellular carcinoma. Ann. Acad. Med. Singap. 1996, 25, 22–30. [Google Scholar]
- Hada, H.; Koide, N.; Morita, T.; Shiraha, H.; Shinji, T.; Nakamura, M.; Ujike, K.; Takayama, N.; Oka, T.; Hanafusa, T.; et al. Promoter-independent loss of mRNA and protein of the Rb gene in a human hepatocellular carcinoma. Hepatogastroenterology 1996, 43, 1185–1189. [Google Scholar] [PubMed]
- Liew, C.T.; Li, H.M.; Lo, K.W.; Leow, C.K.; Chan, J.Y.; Hin, L.Y.; Lau, W.Y.; Lai, P.B.; Lim, B.K.; Huang, J.; et al. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene 1999, 18, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Piao, Z.; Kim, N.G.; Park, C.; Shin, E.C.; Park, J.H.; Jung, H.J.; Kim, C.G.; Kim, H. p16 is a major inactivation target in hepatocellular carcinoma. Cancer 2000, 89, 60–68. [Google Scholar] [PubMed]
- Bollard, J.; Miguela, V.; Ruiz de Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sanchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, F.S.; Chen, Y.L.; Hung, M.H.; Chu, P.Y.; Tsai, M.H.; Chen, L.J.; Hsiao, Y.J.; Shih, C.T.; Chang, M.J.; Chao, T.I.; et al. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK4/6-independent manner. Mol. Oncol. 2017, 11, 1035–1049. [Google Scholar] [CrossRef]
- Rivadeneira, D.B.; Mayhew, C.N.; Thangavel, C.; Sotillo, E.; Reed, C.A.; Grana, X.; Knudsen, E.S. Proliferative suppression by CDK4/6 inhibition: Complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology 2010, 138, 1920–1930. [Google Scholar] [CrossRef]
- Matsuda, Y.; Wakai, T.; Kubota, M.; Takamura, M.; Yamagiwa, S.; Aoyagi, Y.; Osawa, M.; Fujimaki, S.; Sanpei, A.; Genda, T.; et al. Clinical significance of cell cycle inhibitors in hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 185–192. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug. Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Brinkkoetter, P.T.; Pippin, J.W.; Shankland, S.J. Cyclin I-Cdk5 governs survival in post-mitotic cells. Cell Cycle 2014, 9, 1729–1731. [Google Scholar] [CrossRef]
- Ehrlich, S.M.; Liebl, J.; Ardelt, M.A.; Lehr, T.; De Toni, E.N.; Mayr, D.; Brandl, L.; Kirchner, T.; Zahler, S.; Gerbes, A.L.; et al. Targeting cyclin dependent kinase 5 in hepatocellular carcinoma—A novel therapeutic approach. J. Hepatol. 2015, 63, 102–113. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Lujambio, A.; Zuber, J.; Tschaharganeh, D.F.; Doran, M.G.; Evans, M.J.; Kitzing, T.; Zhu, N.; de Stanchina, E.; Sawyers, C.L.; et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014, 28, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Cole, M.D. The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol. Cell. Biol. 2007, 27, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.M.; Villanueva, J.; Nguyen, T.T.; Lioni, M.; Xiao, M.; Kong, J.; Krepler, C.; Vultur, A.; Flaherty, K.T.; Nathanson, K.L.; et al. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLoS ONE 2013, 8, e59588. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Ma, L.; Chu, B.; Wang, X.; Bui, M.M.; Gemmer, J.; Altiok, S.; Pledger, W.J. The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells. Mol. Cancer 2011, 10, 1018–1027. [Google Scholar] [CrossRef]
- Feldmann, G.; Mishra, A.; Bisht, S.; Karikari, C.; Garrido-Laguna, I.; Rasheed, Z.; Ottenhof, N.A.; Dadon, T.; Alvarez, H.; Fendrich, V.; et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol. 2011, 12, 598–609. [Google Scholar] [CrossRef]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Zhu, Y.; Yao, S.L.; Small, K.; et al. Randomized Phase II Trial of the Cyclin-Dependent Kinase Inhibitor Dinaciclib (MK-7965) Versus Capecitabine in Patients with Advanced Breast Cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef]
- Ghia, P.; Scarfo, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.M.; Statkevich, P.; Yao, S.L.; Bannerji, R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11, 259. [Google Scholar] [CrossRef]
- Haider, C.; Grubinger, M.; Reznickova, E.; Weiss, T.S.; Rotheneder, H.; Miklos, W.; Berger, W.; Jorda, R.; Zatloukal, M.; Gucky, T.; et al. Novel inhibitors of cyclin-dependent kinases combat hepatocellular carcinoma without inducing chemoresistance. Mol. Cancer 2013, 12, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; De Savi, C.; Drew, L.; Ferguson, A.D.; Ferguson, D.; Gu, C.; Hawkins, J.; Hird, A.W.; Lamb, M.L.; O’Connell, N.; et al. Discovery of AZD4573, a potent and selective inhibitor of CDK9 that enables transient target engagement for the treatment of hematologic malignancies. Cancer Res. 2018, 78, 1650. [Google Scholar]
- Littman, S.J.; Brus, C.; Burkart, A. A phase II study of palbociclib (PD-0332991) in adult patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2015, 33 (Suppl. 3), 277. [Google Scholar] [CrossRef]
- Hsu, C.; Lin, L.I.; Cheng, Y.C.; Feng, Z.R.; Shao, Y.Y.; Cheng, A.L.; Ou, D.L. Cyclin E1 Inhibition can Overcome Sorafenib Resistance in Hepatocellular Carcinoma Cells Through Mcl-1 Suppression. Clin. Cancer Res. 2016, 22, 2555–2564. [Google Scholar] [CrossRef]
- Ardelt, M.A.; Frohlich, T.; Martini, E.; Muller, M.; Kanitz, V.; Atzberger, C.; Cantonati, P.; Messner, M.; Posselt, L.; Lehr, T.; et al. Inhibition of Cyclin-Dependent Kinase 5: A Strategy to Improve Sorafenib Response in Hepatocellular Carcinoma Therapy. Hepatology 2019, 69, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.M.; Mita, A.C.; Moseley, J.L.; Poon, J.; Small, K.A.; Jou, Y.M.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Statkevich, P.; et al. Phase 1 safety, pharmacokinetic and pharmacodynamic study of the cyclin-dependent kinase inhibitor dinaciclib administered every three weeks in patients with advanced malignancies. Br. J. Cancer 2017, 117, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Shao, Y.Y.; Chan, S.Y.; Huang, C.Y.; Hsu, C.H.; Cheng, A.L. High Serum Transforming Growth Factor-beta1 Levels Predict Outcome in Hepatocellular Carcinoma Patients Treated with Sorafenib. Clin. Cancer Res. 2015, 21, 3678–3684. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, Y.-Y.; Li, Y.-S.; Hsu, H.-W.; Lin, H.; Wang, H.-Y.; Wo, R.R.; Cheng, A.-L.; Hsu, C.-H. Potent Activity of Composite Cyclin Dependent Kinase Inhibition against Hepatocellular Carcinoma. Cancers 2019, 11, 1433. https://doi.org/10.3390/cancers11101433
Shao Y-Y, Li Y-S, Hsu H-W, Lin H, Wang H-Y, Wo RR, Cheng A-L, Hsu C-H. Potent Activity of Composite Cyclin Dependent Kinase Inhibition against Hepatocellular Carcinoma. Cancers. 2019; 11(10):1433. https://doi.org/10.3390/cancers11101433
Chicago/Turabian StyleShao, Yu-Yun, Yong-Shi Li, Hung-Wei Hsu, Hang Lin, Han-Yu Wang, Rita Robin Wo, Ann-Lii Cheng, and Chih-Hung Hsu. 2019. "Potent Activity of Composite Cyclin Dependent Kinase Inhibition against Hepatocellular Carcinoma" Cancers 11, no. 10: 1433. https://doi.org/10.3390/cancers11101433
APA StyleShao, Y.-Y., Li, Y.-S., Hsu, H.-W., Lin, H., Wang, H.-Y., Wo, R. R., Cheng, A.-L., & Hsu, C.-H. (2019). Potent Activity of Composite Cyclin Dependent Kinase Inhibition against Hepatocellular Carcinoma. Cancers, 11(10), 1433. https://doi.org/10.3390/cancers11101433