On the Mechanism of Hyperthermia-Induced BRCA2 Protein Degradation

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
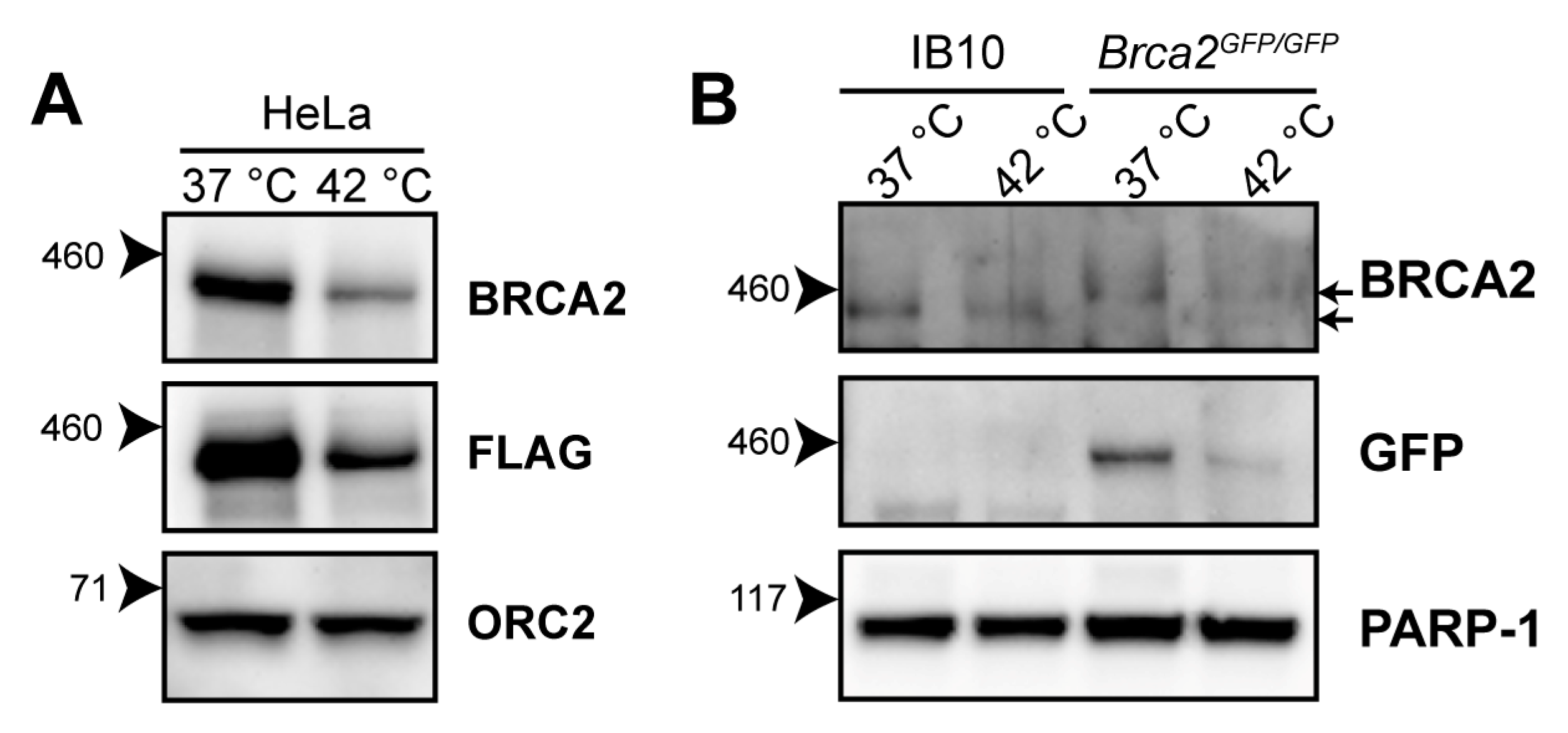
2.1. Heat-Mediated Degradation of BRCA2 and Modulation of HR
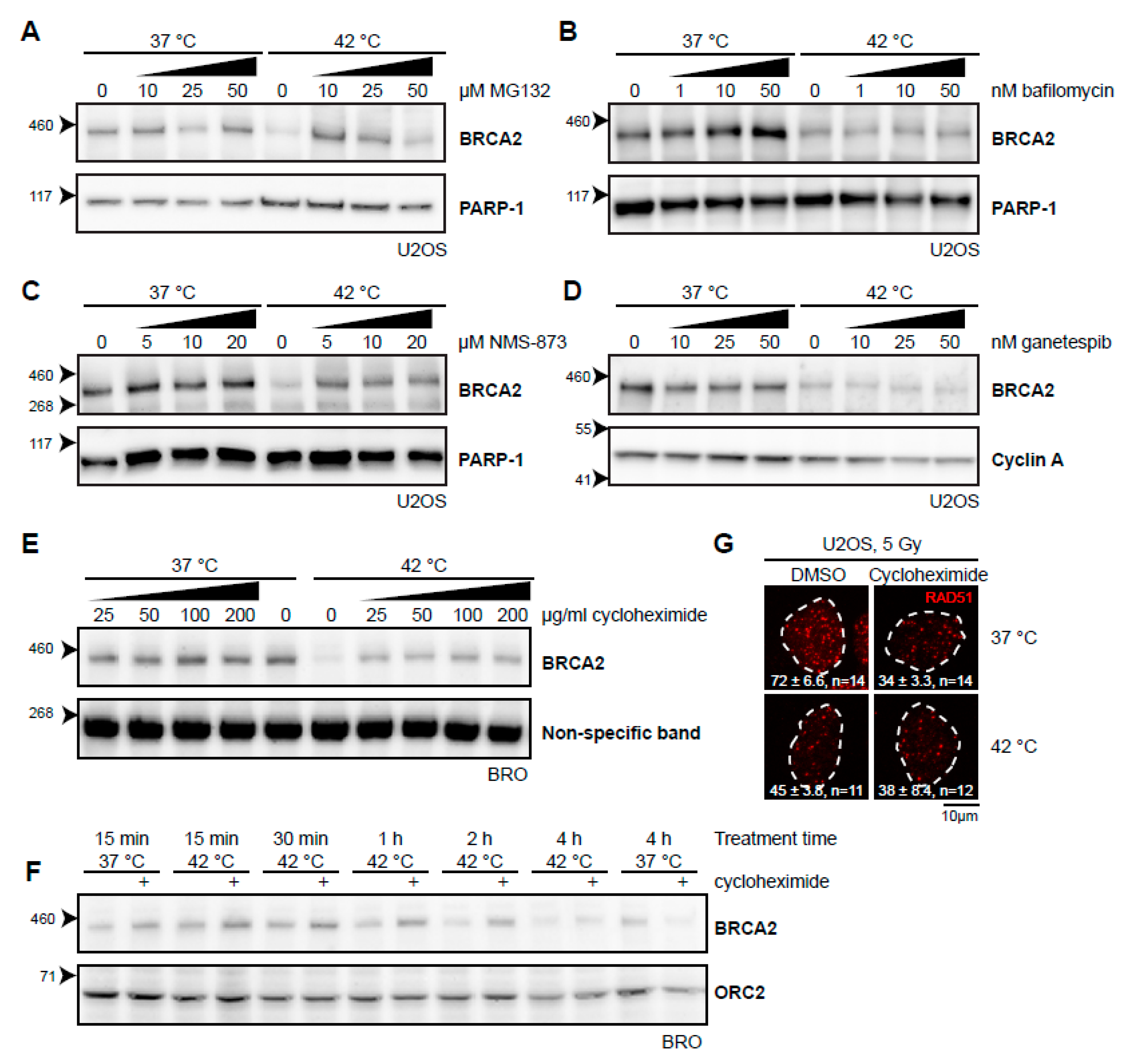
2.2. Various Inhibitors Affect Heat-Mediated BRCA2 Degradation
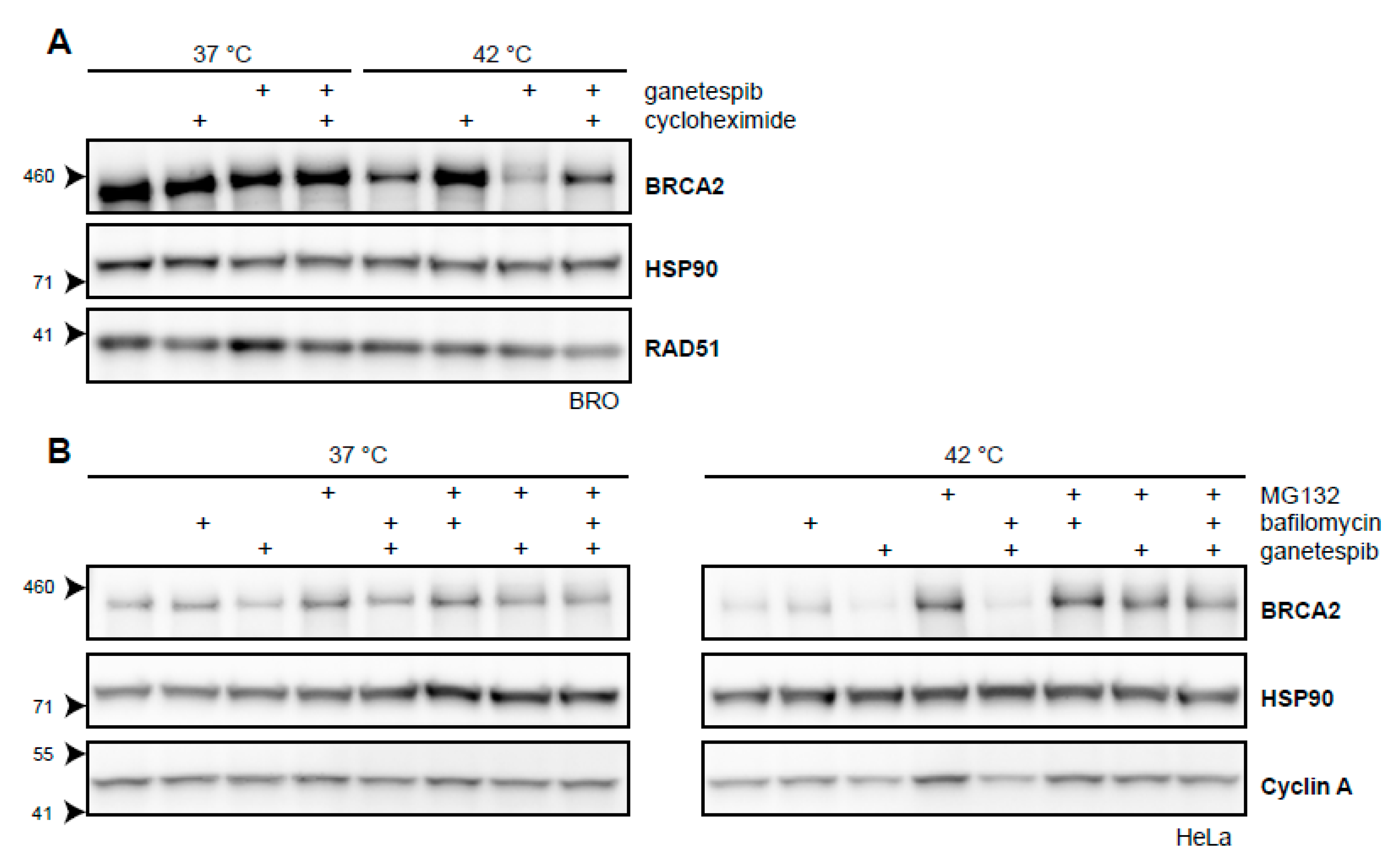
2.3. BRCA2 Stability Is Dependent on HSP90
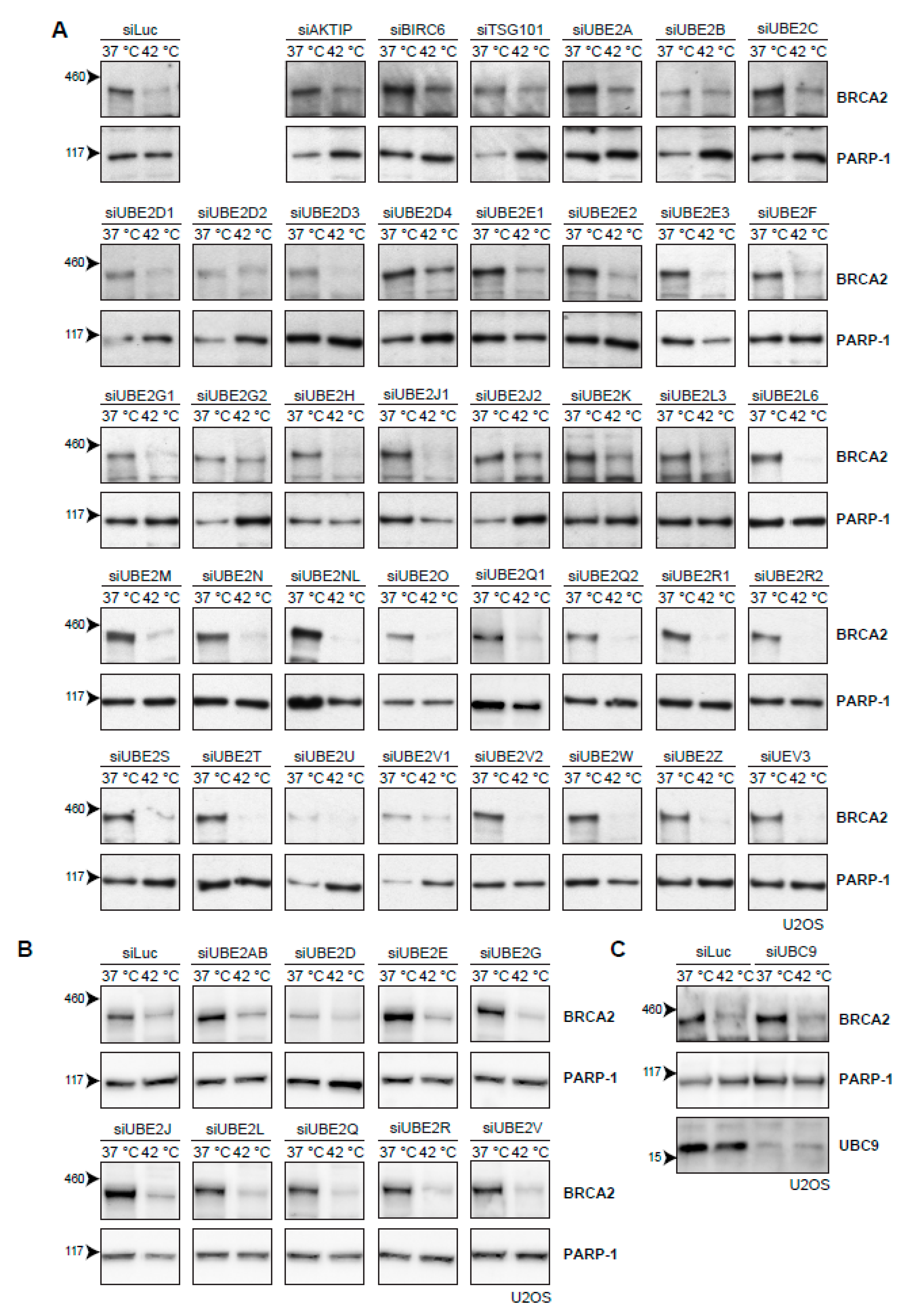
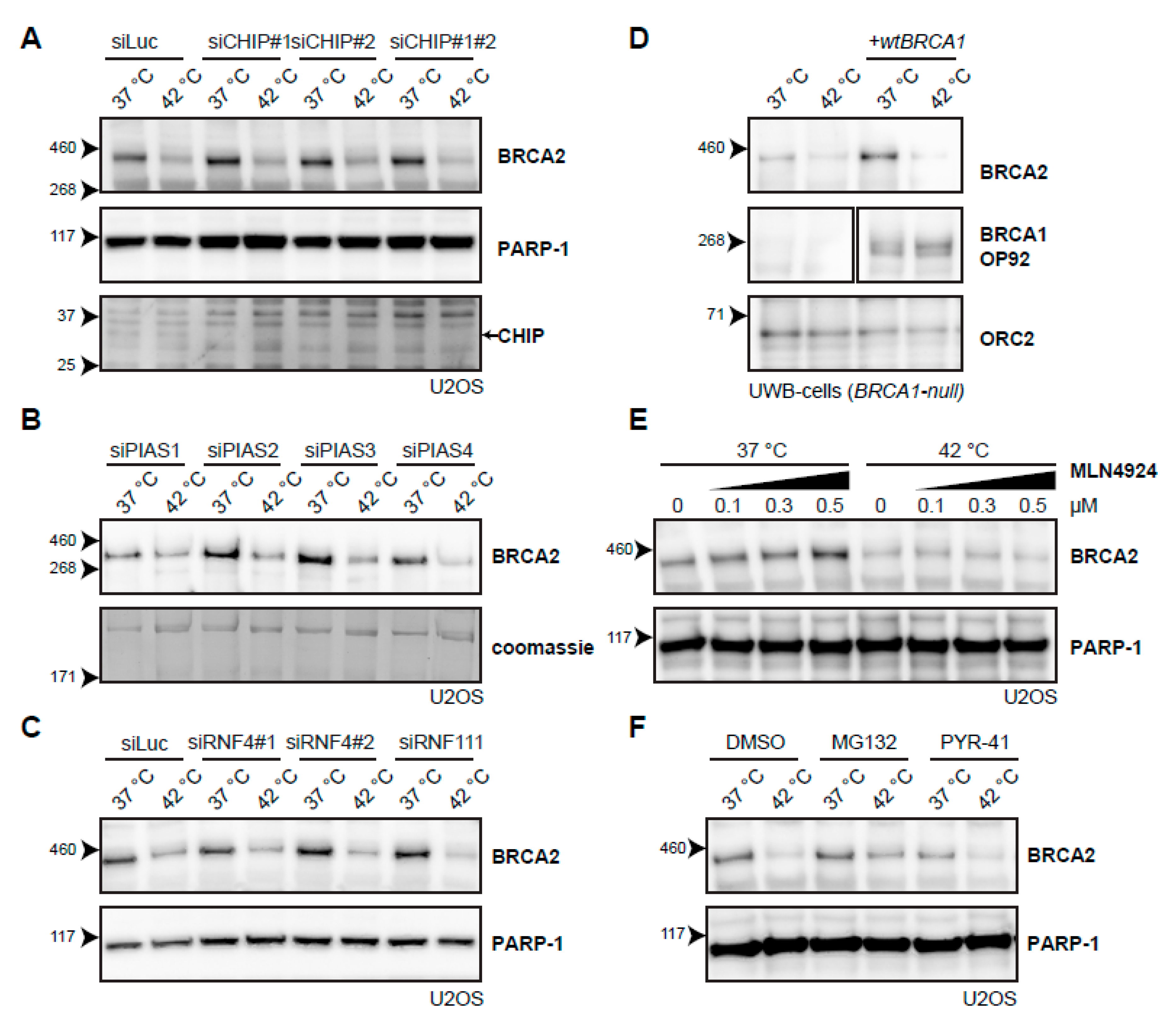
2.4. Searching for the Proteins that Mediate Degradation of BRCA2 upon Hyperthermia
2.5. Heat-Mediated BRCA2 Degradation from the Protein’s Perspective
2.6. Semi-Quantitative Mass Spectrometry Analysis Identifies Putative BRCA2-Interactors upon Hyperthermia
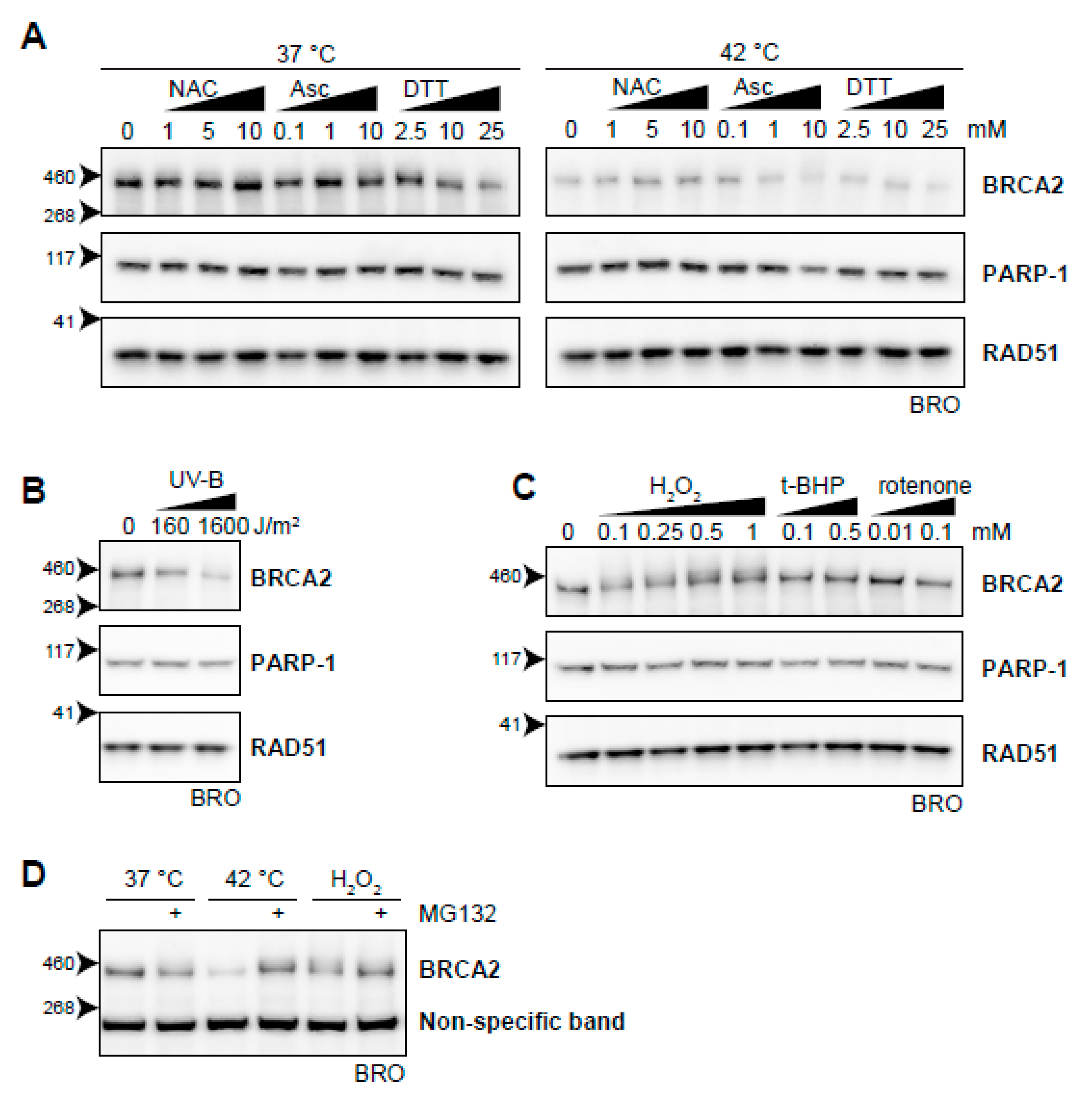
2.7. Oxidative Stress Induces BRCA2 Degradation
3. Discussion
3.1. Heat-Mediated Inhibition of HR from an Evolutionary Perspective
3.2. BRCA2 Protein Stability, HSP90, and Cycloheximide
3.3. HSP90 Is an Attractive Target to Modulate Heat-Induced BRCA2 Degradation
3.4. Ubiquitination May Not Be Required for Heat-Mediated BRCA2-Degradation by the Proteasome
3.5. BRCA2 and the Oxidative Stress Response
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of Constructs and Cell Lines
4.3. Hyperthermia Treatment
4.4. Chemical Agents and UV Irradiation
4.5. siRNA Transfection
4.6. Immunoprecipitation
4.7. Cell Fractionation, Lysis, and Immunoblotting
4.8. Immunofluorescent Staining and Analysis of RAD51-Foci
4.9. Antibodies
4.10. SILAC-Based Mass Spectrometry
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA damage response—A double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. Inhibiting the DNA damage response as a therapeutic manoeuvre in cancer. Br. J. Pharmacol. 2013, 169, 1745–1765. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D’Andrea, A.D. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003, 9, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Brown, J.S.; Kaye, S.B.; Yap, T.A. PARP inhibitors: The race is on. Br. J. Cancer 2016, 114, 713–715. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Tan, T.L.R.; Kanaar, R.; Wyman, C. Rad54, a Jack of all trades in homologous recombination. DNA Repair 2003, 2, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Mazin, A.V.; Mazina, O.M.; Bugreev, D.V.; Rossi, M.J. Rad54, the motor of homologous recombination. DNA Repair 2010, 9, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, S.B.; Game, J.C.; Brown, J.M. Inhibiting homologous recombination for cancer therapy. Cancer Biol. Ther. 2012, 13, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.F.S.; Kanaar, R. Targeting homologous recombination-mediated DNA repair in cancer. Expert Opin. Ther. Targets 2014, 18, 427–458. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Fehrmann, R.S.N.; de Vries, E.G.E.; van Vugt, M.A.T.M. Regulators of homologous recombination repair as novel targets for cancer treatment. Front. Genet. 2015, 6, 96. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef]
- Eppink, B.; Krawczyk, P.M.; Stap, J.; Kanaar, R. Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. Int. J. Hyperth. 2012, 28, 509–517. [Google Scholar] [CrossRef]
- Krawczyk, P.M.; Eppink, B.; Essers, J.; Stap, J.; Rodermond, H.; Odijk, H.; Zelensky, A.; van Bree, C.; Stalpers, L.J.; Buist, M.R.; et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc. Natl. Acad. Sci. USA 2011, 108, 9851–9856. [Google Scholar] [CrossRef] [PubMed]
- Van Den Tempel, N.; Laffeber, C.; Odijk, H.; Van Cappellen, W.A.; van Rhoon, G.C.; Franckena, M.; Kanaar, R. The effect of thermal dose on hyperthermia-mediated inhibition of DNA repair through homologous recombination. Oncotarget 2017, 8, 44593. [Google Scholar] [PubMed]
- Van den Tempel, N.; Odijk, H.; van Holthe, N.; Naipal, K.; Raams, A.; Eppink, B.; van Gent, D.C.; Hardillo, J.; Verduijn, G.M.; Drooger, J.C.; et al. Heat-induced BRCA2 degradation in human tumours provides rationale for hyperthermia-PARP-inhibitor combination therapies. Int. J. Hyperth. 2017, 34, 407–414. [Google Scholar] [CrossRef]
- Van den Tempel, N.; Naipal, K.A.T.; Raams, A.; van Gent, D.C.; Franckena, M.; Boormans, J.L.; Kanaar, R. Ex vivo assays to predict enhanced chemosensitization by hyperthermia in urothelial cancer of the bladder. PLoS ONE 2018, 13, e0209101. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.K.; Bradley, A. Murine Brca2: Sequence, map position, and expression pattern. Genomics 1997, 40, 234–241. [Google Scholar] [CrossRef]
- Reuter, M.; Zelensky, A.; Smal, I.; Meijering, E.; van Cappellen, W.A.; de Gruiter, H.M.; van Belle, G.J.; van Royen, M.E.; Houtsmuller, A.B.; Essers, J.; et al. BRCA2 diffuses as oligomeric clusters with RAD51 and changes mobility after DNA damage in live cells. J. Cell Biol. 2014, 207, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Calderwood, S.K. Autophagy, protein aggregation and hyperthermia: A mini-review. Int. J. Hyperth. 2011, 27, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef]
- Hanzelmann, P.; Schindelin, H. The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97. Front. Mol. Biosci. 2017, 4, 21. [Google Scholar] [CrossRef]
- Noguchi, M.; Yu, D.; Hirayama, R.; Ninomiya, Y.; Sekine, E.; Kubota, N.; Ando, K.; Okayasu, R. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem. Biophys. Res. Commun. 2006, 351, 658–663. [Google Scholar] [CrossRef]
- Vriend, L.E.M.; van den Tempel, N.; Oei, A.L.; L’Acosta, M.; Pieterson, F.J.; Franken, N.A.P.; Kanaar, R.; Krawczyk, P.M. Boosting the effects of hyperthermia-based anticancer treatments by HSP90 inhibition. Oncotarget 2017, 8, 97490. [Google Scholar] [CrossRef] [PubMed]
- Tarsounas, M.; Davies, D.; West, S.C. BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene 2003, 22, 1115–1123. [Google Scholar] [CrossRef]
- Murakawa, Y.; Sonoda, E.; Barber, L.J.; Zeng, W.; Yokomori, K.; Kimura, H.; Niimi, A.; Lehmann, A.; Zhao, G.Y.; Hochegger, H.; et al. Inhibitors of the proteasome suppress homologous DNA recombination in mammalian cells. Cancer Res. 2007, 67, 8536–8543. [Google Scholar] [CrossRef] [PubMed]
- Meerang, M.; Ritz, D.; Paliwal, S.; Garajova, Z.; Bosshard, M.; Mailand, N.; Janscak, P.; Hubscher, U.; Meyer, H.; Ramadan, K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 2011, 13, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.K.; Galanty, Y.; Sczaniecka-Clift, M.; Coates, J.; Jhujh, S.; Demir, M.; Cornwell, M.; Beli, P.; Jackson, S.P. Systematic E2 screening reveals a UBE2D-RNF137-CtIP axis promoting DNA repair. Nat. Cell Biol. 2015, 17, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Castoralova, M.; Brezinova, D.; Sveda, M.; Lipov, J.; Ruml, T.; Knejzlik, Z. SUMO-2/3 conjugates accumulating under heat shock or MG132 treatment result largely from new protein synthesis. Biochim. Biophys. Acta 2012, 1823, 911–919. [Google Scholar] [CrossRef]
- Van Cuijk, L.; van Belle, G.J.; Turkyilmaz, Y.; Poulsen, S.L.; Janssens, R.C.; Theil, A.F.; Sabatella, M.; Lans, H.; Mailand, N.; Houtsmuller, A.B.; et al. SUMO and ubiquitin-dependent XPC exchange drives nucleotide excision repair. Nat. Commun. 2015, 6, 7499. [Google Scholar] [CrossRef] [PubMed]
- Edkins, A.L. CHIP: A co-chaperone for degradation by the proteasome. Subcell. Biochem. 2015, 78, 219–242. [Google Scholar]
- Paul, I.; Ghosh, M.K. The E3 ligase CHIP: Insights into its structure and regulation. Biomed Res. Int. 2014, 2014, 918183. [Google Scholar] [CrossRef] [PubMed]
- Fantini, D.; Moritz, E.; Auvré, F.; Amouroux, R.; Campalans, A.; Epe, B.; Bravard, A.; Radicella, J.P. Rapid inactivation and proteasome-mediated degradation of OGG1 contribute to the synergistic effect of hyperthermia on genotoxic treatments. DNA Repair 2013, 12, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Keeffe, J.R.; Nishikawa, H.; Miyamoto, K.; Fox, D., 3rd; Fukuda, M.; Ohta, T.; Klevit, R. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. USA 2003, 100, 5646–5651. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Ma, J.; Cai, H.; Wu, T.; Sobhian, B.; Huo, Y.; Alcivar, A.; Mehta, M.; Cheung, K.L.; Ganesan, S.; Kong, A.-N.T.; et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 2012, 32, 1506–1517. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Blank, J.L.; Liu, X.J.; Cosmopoulos, K.; Bouck, D.C.; Garcia, K.; Bernard, H.; Tayber, O.; Hather, G.; Liu, R.; Narayanan, U.; et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013, 73, 225–234. [Google Scholar] [CrossRef]
- Yang, Y.; Kitagaki, J.; Dai, R.-M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007, 67, 9472–9481. [Google Scholar] [CrossRef]
- Vaughn, J.P.; Cirisano, F.D.; Huper, G.; Berchuck, A.; Futreal, P.A.; Marks, J.R.; Iglehart, J.D. Cell cycle control of BRCA2. Cancer Res. 1996, 56, 4590–4594. [Google Scholar] [PubMed]
- Bertwistle, D.; Swift, S.; Marston, N.J.; Jackson, L.E.; Crossland, S.; Crompton, M.R.; Marshall, C.J.; Ashworth, A. Nuclear location and cell cycle regulation of the BRCA2 protein. Cancer Res. 1997, 57, 5485–5488. [Google Scholar] [PubMed]
- Sanchez, H.; Paul, M.W.; Grosbart, M.; van Rossum-Fikkert, S.E.; Lebbink, J.H.G.; Kanaar, R.; Houtsmuller, A.B.; Wyman, C. Architectural plasticity of human BRCA2-RAD51 complexes in DNA break repair. Nucleic Acids Res. 2017, 45, 4507–4518. [Google Scholar] [CrossRef] [PubMed]
- Cadinanos, J.; Bradley, A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007, 35, e87. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.-P. Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones 2017, 22, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Sihvola, V.; Levonen, A.-L. Keap1 as the redox sensor of the antioxidant response. Arch. Biochem. Biophys. 2017, 617, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Lee, C.-T.; Ashcraft, K.A. The future of biology in driving the field of hyperthermia. Int. J. Hyperth. 2016, 32, 4–13. [Google Scholar] [CrossRef]
- Moon, E.J.; Sonveaux, P.; Porporato, P.E.; Danhier, P.; Gallez, B.; Batinic-Haberle, I.; Nien, Y.-C.; Schroeder, T.; Dewhirst, M.W. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc. Natl. Acad. Sci. USA 2010, 107, 20477–20482. [Google Scholar] [CrossRef]
- Barth, A.; Bauer, R.; Gedrange, T.; Walter, B.; Linss, C.; Klinger, W. Influence of hypoxia and hyperthermia upon peroxidative and glutathione status in growth-restricted newborn piglets. Exp. Toxicol. Pathol. 1998, 50, 31–33. [Google Scholar] [CrossRef]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell. Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef]
- Stewart, M.S.; Cameron, G.S.; Pence, B.C. Antioxidant nutrients protect against UVB-induced oxidative damage to DNA of mouse keratinocytes in culture. J. Invest. Dermatol. 1996, 106, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.L.; Gonzalez Maglio, D.H.; Weill, F.S.; Bustamante, J.; Leoni, J. Mitochondrial dysfunction and cellular stress progression after ultraviolet B irradiation in human keratinocytes. Photodermatol. Photoimmunol. Photomed. 2008, 24, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Rincheval, V.; Bergeaud, M.; Mathieu, L.; Leroy, J.; Guillaume, A.; Mignotte, B.; Le Floch, N.; Vayssiere, J.-L. Differential effects of Bcl-2 and caspases on mitochondrial permeabilization during endogenous or exogenous reactive oxygen species-induced cell death: A comparative study of H2O2, paraquat, t-BHP, etoposide and TNF-alpha-induced cell death. Cell Biol. Toxicol. 2012, 28, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Makino, K.; Su, L.K.; Pao, A.Y.; Kim, J.S.; Hung, M.C. Ultraviolet irradiation induces BRCA2 protein depletion through a p53-independent and protein synthesis-dependent pathway. Cancer Res. 2001, 61, 2838–2842. [Google Scholar] [PubMed]
- Spector, A.; Ma, W.; Sun, F.; Li, D.; Kleiman, N.J. The effect of H2O2 and tertiary butyl hydroperoxide upon a murine immortal lens epithelial cell line, alphaTN4-1. Exp. Eye Res. 2002, 75, 573–582. [Google Scholar] [CrossRef]
- Van den Tempel, N.; Horsman, M.R.; Kanaar, R. Improving efficacy of hyperthermia in oncology by exploiting biological mechanisms. Int. J. Hyperth. 2016, 32, 446–454. [Google Scholar] [CrossRef]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. IOWA Orthop. J. 2006, 26, 154–158. [Google Scholar]
- Westermark, K.F. Uber die Behandlung des Ulcerirenden Cervix Carcinoma Mittels Konstanter Warme. Zentralbl Gynaekol 1898, 22, 1335–1339. [Google Scholar]
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the thermal regulation of immunity: The immune system feels the heat. Nat. Rev. Immunol. 2015, 15, 335–349. [Google Scholar] [CrossRef]
- Kudoh, A.; Iwahori, S.; Sato, Y.; Nakayama, S.; Isomura, H.; Murata, T.; Tsurumi, T. Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in Epstein-Barr virus replication compartments. J. Virol. 2009, 83, 6641–6651. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.I.; McBee, R.M.; Le, U.Q.; Stone, A.C.; Wilkerson, G.K.; Demogines, A.M.; Sawyer, S.L. Rapid evolution of BRCA1 and BRCA2 in humans and other primates. BMC Evol. Biol. 2014, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Tachiiri, S.; Fujimori, A.; Thompson, L.H.; Miki, Y.; Hiraoka, M.; Takeda, S.; Yamazoe, M. Conserved domains in the chicken homologue of BRCA2. Oncogene 2002, 21, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.A.; Marais, M.; Maloney, S.K. A review of the physiology of fever in birds. J. Comp. Physiol. B 2013, 183, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Armour, E.P.; Lee, Y.J.; Corry, P.M.; Borrelli, M.J. Protection from heat-induced protein migration and DNA repair inhibition by cycloheximide. Biochem. Biophys. Res. Commun. 1988, 157, 611–617. [Google Scholar] [CrossRef]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Bullock, A.N. New strategies to inhibit KEAP1 and the Cul3-based E3 ubiquitin ligases. Biochem. Soc. Trans. 2014, 42, 103–107. [Google Scholar] [CrossRef]
- Walters, K.J.; Lech, P.J.; Goh, A.M.; Wang, Q.; Howley, P.M. DNA-repair protein hHR23a alters its protein structure upon binding proteasomal subunit S5a. Proc. Natl. Acad. Sci. USA 2003, 100, 12694–12699. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Franke, W.W.; Kleinschmidt, J.A. Distinct 19 S and 20 S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm. J. Biol. Chem. 1994, 269, 7709–7718. [Google Scholar] [PubMed]
- Raynes, R.; Pomatto, L.C.D.; Davies, K.J.A. Degradation of oxidized proteins by the proteasome: Distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Mol. Asp. Med. 2016, 50, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.-X.; Pang, Y.; Liu, C.H.; Haratake, K.; Du, B.-Y.; Ji, D.-Y.; Wang, G.-F.; Zhu, Q.-Q.; Song, W.; Yu, Y.; et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 2013, 153, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Pickering, A.M.; Koop, A.L.; Teoh, C.Y.; Ermak, G.; Grune, T.; Davies, K.J.A. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010, 432, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Reuven, N.; Shaul, Y. 20S proteasomes and protein degradation “by default”. Bioessays 2006, 28, 844–849. [Google Scholar] [CrossRef]
- Shringarpure, R.; Grune, T.; Mehlhase, J.; Davies, K.J.A. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J. Biol. Chem. 2003, 278, 311–318. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Hartford, S.A.; Chittela, R.; Ding, X.; Vyas, A.; Martin, B.; Burkett, S.; Haines, D.C.; Southon, E.; Tessarollo, L.; Sharan, S.K. Interaction with PALB2 Is Essential for Maintenance of Genomic Integrity by BRCA2. PLoS Genet. 2016, 12, e1006236. [Google Scholar] [CrossRef]
- Wong, A.K.C.; Pero, R.; Ormonde, P.A.; Tavtigian, S.V.; Bartel, P.L. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem. 1997, 272, 31941–31944. [Google Scholar] [CrossRef]
- Grune, T.; Botzen, D.; Engels, M.; Voss, P.; Kaiser, B.; Jung, T.; Grimm, S.; Ermak, G.; Davies, K.J.A. Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch. Biochem. Biophys. 2010, 500, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; Russo, A. Thiols, thiol depletion, and thermosensitivity. Radiat. Res. 1983, 95, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.F.; Whyte, B.; Bissinger, P.H.; Schiestl, R.H. Oxidative stress is involved in heat-induced cell death in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1996, 93, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef] [PubMed]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, K.; Fu, J.; Patsch, C.; Hu, S.; Weidlich, S.; Duerschke, K.; Buchholz, F.; Edenhofer, F.; Stewart, A.F. Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Dis. Model. Mech. 2009, 2, 508–515. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef]
- Shimazu, T.; Barjau, J.; Sohtome, Y.; Sodeoka, M.; Shinkai, Y. Selenium-based S-adenosylmethionine analog reveals the mammalian seven-beta-strand methyltransferase METTL10 to be an EF1A1 lysine methyltransferase. PLoS ONE 2014, 9, e105394. [Google Scholar] [CrossRef]
- Essers, J.; Hendriks, R.W.; Wesoly, J.; Beerens, C.E.M.T.; Smit, B.; Hoeijmakers, J.H.J.; Wyman, C.; Dronkert, M.L.G.; Kanaar, R. Analysis of mouse Rad54 expression and its implications for homologous recombination. DNA Repair 2002, 1, 779–793. [Google Scholar] [CrossRef]
- Masuda, T.; Saito, N.; Tomita, M.; Ishihama, Y. Unbiased quantitation of Escherichia coli membrane proteome using phase transfer surfactants. Mol. Cell. Proteom. 2009, 8, 2770–2777. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Peptides | Coverage | Maxquant Pep | 20 Min HT | 60 Min HT | ||
|---|---|---|---|---|---|---|---|
| Exp 1 | Exp 2 | Exp 1 | Exp 2 | ||||
| BRCA2 | 211 | 65 | 0.0E + 00 | 1.20 | 0.90 | 0.28 | 0.53 |
| PALB2 | 36 | 44.5 | 0.0E + 00 | 1.49 | 0.92 | 0.30 | 0.37 |
| RAD51 | 16 | 57.5 | 0.0E + 00 | 1.24 | 0.57 | 0.18 | 0.22 |
| BRCA1 | 19 | 12.9 | 2.5E − 58 | 1.25 | 1.39 | 0.15 | 0.83 |
| KEAP1 | 21 | 39.6 | 3.5E − 164 | 1.42 | 1.02 | 0.29 | 0.42 |
| MORF4L1 | 17 | 61.9 | 1.1E − 271 | 1.44 | 0.89 | 0.29 | 0.42 |
| MORF4L2 | 11 | 43.1 | 2.2E − 78 | 1.47 | 0.79 | 0.71 | 0.49 |
| Ubiquitin | 8 | 56.4 | 8.3E − 87 | 3.42 | 4.60 | 4.06 | 3.54 |
| HSPB1 | 10 | 67.9 | 4.3E − 69 | 1.45 | 2.26 | 3.19 | 1.87 |
| USP28 | 6 | 6 | 1.0E − 12 | 1.61 | 3.75 | 1.00 | 2.74 |
| % | SILAC ratio treated to untreated | ||||||
| Name | BRCA2-region (Amino Acid) | Molecular Weight (kDa) | Vector | Pro-Motor | Code/Ref |
|---|---|---|---|---|---|
| FLAG-BRCA2 | Met 1–Ile 3418 | 382 | pGb-LPL | CAG | pAZ148 |
| GFP-BRCA2 | Met 1–Ile 3418 | 406 | pGb-LPL | CAG | pAZ114 [26] |
| BRCA2-N-GFP | Met 1–Thr 939 | 131 | pGb-LPL | CAG | pAZ108 |
| BRCA2-M-GFP | Gln 940–Glu 2198 | 167 | pGb-LPL | CAG | pAZ109 |
| BRCA2-C-GFP | Thr 2199–Ile 3418 | 162 | pGb-LPL | CAG | pAZ110 |
| FLAG-BRCA2-N | Met 1–Thr 939 | 107 | pGb-LPL | CMV | pAZ97 |
| FLAG-BRCA2-M | Gln 940–Glu 2198 | 123 | pGb-LPL | CMV | pAZ98 |
| FLAG-BRCA2-C | Thr 2199–Ile 3418 | 118 | pGb-LPL | CMV | pAZ104 |
| FLAG-BRCA2-ΔM | Met 1–Thr 939 Thr 2199–Ile 3418 | 250 | pGb-LPL | CAG | pAZ253 |
| Clover-HSP90 | n.a. | 109 | pGb-LPL | CAG | n.a. |
| FLAG-HSP90 | n.a. | 82 | pGb-LPL | CMV | [99] |
| Set | Name | Sequence |
|---|---|---|
| 1F | GA15N-F | 5’-GCTCCTGGGCAACGTGCCTCGAGATGCCTATTGGATCCAAA GAGAGGCCAAC-3’ |
| 1R | GA15N-R | 5’-TTGCTCACCATGGTGGCCTCGAGGGTTGCTTGTTTATCACCT GTGT-3’ |
| 2F | GA15M-F | 5’-GCTCCTGGGCAACGTGCCTCGAGATGCAAGTGTCAATTAAAA AAGATTTGGTTTATGTTCTTGC-3’ |
| 2R | GA15M-R | 5’-TTGCTCACCATGGTGGCCTCGAAAGTTTCAGTTTTACCAATTT CCATTTTTACGTT-3’ |
| 3F | GA15C-F | 5’-GCTCCTGGGCAACGTGCCTCGAGATGACTTTTTCTGATGTTC CTGTGAAAACAAATATAGAAG-3’ |
| 3R | GA15C-R | 5’-TTGCTCACCATGGTGGCCTCGAGGATATATTTTTTAGTTGTAA TTGTGTCCTGCTTATTTTTCTCACA-3’ |
| 4F | BRCA2-Nterm-F | 5’-CAAGGATGACGACGACAAGAGCCCTATTGGATCCAAAGAGA GGC-3’ |
| 4R | BRCA2-Nterm-R | 5’-GCTGATTATGATCTAGAGTCAGGTTGCTTGTTTATCACCTGT GTCT-3’ |
| 5F | GA04M-F | 5’-CAAGGATGACGACGACAAGAGATCTACCCAAGTGTCAATTA AAAAAGATTTGGTTTATGT-3’ |
| 5R | GA04M-R | 5’-GCTGATTATGATCTAGAGTCAGATCTTTTCAGTTTTACCAATT TCCATTTTTACGTTTTTAGGT-3’ |
| 6F | GA04C-F | 5’-CAAGGATGACGACGACAAGAGATCTACTTTTTCTGATGTTCC TGTGAAAACAAATATAGAAG-3’ |
| 6R | GA04C-R | 5’-GCTGATTATGATCTAGAGTCAGATCTTTAGATATATTTTTTAGT TGTAA TTGTGTCCTGCTTATTTTTCTCACAT-3’ |
| 7F | B2flFLAG-F1 | 5’-CCTGGGCAACGTGCCGATTATAAAGACCACGATGGAGACT ATAAAGATCATGACATTGACT-3’ |
| 7R | B2flFLAG-R2 | 5’-CACTGTCCTTCCTGCAGGCATGACAGAGAA-3’ |
| 8F | B2intdel-GA-F1 | 5’-TTCTCTGTCATGCCTGCAGGAAGGAC-3’ |
| 8R | B2intdel-GA-R1 | 5’-CAACATTTAAGTTATTTGATAATTTGGTTGCTTGTTTATCACC TGTGTCT-3’ |
| Name | Reference/Suppliers |
|---|---|
| Bafilomycin | Sigma-Aldrich |
| NMS-873 | Selleckchem |
| Ganetespib | Syntha Pharmaceuticals |
| MG132 | Merck Millipore |
| Cycloheximide | Sigma-Aldrich |
| MLN4924 | MedChem Express |
| PYR-41 | Calbiochem |
| NAC | Sigma-Aldrich |
| Ascorbic Acid | Sigma-Aldrich |
| DTT | Sigma-Aldrich |
| H2O2 | Sigma-Aldrich |
| t-BHP | Sigma-Aldrich |
| Rotenone | MP Biomedicals |
| siRNA | SenseSequence | Reference/Suppliers |
|---|---|---|
| Luc | CGUACGCGGAAUACUUCGA | Thermo Scientific |
| siAKTIP#1 | GAAUUUACCUUGGUUGUGA | [37] |
| siAKTIP#2 | AGAAAACAGUGGCGACUUA | [37] |
| siBIRC6#1 | UCAUUGCCUUACUCACAUA | [37] |
| siBIRC6#2 | GGUCAAAGAUCACUUAGUA | [37] |
| siTSG101#1 | AGUAGCCGAGGUUGAUAAA | [37] |
| siTSG101#2 | AAACUGAGAUGGCGGAUGA | [37] |
| siUBE2A#1 | UGAUGUGUCUUCCAUUCUA | [37] |
| siUBE2A#2 | GAUGAACCCAAUCCCAAUA | [37] |
| siUBE2B#1 | AUAGACAACUGGUCUGUUA | [37] |
| siUBE2B#2 | UUGGACCAGAAGGGACACC | [37] |
| siUBE2C#1 | GCAAGAAACCUACUCAAAG | [37] |
| siUBE2C#2 | UAAAUUAAGCCUCGGUUGA | [37] |
| siUBE2D1#1 | UACUGUAUGUGUUGUCUAA | [37] |
| siUBE2D1#2 | CAACAGACAUGCAAGAGAA | [37] |
| siUBE2D2#1 | CAGUAAUGGCAGCAUUUGU | [37] |
| siUBE2D2#2 | CCAACCAGAUUAAACUCUA | [37] |
| siUBE2D3#1 | UGAUGUAAAGUUCGAAAGA | [37] |
| siUBE2D3#2 | CCACAAUUAUGGGACCUAA | [37] |
| siUBE2D4#1 | CAGCGUUGACUGUGUCAAA | [37] |
| siUBE2D4#2 | GGAAUUAACCGACUUGCAG | [37] |
| siUBE2E1#1 | GCGAUAACAUCUAUGAAUG | [37] |
| siUBE2E1#2 | GGUGUAUUCUUUCUCGAUA | [37] |
| siUBE2E2#1 | ACUUGAAAGAUUUGGGAUU | [37] |
| siUBE2E2#2 | UCACCAGACUAUCCGUUUA | [37] |
| siUBE2E3#1 | GCAUAGCCACUCAGUAUUU | [37] |
| siUBE2E3#2 | GCUAAGUUAUCCACUAGUG | [37] |
| siUBE2F#1 | GGAAUAAAGUGGAUGACUA | [37] |
| siUBE2F#2 | CAACAUAAAUACAGCAAGA | [37] |
| siUBE2G1#1 | UGUUGAUGCUGCGAAAGAA | [37] |
| siUBE2G1#2 | GGGAAGAUAAGUAUGGUUA | [37] |
| siUBE2G2#1 | AUGAUGACUUAAUGUCGAA | [37] |
| siUBE2G2#2 | UGACGAAAGUGGAGCUAAC | [37] |
| siUBE2H#1 | CGAGAGUAAACAUGAGGUU | [37] |
| siUBE2H#2 | CUACUGAACUGUCGAAGGA | [37] |
| siUBE2J1#1 | GAACUGGCUAGGCAAAUAA | [37] |
| siUBE2J1#2 | GAAAGAAGCGGCAGAAUUG | [37] |
| siUBE2J2#1 | GAAGGUGGCUAUUAUCAUG | [37] |
| siUBE2J2#2 | GCACAAGACGAACUCAGUA | [37] |
| siUBE2K#1 | CUCUCCGCACGGUAUUAUU | [37] |
| siUBE2K#2 | GAAUCAAGCGGGAGUUCAA | [37] |
| siUBE2L3#1 | UGAAGAGUUUACAAAGAAA | [37] |
| siUBE2L3#2 | GGGCUGACCUAGCUGAAGA | [37] |
| siUBE2L6#1 | UGAUCAAAUUCACAACCAA | [37] |
| siUBE2L6#2 | UCAAUGUGCUGGUGAAUAG | [37] |
| siUBE2M#1 | AGCCAGUCCUUACGAUAAA | [37] |
| siUBE2M#2 | GAUGAGGGCUUCUACAAGA | [37] |
| siUBE2N#1 | GCGGAGCAGUGGAAGACCA | [37] |
| siUBE2N#2 | CUAUCUAGCUUGUGUGUCA | [37] |
| siUBE2NL#1 | AAACGUGAACUAUUACUUG | [37] |
| siUBE2NL#2 | GACAAGUUGGAAAGAAUAA | [37] |
| siUBE2O#1 | ACAUCGACUGUGCCGUCAA | [37] |
| siUBE2O#2 | GGGACUACAUUGCCUAUGA | [37] |
| siUBE2Q1#1 | UCAUCUCCGACCUGUGUAA | [37] |
| siUBE2Q1#2 | GAAAGGGAAUACUCUGCUA | [37] |
| siUBE2Q2#1 | UACAGAUCACAGAGUUAUA | [37] |
| siUBE2Q2#2 | GUAUGGAACUUCUCACAAA | [37] |
| siUBE2R1#1 | CGCAGAACGUCAGGACCAU | [37] |
| siUBE2R1#2 | GGAAGUGGAAAGAGAGCAA | [37] |
| siUBE2R2#1 | UGUGAGGACUAUCCUAUUA | [37] |
| siUBE2R2#2 | CCACAACCCUGGCGGAAUA | [37] |
| siUBE2S#1 | AUGGCGAGAUCUGCGUCAA | [37] |
| siUBE2S#2 | ACAAGGAGGUGACGACACU | [37] |
| siUBE2T#1 | AGAGAGAGCUGCACAUGUU | [37] |
| siUBE2T#2 | CCUGCGAGCUCAAAUAUUA | [37] |
| siUBE2U#1 | ACAGGCCAUUACAAAUGAA | [37] |
| siUBE2U#2 | GAAGUGGAAUACAAACUAU | [37] |
| siUBE2V1#1 | GGACAGUGUUACAGCAAUU | [37] |
| siUBE2V1#2 | GUGGAUGCAUACCGAAAUA | [37] |
| siUBE2V2#1 | AGUUGUACUUCAAGAGCUA | [37] |
| siUBE2V2#2 | GUUAAAGUUCCUCGUAAUU | [37] |
| siUBE2W#1 | CGACCACCGGAUAAUUCUU | [37] |
| siUBE2W#2 | GCGAACAUGUAACAAGAAU | [37] |
| siUBE2Z#1 | AUGUUCGUUGUACCUGAUA | [37] |
| siUBE2Z#2 | GGGAAAGUCUGCUUGAGUA | [37] |
| siUEV3#1 | CGAUGGACCUUGAAAUCUU | [37] |
| siUEV3#2 | AGAAAGACCUGCUGAAUUU | [37] |
| UBC9 | GGGAUUGGUUUGGCAAGAA | [39] |
| CHIP#1 | GCGCUCUUCGAAUCGCGAAGA | Invitrogen |
| CHIP#2 | UGCCGCCACUAUCUGUGUAAU | Invitrogen |
| PIAS1 | GGAUCAUUCUAGAGCUUUA | [43] |
| PIAS2 | CUUGAAUAUUACAUCUUUA | [43] |
| PIAS3 | CCCUGAUGUCACCAUGAAA | [43] |
| PIAS4 | GGAGUAAGAGUGGACUGAA | [43] |
| RNF4#1 | GAAUGGACGUCUCAUCGUU | [39] |
| RNF4#2 | GACAGAGACGUAUAUGUGA | Thermo Scientific |
| RNF111 | GGAUAUUAAUGCAGAGGAA | [39] |
| Host | Epitope | Dilutions Used | Reference/Suppliers |
|---|---|---|---|
| Mouse | BRCA2 (OP95) | WB 1:1000 | EMD Millipore |
| Mouse | FLAG (M2) | WB 1:1000–1:5000 | Sigma-Aldrich |
| Mouse | ORC2 (68348) | WB 1:1000 | Abcam |
| Rabbit | Brca2 (27976) | WB 1:500 | Abcam |
| Mouse | GFP (clones 7.1 and 13.1) | WB 1:1000–1:5000 | Sigma-Aldrich |
| Mouse | PARP-1 (C2-10) | WB 1:5000 | Enzo Lifesciences |
| Mouse | BRCA1 (OP92) | WB 1:250 | EMD Millipore |
| Rabbit | Cyclin A (C-19) | WB 1:5000 | Santa Cruz |
| Goat | RAD54 (D-18) | WB 1:1000 | Santa Cruz |
| Rabbit | RAD51 (2307) | WB 1:10,000 | Home-made [100] |
| Rabbit | RAD51 (2308) | IF 1:10,000 | Home-made [100] |
| Mouse | GRB2 | WB 1:1000 | BD Pharmingen |
| Mouse | HSP90 (AC88, 13492) | WB 1:5000 | Abcam |
| Rabbit | CDC37 (3618S) | WB 1:1000 | Cell Signaling |
| Goat | UBC9 (N-15) | WB 1:1000 | Santa Cruz |
| Rabbit | CHIP (PA1-015) | WB 1:1000 | Thermo Scientific |
| Sheep | Peroxidase anti-Mouse IgG (H+L) | WB 1:2000 | Jackson Immunoresearch |
| Donkey | Peroxidase anti-Rabbit IgG (H+L) | WB 1:2000 | Jackson Immunoresearch |
| Donkey | Peroxidase anti-Goat IgG (H+L) | WB 1:2000 | Jackson Immunoresearch |
| Goat | Alexa-Fluor-594 (red) | IF 1:1000 | Thermo Scientific |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van den Tempel, N.; Zelensky, A.N.; Odijk, H.; Laffeber, C.; Schmidt, C.K.; Brandsma, I.; Demmers, J.; Krawczyk, P.M.; Kanaar, R. On the Mechanism of Hyperthermia-Induced BRCA2 Protein Degradation. Cancers 2019, 11, 97. https://doi.org/10.3390/cancers11010097
van den Tempel N, Zelensky AN, Odijk H, Laffeber C, Schmidt CK, Brandsma I, Demmers J, Krawczyk PM, Kanaar R. On the Mechanism of Hyperthermia-Induced BRCA2 Protein Degradation. Cancers. 2019; 11(1):97. https://doi.org/10.3390/cancers11010097
Chicago/Turabian Stylevan den Tempel, Nathalie, Alex N. Zelensky, Hanny Odijk, Charlie Laffeber, Christine K. Schmidt, Inger Brandsma, Jeroen Demmers, Przemek M. Krawczyk, and Roland Kanaar. 2019. "On the Mechanism of Hyperthermia-Induced BRCA2 Protein Degradation" Cancers 11, no. 1: 97. https://doi.org/10.3390/cancers11010097
APA Stylevan den Tempel, N., Zelensky, A. N., Odijk, H., Laffeber, C., Schmidt, C. K., Brandsma, I., Demmers, J., Krawczyk, P. M., & Kanaar, R. (2019). On the Mechanism of Hyperthermia-Induced BRCA2 Protein Degradation. Cancers, 11(1), 97. https://doi.org/10.3390/cancers11010097