EphA3 Pay-Loaded Antibody Therapeutics for the Treatment of Glioblastoma

 , ,
, ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
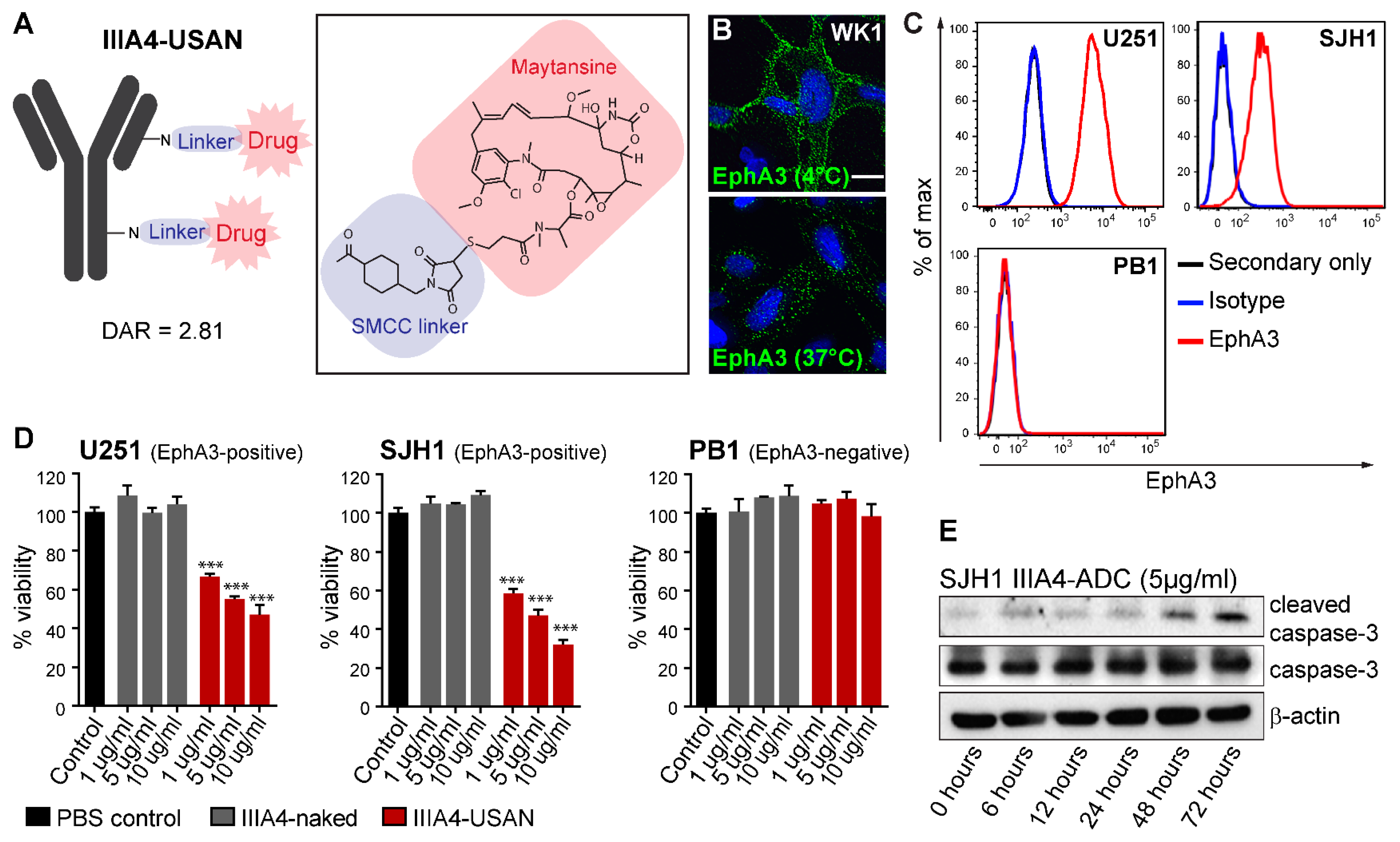
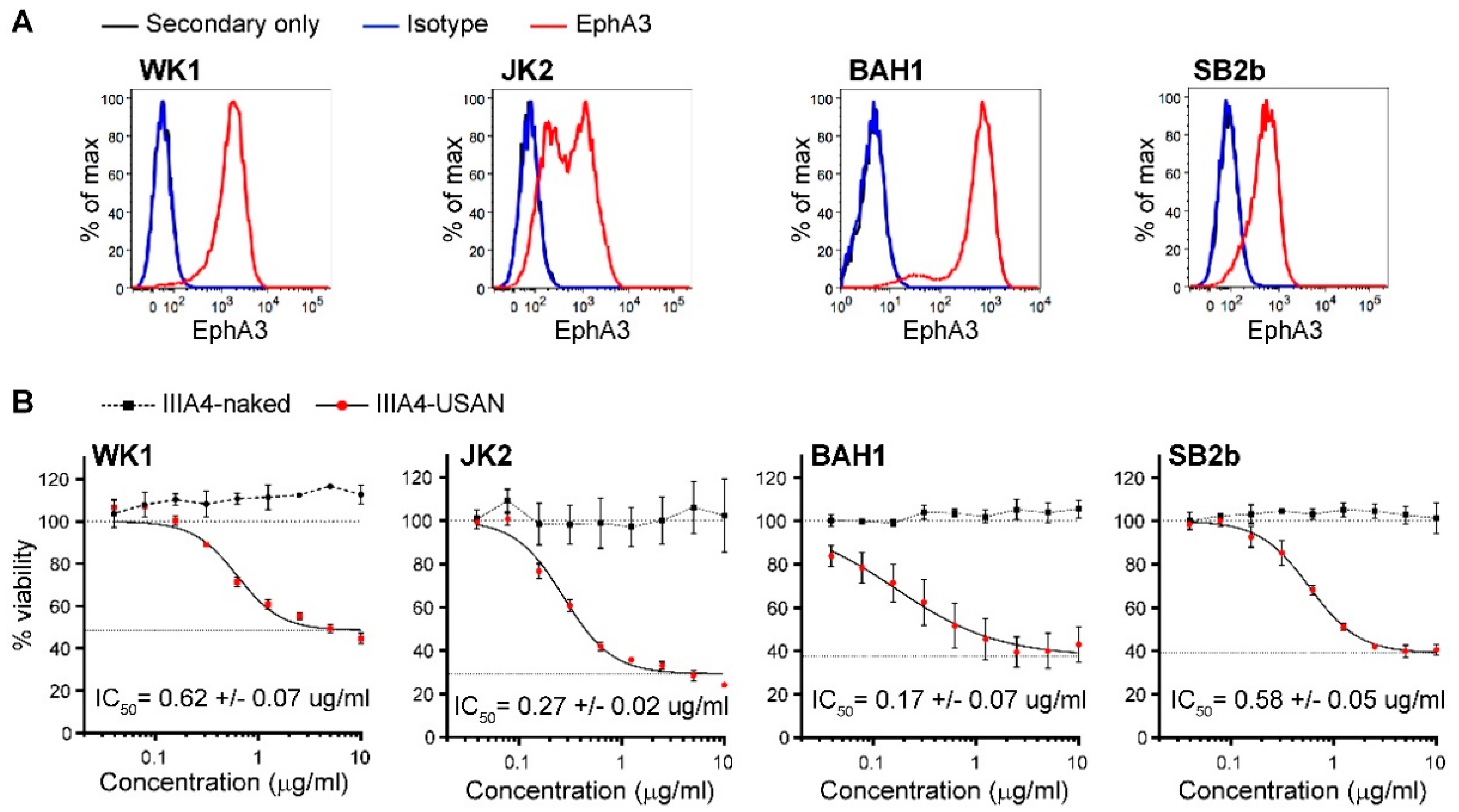
2.1. The EphA3-Maytansine ADC Induces Potent Cell Killing In Vitro
2.2. IIIA4 Crosses the Blood-Tumour Barrier (BTB) and Accumulates Specifically in GBM
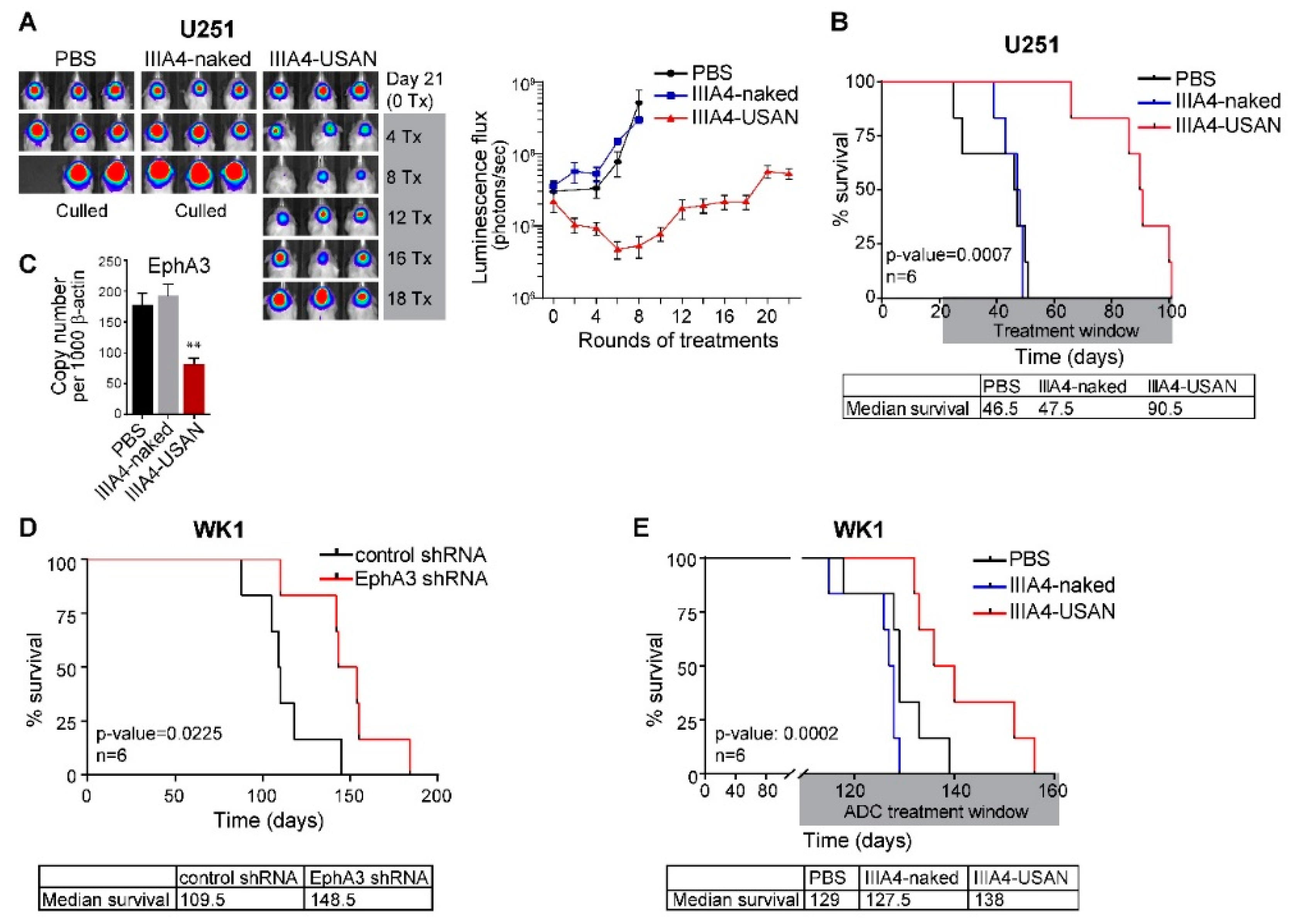
2.3. The EphA3-Maytansine ADC Induces A Potent GBM Anti-Tumour Response In Vivo
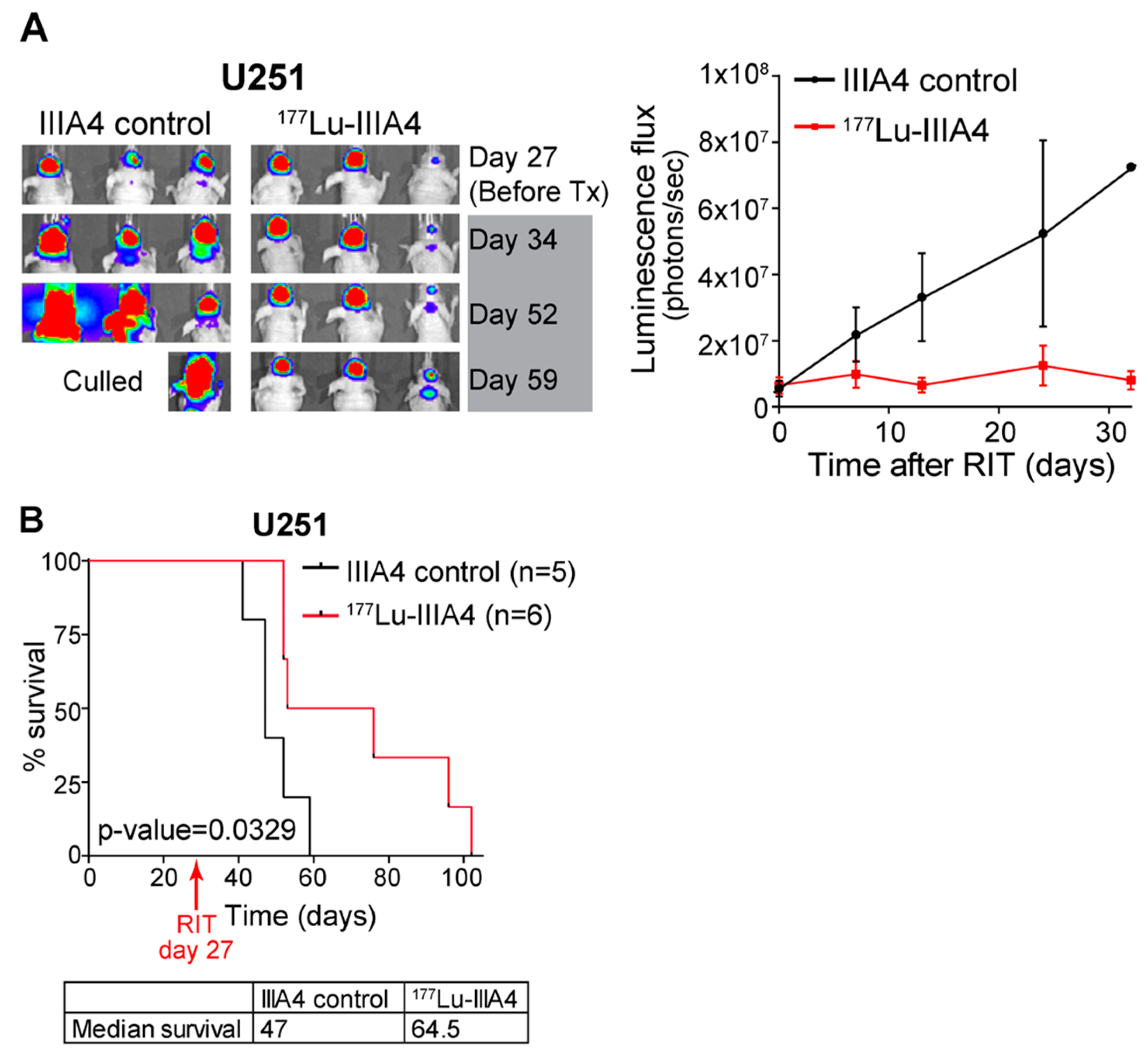
2.4. EphA3-Targeting RIT Induces A GBM Anti-Tumour Response In Vivo
3. Discussion
4. Materials and Methods
4.1. Primary Cell Cultures
4.2. ADC Preparation
4.3. Bio-Layer Interferometry (BLI)
4.4. Cell Viability Assay
4.5. Caspase-3 Activation
4.6. Immunofluorescence Analysis
4.7. Flow Cytometry
4.8. Animal Studies
4.9. Imaging
4.9.1. Generation of IIIA4-NOTA
4.9.2. Radiolabelling
4.9.3. PET/CT Imaging
4.9.4. Magnetic Resonance Imaging (MRI)
4.9.5. Image Analysis
4.10. Radioimmunotherapy (RIT)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 2005, 6, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.W.; Bartlett, P.F.; Lackmann, M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 2013, 13, 39–62. [Google Scholar] [CrossRef]
- Boyd, A.W.; Ward, L.D.; Wicks, I.P.; Simpson, R.J.; Salvaris, E.; Wilks, A.; Welch, K.; Loudovaris, M.; Rockman, S.; Busmanis, I. Isolation and characterization of a novel receptor-type protein tyrosine kinase (hek) from a human pre-B cell line. J. Biol. Chem. 1992, 267, 3262–3267. [Google Scholar] [PubMed]
- Pasquale, E.B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhauser, C.; et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef]
- Day, B.W.; Stringer, B.W.; Boyd, A.W. Eph receptors as therapeutic targets in glioblastoma. Br. J. Cancer 2014, 111, 1255–1261. [Google Scholar] [CrossRef]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Adams, J.; Singh, M.; Hu, A.; Gorelik, M.; Subapanditha, M.K.; Savage, N.; Yang, J.; et al. Cotargeting Ephrin Receptor Tyrosine Kinases A2 and A3 in Cancer Stem Cells Reduces Growth of Recurrent Glioblastoma. Cancer Res. 2018, 78, 5023–5037. [Google Scholar] [CrossRef]
- Ferluga, S.; Tome, C.M.; Herpai, D.M.; D’Agostino, R.; Debinski, W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget 2016, 7, 59860–59876. [Google Scholar] [CrossRef] [PubMed]
- Vail, M.E.; Murone, C.; Tan, A.; Hii, L.; Abebe, D.; Janes, P.W.; Lee, F.T.; Baer, M.; Palath, V.; Bebbington, C.; et al. Targeting EphA3 Inhibits Cancer Growth by Disrupting the Tumor Stromal Microenvironment. Cancer Res. 2014, 74, 4470–4481. [Google Scholar] [CrossRef] [PubMed]
- Lackmann, M.; Mann, R.J.; Kravets, L.; Smith, F.M.; Bucci, T.A.; Maxwell, K.F.; Howlett, G.J.; Olsson, J.E.; Vanden Bos, T.; Cerretti, D.P.; et al. Ligand for EPH-related kinase (LERK) 7 is the preferred high affinity ligand for the HEK receptor. J. Biol. Chem. 1997, 272, 16521–16530. [Google Scholar] [CrossRef] [PubMed]
- Lackmann, M. Isolation and characterization of “orphan-RTK” ligands using an integrated biosensor approach. Methods Mol. Biol. 2001, 124, 335–359. [Google Scholar]
- Smith, F.M.; Vearing, C.; Lackmann, M.; Treutlein, H.; Himanen, J.; Chen, K.; Saul, A.; Nikolov, D.; Boyd, A.W. Dissecting the EphA3/Ephrin-A5 interactions using a novel functional mutagenesis screen. J. Biol. Chem. 2004, 279, 9522–9531. [Google Scholar] [CrossRef] [PubMed]
- Vearing, C.; Lee, F.T.; Wimmer-Kleikamp, S.; Spirkoska, V.; To, C.; Stylianou, C.; Spanevello, M.; Brechbiel, M.; Boyd, A.W.; Scott, A.M.; et al. Concurrent binding of anti-EphA3 antibody and ephrin-A5 amplifies EphA3 signaling and downstream responses: Potential as EphA3-specific tumor-targeting reagents. Cancer Res. 2005, 65, 6745–6754. [Google Scholar] [CrossRef]
- Swords, R.T.; Greenberg, P.L.; Wei, A.H.; Durrant, S.; Advani, A.S.; Hertzberg, M.S.; Jonas, B.A.; Lewis, I.D.; Rivera, G.; Gratzinger, D.; et al. KB004, a first in class monoclonal antibody targeting the receptor tyrosine kinase EphA3, in patients with advanced hematologic malignancies: Results from a phase 1 study. Leuk. Res. 2016, 50, 123–131. [Google Scholar] [CrossRef]
- Gan, H.K.; Reardon, D.A.; Lassman, A.B.; Merrell, R.; van den Bent, M.; Butowski, N.; Lwin, Z.; Wheeler, H.; Fichtel, L.; Scott, A.M.; et al. Safety, pharmacokinetics, and antitumor response of depatuxizumab mafodotin as monotherapy or in combination with temozolomide in patients with glioblastoma. Neuro Oncol. 2018, 20, 838–847. [Google Scholar] [CrossRef]
- Lassman, A.B.; van den Bent, M.J.; Gan, H.K.; Reardon, D.A.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro Oncol. 2018. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Day, B.W.; Stringer, B.W.; Wilson, J.; Jeffree, R.L.; Jamieson, P.J.; Ensbey, K.S.; Bruce, Z.C.; Inglis, P.; Allan, S.; Winter, C.; et al. Glioma Surgical Aspirate: A Viable Source of Tumor Tissue for Experimental Research. Cancers 2013, 5, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J. Clin. Oncol. 2011, 29, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef]
- Janes, P.W.; Slape, C.I.; Farnsworth, R.H.; Atapattu, L.; Scott, A.M.; Vail, M.E. EphA3 biology and cancer. Growth Factors 2014, 32, 176–189. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Sung, M.; Gerber, H.P. Mechanisms of Resistance to Antibody-Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Day, B.W.; Stringer, B.W.; Spanevello, M.D.; Charmsaz, S.; Jamieson, P.R.; Ensbey, K.S.; Carter, J.C.; Cox, J.M.; Ellis, V.J.; Brown, C.L.; et al. ELK4 neutralization sensitizes glioblastoma to apoptosis through downregulation of the anti-apoptotic protein Mcl-1. Neuro Oncol. 2011, 13, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Shi, W.; Miranda, M.; Simpson, P.T.; Vargas, A.C.; Song, S.; Wiegmans, A.P.; Swarbrick, A.; Welm, A.L.; Brown, M.P.; et al. Treatment of triple-negative breast cancer using anti-EGFR-directed radioimmunotherapy combined with radiosensitizing chemotherapy and PARP inhibitor. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2013, 54, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Pajic, M.; Shi, W.; Kalimutho, M.; Miranda, M.; Nagrial, A.M.; Chou, A.; Biankin, A.V.; Grimmond, S.M.; Australian Pancreatic Cancer Genome Initiative; et al. Gemcitabine and CHK1 inhibition potentiate EGFR-directed radioimmunotherapy against pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2014, 20, 3187–3197. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Offenhäuser, C.; Al-Ejeh, F.; Puttick, S.; Ensbey, K.S.; Bruce, Z.C.; Jamieson, P.R.; Smith, F.M.; Stringer, B.W.; Carrington, B.; Fuchs, A.V.; et al. EphA3 Pay-Loaded Antibody Therapeutics for the Treatment of Glioblastoma. Cancers 2018, 10, 519. https://doi.org/10.3390/cancers10120519
Offenhäuser C, Al-Ejeh F, Puttick S, Ensbey KS, Bruce ZC, Jamieson PR, Smith FM, Stringer BW, Carrington B, Fuchs AV, et al. EphA3 Pay-Loaded Antibody Therapeutics for the Treatment of Glioblastoma. Cancers. 2018; 10(12):519. https://doi.org/10.3390/cancers10120519
Chicago/Turabian StyleOffenhäuser, Carolin, Fares Al-Ejeh, Simon Puttick, Kathleen S. Ensbey, Zara C. Bruce, Paul R. Jamieson, Fiona M. Smith, Brett W. Stringer, Benjamin Carrington, Adrian V. Fuchs, and et al. 2018. "EphA3 Pay-Loaded Antibody Therapeutics for the Treatment of Glioblastoma" Cancers 10, no. 12: 519. https://doi.org/10.3390/cancers10120519
APA StyleOffenhäuser, C., Al-Ejeh, F., Puttick, S., Ensbey, K. S., Bruce, Z. C., Jamieson, P. R., Smith, F. M., Stringer, B. W., Carrington, B., Fuchs, A. V., Bell, C. A., Jeffree, R., Rose, S., Thurecht, K. J., Boyd, A. W., & Day, B. W. (2018). EphA3 Pay-Loaded Antibody Therapeutics for the Treatment of Glioblastoma. Cancers, 10(12), 519. https://doi.org/10.3390/cancers10120519