Finding the Right Way to Target EGFR in Glioblastomas; Lessons from Lung Adenocarcinomas


Abstract
1. Introduction
2. EGFR Mutations in LUAD and Gliomas
3. Clinical Activity of EGFR-TKIs in LUAD, But Not GBM Patients
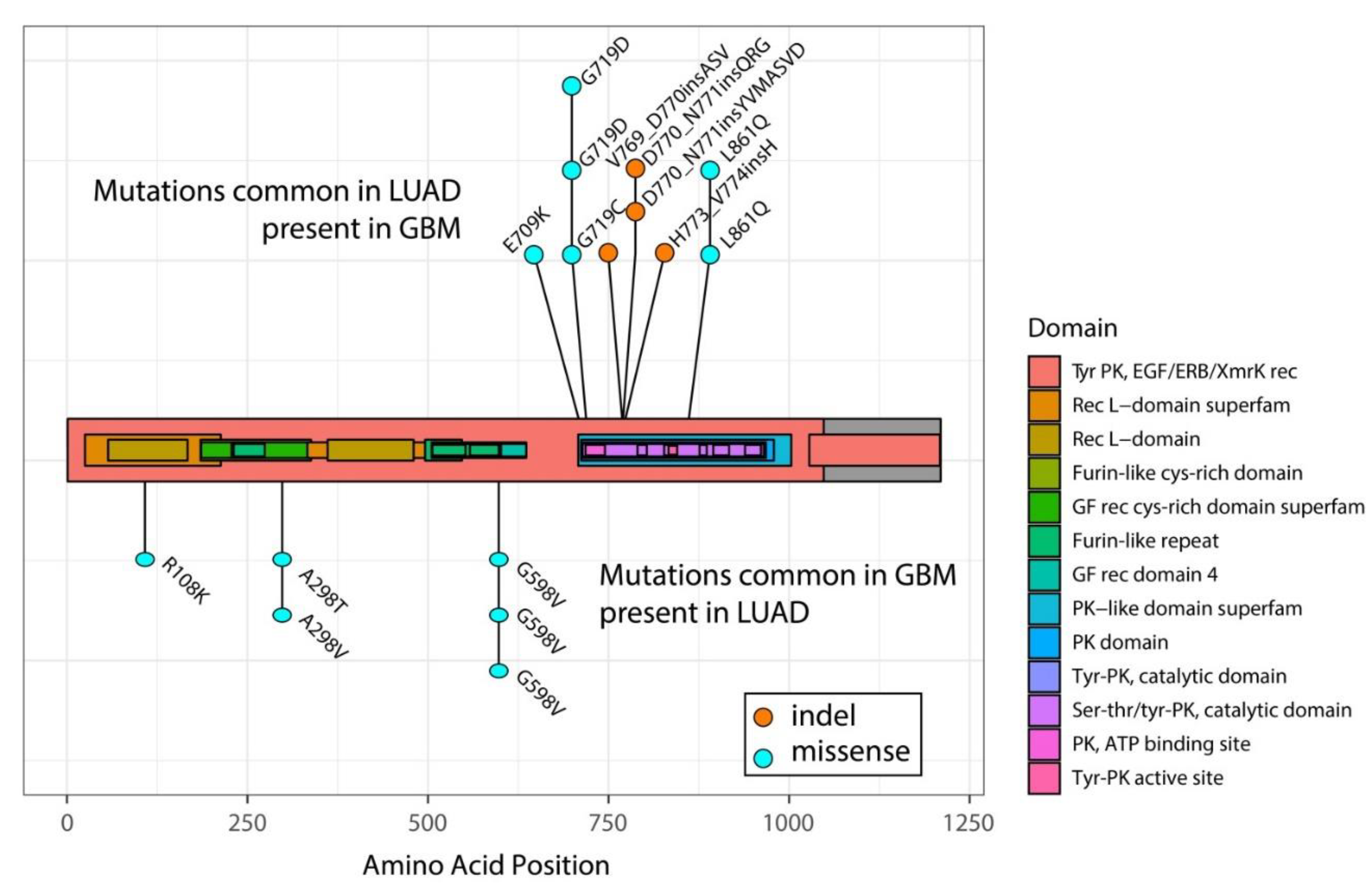
4. Different Mutations Activate Different Pathways and May Explain Refractoriness to EGFR-TKIs
5. Lessons from LUAD: Not all LUAD Respond to EGFR-TKIs and Not All TKIs are Effective in LUAD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cohen, S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 1962, 237, 1555–1562. [Google Scholar] [PubMed]
- Cohen, S.; Elliott, G.A. The stimulation of epidermal keratinization by a protein isolated from the submaxillary gland of the mouse. J. Investig. Dermatol. 1963, 40, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S. The stimulation of epidermal proliferation by a specific protein (EGF). Dev. Biol. 1965, 12, 394–407. [Google Scholar] [CrossRef]
- Carpenter, G.; Lembach, K.J.; Morrison, M.M.; Cohen, S. Characterization of the binding of 125-I-labeled epidermal growth factor to human fibroblasts. J. Biol. Chem. 1975, 250, 4297–4304. [Google Scholar] [PubMed]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, I.N. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The Biology of Cancer, 2nd ed.; Ww Norton & Co.: New York, NY, USA, 2013. [Google Scholar]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel) 2017, 9. [Google Scholar] [CrossRef]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Wilson, K.J.; Gilmore, J.L.; Foley, J.; Lemmon, M.A.; Riese, D.J., 2nd. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol. Ther. 2009, 122, 1–8. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; Barron, D.; Chakravarty, D.; Gao, J.; Chang, M.T.; Ni, A.; Kundra, R.; Jonsson, P.; et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017, 7, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. 1), S24–S31. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Fu, Y.N.; Lin, C.H.; Yang, S.T.; Hu, S.F.; Chen, Y.T.; Tsai, S.F.; Huang, S.F. Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene 2006, 25, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y.; et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006, 3, e485. [Google Scholar] [CrossRef]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Klingler, S.; Guo, B.; Yao, J.; Yan, H.; Zhang, L.; Vaseva, A.V.; Chen, S.; Canoll, P.; Horner, J.W.; Wang, Y.A.; et al. Development of Resistance to EGFR-Targeted Therapy in Malignant Glioma Can Occur through EGFR-Dependent and -Independent Mechanisms. Cancer Res. 2015, 75, 2109–2119. [Google Scholar] [CrossRef]
- Vivanco, I.; Robins, H.I.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N.; et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, D.; Chen, L.; Shimamura, T.; Kobayashi, S.; McNamara, K.; Mahmood, U.; Mitchell, A.; Sun, Y.; Al-Hashem, R.; et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 2006, 9, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.C.; Buckner, J.C.; James, C.D.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 347–353. [Google Scholar] [CrossRef]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.K.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro. Oncol. 2010, 12, 95–103. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Krishnan, S.; Sarkaria, J.N.; Wu, W.; Jaeckle, K.A.; Uhm, J.H.; Geoffroy, F.J.; Arusell, R.; Kitange, G.; Jenkins, R.B.; et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J. Clin. Oncol. 2008, 26, 5603–5609. [Google Scholar] [CrossRef] [PubMed]
- Sathornsumetee, S.; Desjardins, A.; Vredenburgh, J.J.; McLendon, R.E.; Marcello, J.; Herndon, J.E.; Mathe, A.; Hamilton, M.; Rich, J.N.; Norfleet, J.A.; et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro. Oncol. 2010, 12, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A.; et al. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro. Oncol. 2013, 15, 490–496. [Google Scholar] [CrossRef]
- Clarke, J.L.; Molinaro, A.M.; Phillips, J.J.; Butowski, N.A.; Chang, S.M.; Perry, A.; Costello, J.F.; DeSilva, A.A.; Rabbitt, J.E.; Prados, M.D. A single-institution phase II trial of radiation, temozolomide, erlotinib, and bevacizumab for initial treatment of glioblastoma. Neuro. Oncol. 2014, 16, 984–990. [Google Scholar] [CrossRef]
- Prados, M.D.; Chang, S.M.; Butowski, N.; DeBoer, R.; Parvataneni, R.; Carliner, H.; Kabuubi, P.; Ayers-Ringler, J.; Rabbitt, J.; Page, M.; et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J. Clin. Oncol. 2009, 27, 579–584. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Herndon, J.E., 2nd; Marcello, J.; Norfleet, J.A.; McLendon, R.E.; Sampson, J.H.; et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neurooncol. 2010, 96, 219–230. [Google Scholar] [CrossRef]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P.; et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro. Oncol. 2014, 16, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; McBain, C.; Nash, S.; Hopkins, K.; Sanghera, P.; Saran, F.; Phillips, M.; Dungey, F.; Clifton-Hadley, L.; Wanek, K.; et al. Multi-Center Randomized Phase II Study Comparing Cediranib plus Gefitinib with Cediranib plus Placebo in Subjects with Recurrent/Progressive Glioblastoma. PLoS ONE 2016, 11, e0156369. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro. Oncol. 2015, 17, 430–439. [Google Scholar] [CrossRef]
- Sepulveda-Sanchez, J.M.; Vaz, M.A.; Balana, C.; Gil-Gil, M.; Reynes, G.; Gallego, O.; Martinez-Garcia, M.; Vicente, E.; Quindos, M.; Luque, R.; et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro. Oncol. 2017, 19, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Smith, P.; Sul, J.; Salgado, C.; Iwamoto, F.M.; Shih, J.H.; Fine, H.A. Continuous daily sunitinib for recurrent glioblastoma. J. Neurooncol. 2013, 111, 41–48. [Google Scholar] [CrossRef]
- Reardon, D.A.; Quinn, J.A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Desjardins, A.; Sathornsumetee, S.; Herndon, J.E., 2nd; Dowell, J.M.; McLendon, R.E.; et al. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin. Cancer Res. 2006, 12, 860–868. [Google Scholar] [CrossRef]
- Franceschi, E.; Cavallo, G.; Lonardi, S.; Magrini, E.; Tosoni, A.; Grosso, D.; Scopece, L.; Blatt, V.; Urbini, B.; Pession, A.; et al. Gefitinib in patients with progressive high-grade gliomas: A multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br. J. Cancer 2007, 96, 1047–1051. [Google Scholar] [CrossRef]
- Yung, W.K.; Vredenburgh, J.J.; Cloughesy, T.F.; Nghiemphu, P.; Klencke, B.; Gilbert, M.R.; Reardon, D.A.; Prados, M.D. Safety and efficacy of erlotinib in first-relapse glioblastoma: A phase II open-label study. Neuro. Oncol. 2010, 12, 1061–1070. [Google Scholar] [CrossRef]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Del Vecchio, C.A.; Giacomini, C.P.; Vogel, H.; Jensen, K.C.; Florio, T.; Merlo, A.; Pollack, J.R.; Wong, A.J. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013, 32, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Gao, Y.; Kerkhof, M.; Kros, J.M.; Gorlia, T.; van Zwieten, K.; Prince, J.; van Duinen, S.; Sillevis Smitt, P.A.; Taphoorn, M.; et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro. Oncol. 2015, 17, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Antonyak, M.A.; Moscatello, D.K.; Wong, A.J. Constitutive activation of c-Jun N-terminal kinase by a mutant epidermal growth factor receptor. J. Biol. Chem. 1998, 273, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Everiss, K.D.; Wikstrand, C.J.; Batra, S.K.; Kung, H.J.; Bigner, D.D. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). Biochem. J. 1997, 324 Pt 3, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Chumbalkar, V.; Latha, K.; Hwang, Y.; Maywald, R.; Hawley, L.; Sawaya, R.; Diao, L.; Baggerly, K.; Cavenee, W.K.; Furnari, F.B.; et al. Analysis of phosphotyrosine signaling in glioblastoma identifies STAT5 as a novel downstream target of DeltaEGFR. J. Proteome Res. 2011, 10, 1343–1352. [Google Scholar] [CrossRef]
- Latha, K.; Li, M.; Chumbalkar, V.; Gururaj, A.; Hwang, Y.; Dakeng, S.; Sawaya, R.; Aldape, K.; Cavenee, W.K.; Bogler, O.; et al. Nuclear EGFRvIII-STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl-XL promoter. Int. J. Cancer 2012. [Google Scholar] [CrossRef]
- Pedersen, M.W.; Pedersen, N.; Damstrup, L.; Villingshoj, M.; Sonder, S.U.; Rieneck, K.; Bovin, L.F.; Spang-Thomsen, M.; Poulsen, H.S. Analysis of the epidermal growth factor receptor specific transcriptome: Effect of receptor expression level and an activating mutation. J. Cell. Biochem. 2005, 96, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Erdem-Eraslan, L.; Gao, Y.; Kloosterhof, N.K.; Atlasi, Y.; Demmers, J.; Sacchetti, A.; Kros, J.M.; Sillevis Smitt, P.; Aerts, J.; French, P.J. Mutation specific functions of EGFR result in a mutation-specific downstream pathway activation. Eur. J. Cancer 2015, 51, 893–903. [Google Scholar] [CrossRef]
- Lin, S.Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell. Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Mikula, M.; Skrzypczak, M.; Goryca, K.; Paczkowska, K.; Ledwon, J.K.; Statkiewicz, M.; Kulecka, M.; Grzelak, M.; Dabrowska, M.; Kuklinska, U.; et al. Genome-wide co-localization of active EGFR and downstream ERK pathway kinases mirrors mitogen-inducible RNA polymerase 2 genomic occupancy. Nucleic. Acids Res. 2016, 44, 10150–10164. [Google Scholar] [CrossRef] [PubMed]
- Liccardi, G.; Hartley, J.A.; Hochhauser, D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011, 71, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005, 7, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Marti, U.; Ruchti, C.; Kampf, J.; Thomas, G.A.; Williams, E.D.; Peter, H.J.; Gerber, H.; Burgi, U. Nuclear localization of epidermal growth factor and epidermal growth factor receptors in human thyroid tissues. Thyroid 2001, 11, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Gururaj, A.E.; Gibson, L.; Panchabhai, S.; Bai, M.; Manyam, G.; Lu, Y.; Latha, K.; Rojas, M.L.; Hwang, Y.; Liang, S.; et al. Access to the nucleus and functional association with c-Myc is required for the full oncogenic potential of DeltaEGFR/EGFRvIII. J. Biol. Chem. 2013, 288, 3428–3438. [Google Scholar] [CrossRef] [PubMed]
- Bollig-Fischer, A.; Chen, W.; Gadgeel, S.M.; Wenzlaff, A.S.; Cote, M.L.; Schwartz, A.G.; Bepler, G. Racial diversity of actionable mutations in non-small cell lung cancer. J. Thorac. Oncol. 2015, 10, 250–255. [Google Scholar] [CrossRef]
- Seo, J.S.; Ju, Y.S.; Lee, W.C.; Shin, J.Y.; Lee, J.K.; Bleazard, T.; Lee, J.; Jung, Y.J.; Kim, J.O.; Shin, J.Y.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Iyevleva, A.G.; Novik, A.V.; Moiseyenko, V.M.; Imyanitov, E.N. EGFR mutation in kidney carcinoma confers sensitivity to gefitinib treatment: A case report. Urol. Oncol. 2009, 27, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Masago, K.; Asato, R.; Fujita, S.; Hirano, S.; Tamura, Y.; Kanda, T.; Mio, T.; Katakami, N.; Mishima, M.; Ito, J. Epidermal growth factor receptor gene mutations in papillary thyroid carcinoma. Int. J. Cancer 2009, 124, 2744–2749. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.M.; Alpaugh, R.K.; Buell, J.K.; Stephens, P.J.; Yu, J.Q.; Wu, H.; Hiemstra, C.N.; Miller, V.A.; Lipson, D.; Palmer, G.A.; et al. Antitumor response of an ERBB2 amplified inflammatory breast carcinoma with EGFR mutation to the EGFR-TKI erlotinib. Clin. Breast Cancer 2014, 14, e14–e16. [Google Scholar] [CrossRef] [PubMed]
- Voss, J.S.; Holtegaard, L.M.; Kerr, S.E.; Fritcher, E.G.; Roberts, L.R.; Gores, G.J.; Zhang, J.; Highsmith, W.E.; Halling, K.C.; Kipp, B.R. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum. Pathol. 2013, 44, 1216–1222. [Google Scholar] [CrossRef]
- Agatsuma, N.; Yasuda, Y.; Ozasa, H. Malignant Pleural Mesothelioma Harboring Both G719C and S768I Mutations of EGFR Successfully Treated with Afatinib. J. Thorac. Oncol. 2017, 12, e141–e143. [Google Scholar] [CrossRef]
- Chi, A.S.; Batchelor, T.T.; Dias-Santagata, D.; Borger, D.; Stiles, C.D.; Wang, D.L.; Curry, W.T.; Wen, P.Y.; Ligon, K.L.; Ellisen, L.; et al. Prospective, high-throughput molecular profiling of human gliomas. J. Neurooncol. 2012, 110, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Byeon, S.; Kim, Y.; Lim, S.W.; Cho, J.H.; Park, S.; Lee, J.; Sun, J.M.; Choi, Y.L.; Lee, S.H.; Ahn, J.S.; et al. Clinical Outcomes of EGFR Exon 20 Insertion Mutations in Advanced Non-small Cell Lung Cancer in Korea. Cancer Res. Treat. 2018. [Google Scholar] [CrossRef]
- Yasuda, H.; Kobayashi, S.; Costa, D.B. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012, 13, e23–e31. [Google Scholar] [CrossRef]
- Hirano, T.; Yasuda, H.; Tani, T.; Hamamoto, J.; Oashi, A.; Ishioka, K.; Arai, D.; Nukaga, S.; Miyawaki, M.; Kawada, I.; et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget 2015, 6, 38789–38803. [Google Scholar] [CrossRef]
- Yang, M.; Xu, X.; Cai, J.; Ning, J.; Wery, J.P.; Li, Q.X. NSCLC harboring EGFR exon-20 insertions after the regulatory C-helix of kinase domain responds poorly to known EGFR inhibitors. Int. J. Cancer 2016, 139, 171–176. [Google Scholar] [CrossRef]
- Ruan, Z.; Kannan, N. Altered conformational landscape and dimerization dependency underpins the activation of EGFR by alphaC-beta4 loop insertion mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E8162–E8171. [Google Scholar] [CrossRef] [PubMed]
- Hasako, S.; Terasaka, M.; Abe, N.; Uno, T.; Ohsawa, H.; Hashimoto, A.; Fujita, R.; Tanaka, K.; Okayama, T.; Wadhwa, R.; et al. TAS6417, A Novel EGFR Inhibitor Targeting Exon 20 Insertion Mutations. Mol. Cancer Ther. 2018, 17, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Ross, H.J.; Blumenschein, G.R., Jr.; Aisner, J.; Damjanov, N.; Dowlati, A.; Garst, J.; Rigas, J.R.; Smylie, M.; Hassani, H.; Allen, K.E.; et al. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clin. Cancer Res. 2010, 16, 1938–1949. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Phase | Clinical Trial ID | Comparator | Histology | n | Ref. |
|---|---|---|---|---|---|---|
| Erl + TMZ/RT | I/II | NCT00039494 | single arm | GBM | 97 | [35] |
| Erl + Bev | II | NCT00671970 | single arm | GBM, AG | 57 | [36] |
| Erl + Soraf | II | NCT00445588 | single arm | rGBM | 56 | [37] |
| Erl + Bev + TMZ | II | NCT00525525 | single arm | GBM, GSC | 74 | [38] |
| Erl + TMZ | II | NCT00187486 | single arm | GBM, GLS | 65 | [39] |
| Erl | I/II | NCT00301418 | single arm | GBM, AA | 11 | [40] |
| Erl + Sirol | II | NCT00672243 | single arm | rGBM | 32 | [41] |
| Erl + Sirol | I/II | NCT00112736 | single arm | rGlioma | 69 | [42] |
| Erl | II | NCT00250887 | TMZ/BCNU | rGBM | 110 | [34] |
| Gef | II | NCT00250887 | single arm | rGBM | 22 | [43] |
| Gef + Cedir | II | NCT01310855 | Cedir | rGBM | 38 | [44] |
| Gef | II | NCT00016991 | Single arm | rGBM | 57 | [45] |
| Lap | I/II | NCT00099060 | single arm | rGBM | 17 | [46] |
| Afa | I/II | NCT00727506 | TMZ/afa, TMZ | rGBM | 119 | [47] |
| Dac | II | NCT01520870 | single arm | rGBM | 49 | [48] |
| Sun | II | NCT00923117 | Bev naïve/resistant | GBM | 72 | [49] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Vallentgoed, W.R.; French, P.J. Finding the Right Way to Target EGFR in Glioblastomas; Lessons from Lung Adenocarcinomas. Cancers 2018, 10, 489. https://doi.org/10.3390/cancers10120489
Gao Y, Vallentgoed WR, French PJ. Finding the Right Way to Target EGFR in Glioblastomas; Lessons from Lung Adenocarcinomas. Cancers. 2018; 10(12):489. https://doi.org/10.3390/cancers10120489
Chicago/Turabian StyleGao, Ya, Wies R. Vallentgoed, and Pim J. French. 2018. "Finding the Right Way to Target EGFR in Glioblastomas; Lessons from Lung Adenocarcinomas" Cancers 10, no. 12: 489. https://doi.org/10.3390/cancers10120489
APA StyleGao, Y., Vallentgoed, W. R., & French, P. J. (2018). Finding the Right Way to Target EGFR in Glioblastomas; Lessons from Lung Adenocarcinomas. Cancers, 10(12), 489. https://doi.org/10.3390/cancers10120489