Intronic miR-744 Inhibits Glioblastoma Migration by Functionally Antagonizing Its Host Gene MAP2K4


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
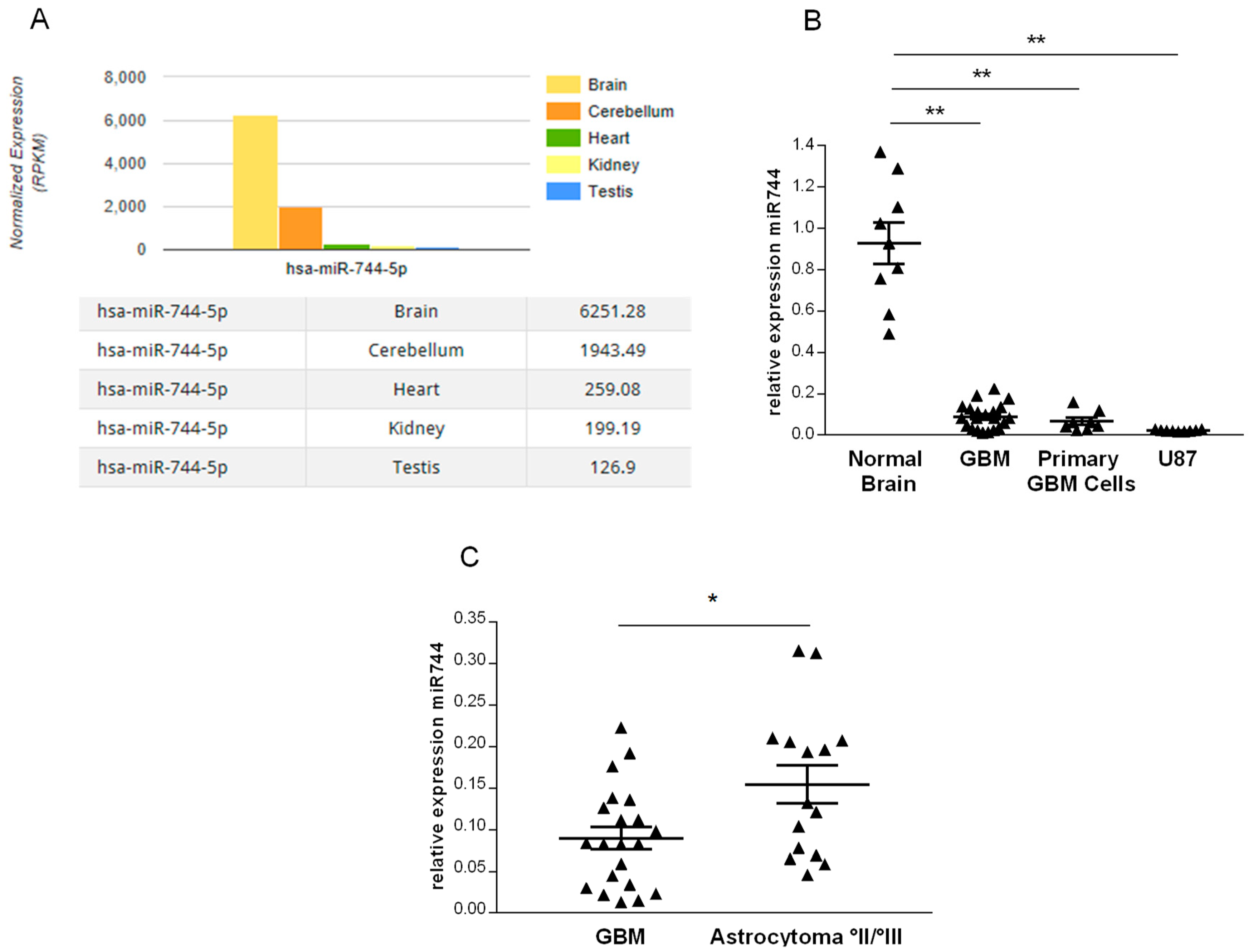
2.1. miR-744 Is Strongly Repressed in Human Glioma
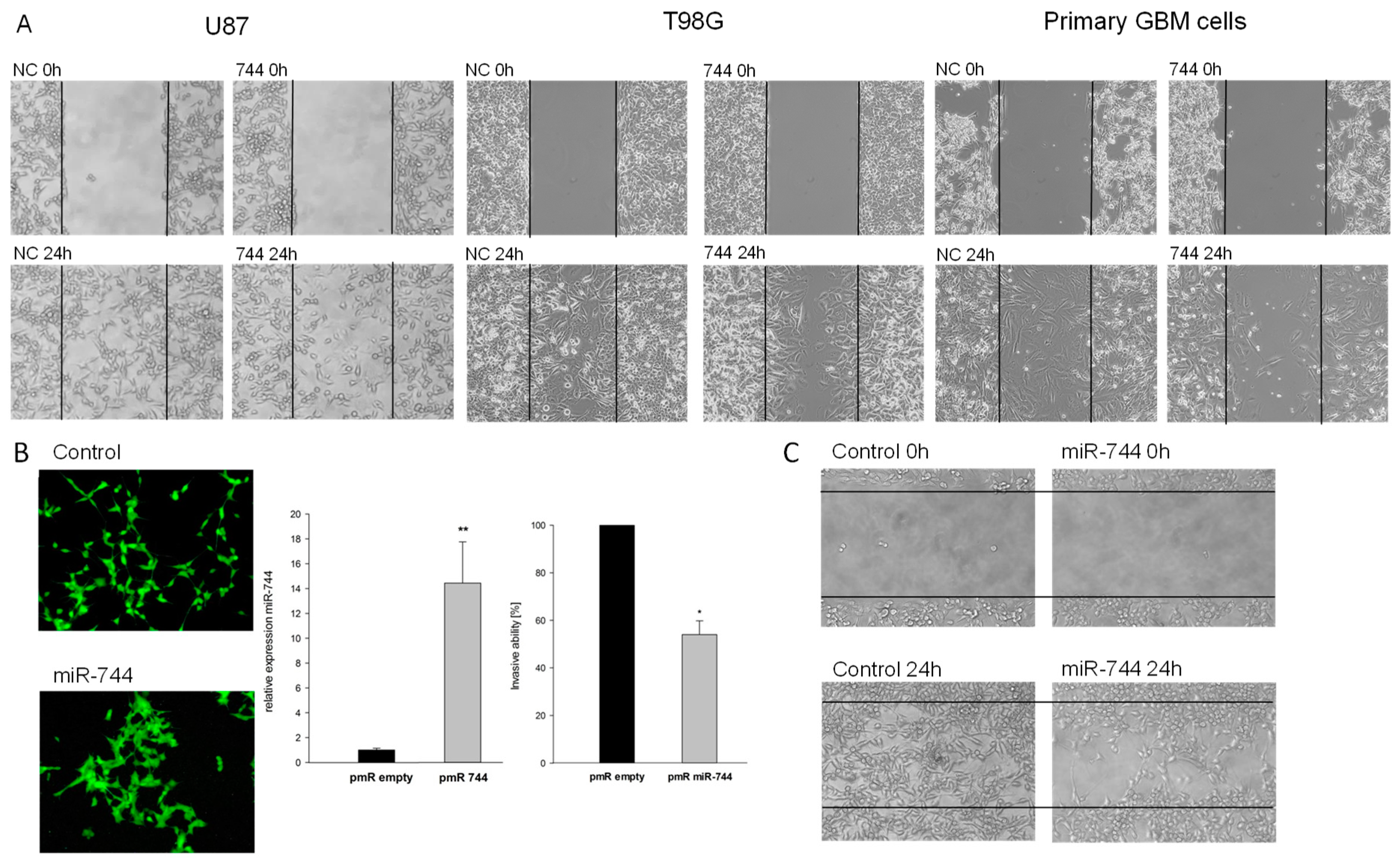
2.2. Overexpression of miR-744 Reduces Migration of GBM Cells
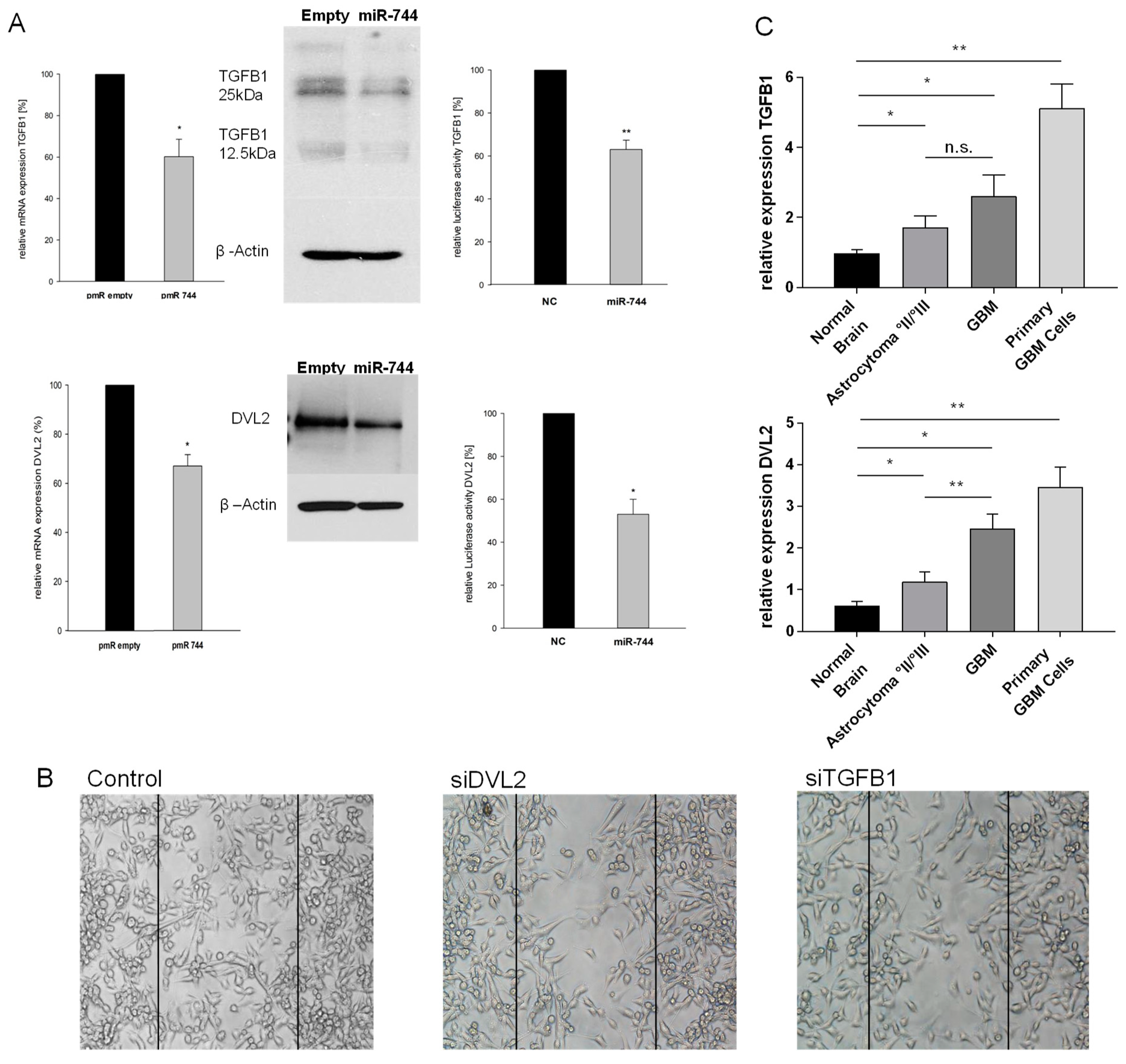
2.3. TGFB1 and DVL2 are Direct Targets of miR-744, Regulate Migration in GBM Cells, and are Induced in Tissue of Human Malignant Glioma
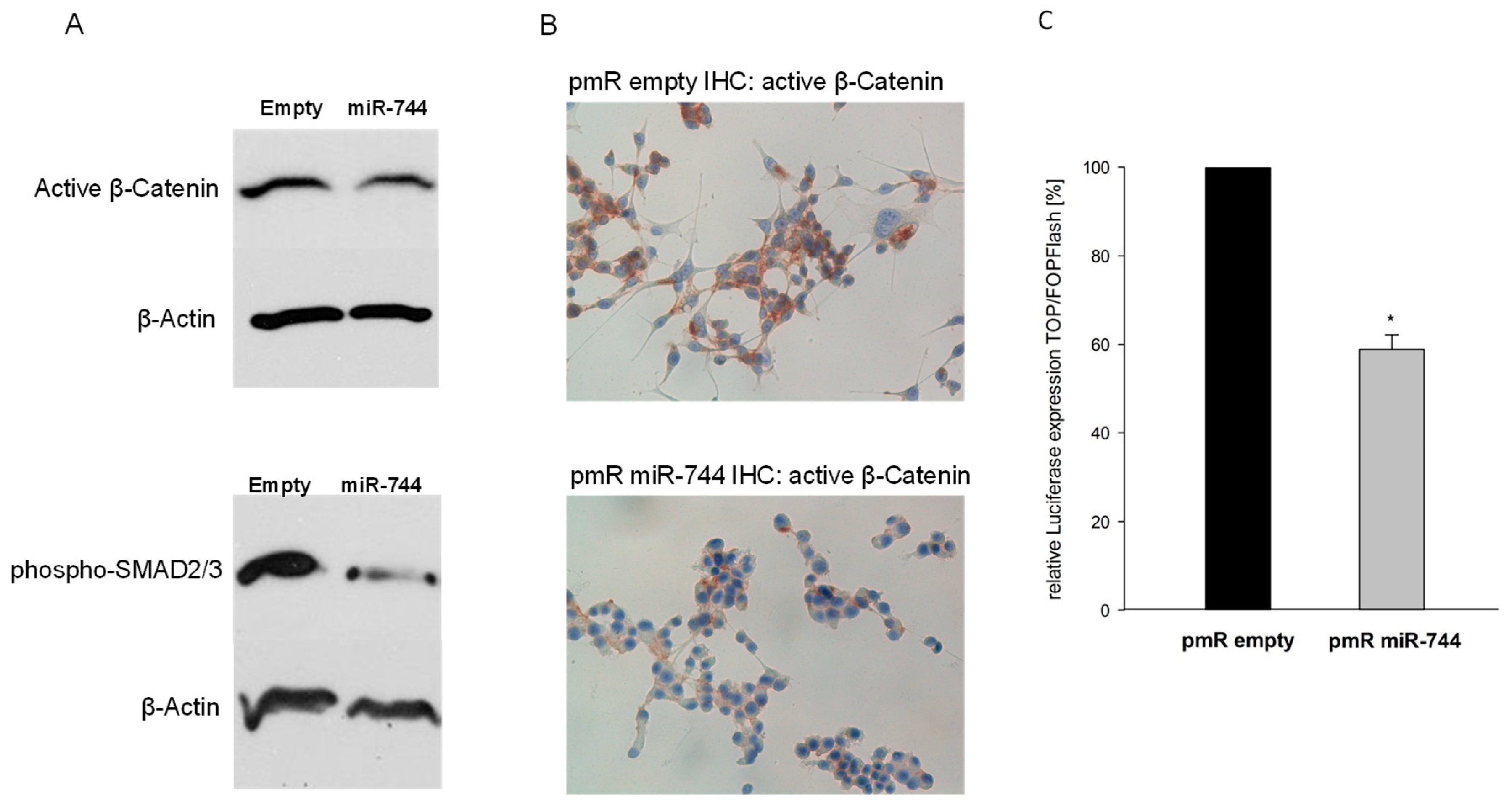
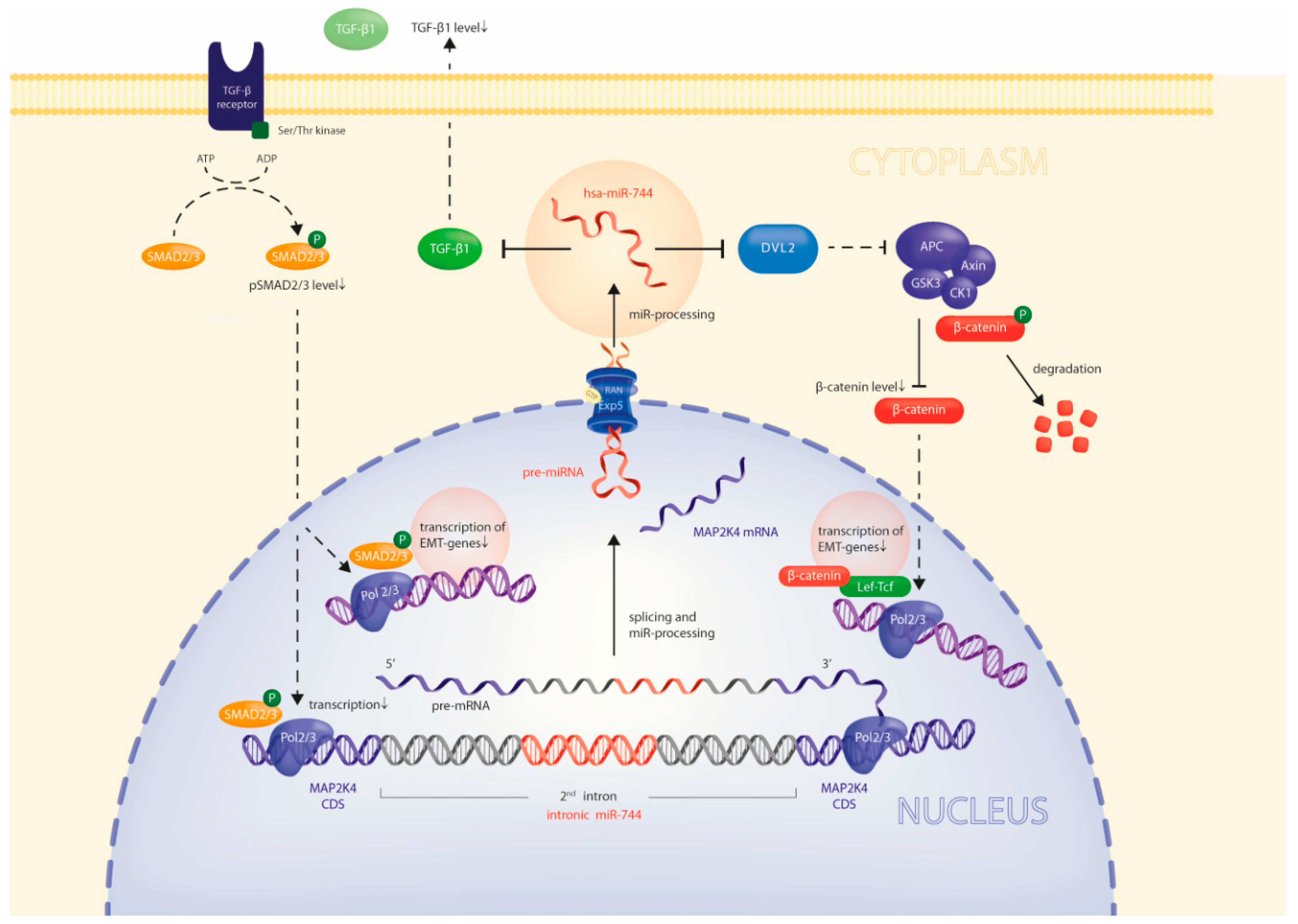
2.4. Via Repression of DVL2 and TGFB1, miR-744 Regulates Beta-Catenin and SMAD-Signaling Pathways
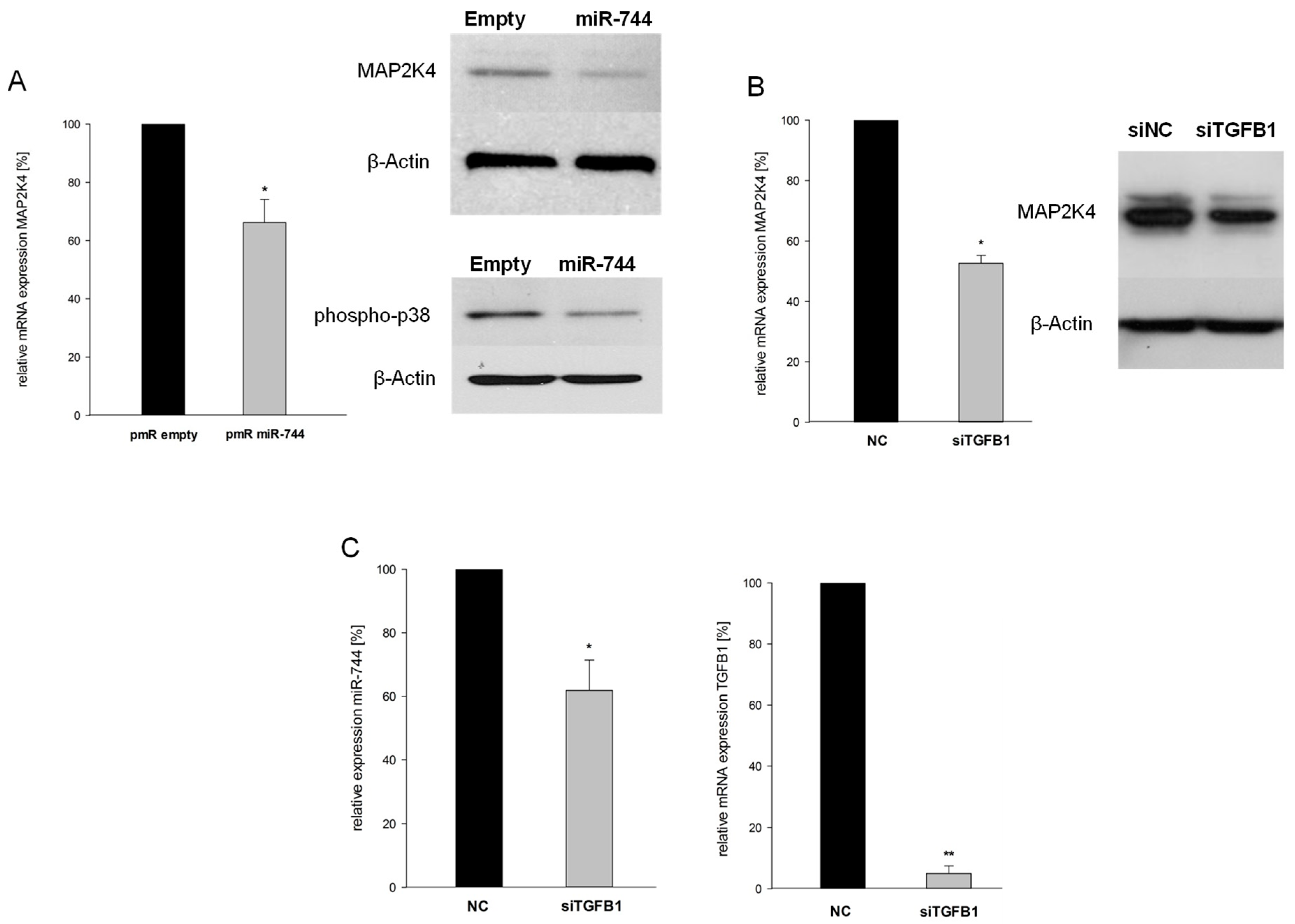
2.5. miR-744 Reduces It’s Host Gene MAP2K4 Through TGFB1-Mediated Negative Feedback
3. Discussion
4. Materials and Methods
4.1. Bioinformatics
4.2. Human Tissue Samples
4.3. RNA Extraction and cDNA Synthesis
4.4. Quantitative RT-PCR
4.5. Quantification of miRNA Expression
4.6. SDS-PAGE
4.7. Cell Culture
4.8. Cloning of Reporter Constructs
4.9. Cloning of miRNA Expression Vector
4.10. Transfections
4.11. Reporter Gene Assays
4.12. Migration Assays
4.13. Immunohistochemistry
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kreth, S.; Hübner, M.; Hinske, L.C. MicroRNAs as Clinical Biomarkers and Therapeutic Tools in Perioperative Medicine. Anesth. Analg. 2018, 126, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Hinske, L.C.; Dos Santos, F.R.C.; Ohara, D.T.; Ohno-Machado, L.; Kreth, S.; Galante, P.A.F. MiRIAD update: using alternative polyadenylation, protein interaction network analysis and additional species to enhance exploration of the role of intragenic miRNAs and their host genes. Database 2017. [Google Scholar] [CrossRef] [PubMed]
- Hinske, L.C.; França, G.S.; Torres, H.A.M.; Ohara, D.T.; Lopes-Ramos, C.M.; Heyn, J.; Reis, L.F.L.; Ohno-Machado, L.; Kreth, S.; Galante, P.A.F. miRIAD-integrating microRNA inter- and intragenic data. Database 2014. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Kim, V.N. Processing of intronic microRNAs. EMBO J. 2007, 26, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, D.C.; Madeira da Silva, L.; Zhang, W.; Liu, Z.; Arora, R.; Lim, S.; Schuler, A.M.; McClellan, S.; Andrews, J.F.; Kahn, A.G.; et al. ErbB2-intronic microRNA-4728: a novel tumor suppressor and antagonist of oncogenic MAPK signaling. Cell Death Dis. 2015, 6, e1742. [Google Scholar] [CrossRef] [PubMed]
- Hinske, L.C.; Heyn, J.; Hübner, M.; Rink, J.; Hirschberger, S.; Kreth, S. Intronic miRNA-641 controls its host Gene’s pathway PI3K/AKT and this relationship is dysfunctional in glioblastoma multiforme. Biochem. Biophys. Res. Commun. 2017, 489, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Pavese, J.M.; Ogden, I.M.; Voll, E.A.; Huang, X.; Xu, L.; Jovanovic, B.; Bergan, R.C. Mitogen-activated protein kinase kinase 4 (MAP2K4) promotes human prostate cancer metastasis. PLoS ONE 2014, 9, e102289. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pan, Y.; Dai, J.L. Evidence of MKK4 pro-oncogenic activity in breast and pancreatic tumors. Oncogene 2004, 23, 5978–5985. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gong, Z.; Sun, L.; Ma, L.; Wang, Q. MicroRNA-802 plays a tumour suppressive role in tongue squamous cell carcinoma through directly targeting MAP2K4. Cell Prolif. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Khatlani, T.S.; Wislez, M.; Sun, M.; Srinivas, H.; Iwanaga, K.; Ma, L.; Hanna, A.E.; Liu, D.; Girard, L.; Kim, Y.H.; et al. c-Jun N-terminal kinase is activated in non-small-cell lung cancer and promotes neoplastic transformation in human bronchial epithelial cells. Oncogene 2007, 26, 2658–2666. [Google Scholar] [CrossRef] [PubMed]
- Finegan, K.G.; Tournier, C. The mitogen-activated protein kinase kinase 4 has a pro-oncogenic role in skin cancer. Cancer Res. 2010, 70, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Pulvirenti, T.; Van Der Heijden, M.; Droms, L.A.; Huse, J.T.; Tabar, V.; Hall, A. Dishevelled 2 signaling promotes self-renewal and tumorigenicity in human gliomas. Cancer Res. 2011, 71, 7280–7290. [Google Scholar] [CrossRef] [PubMed]
- Frei, K.; Gramatzki, D.; Tritschler, I.; Schroeder, J.J.; Espinoza, L.; Rushing, E.J.; Weller, M. Transforming growth factor-β pathway activity in glioblastoma. Oncotarget 2015, 6, 5963–5977. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Mishra, B. TGF-beta, neuronal stem cells and glioblastoma. Oncogene 2005, 24, 5722–5730. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Lo Cascio, C. Developmentally regulated signaling pathways in glioma invasion. Cell. Mol. Life Sci. 2018, 75. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, J.-K.; Ahn, S.H.; Lee, J.; Nam, D.-H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Invest. 2016, 96, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Castro-Piedras, I.; Simmons, G.E., Jr.; Pruitt, K. Dishevelled: A masterful conductor of complex Wnt signals. Cell. Signal. 2018, 47, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Dill, H.; Linder, B.; Fehr, A.; Fischer, U. Intronic miR-26b controls neuronal differentiation by repressing its host transcript, ctdsp2. Genes Dev. 2012, 26, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Hinske, L.C.; Galante, P.A.F.; Limbeck, E.; Möhnle, P.; Parmigiani, R.B.; Ohno-Machado, L.; Camargo, A.A.; Kreth, S. Alternative polyadenylation allows differential negative feedback of human miRNA miR-579 on its host gene ZFR. PLoS ONE 2015, 10, e0121507. [Google Scholar] [CrossRef] [PubMed]
- Steiman-Shimony, A.; Shtrikman, O.; Margalit, H. Assessing the functional association of intronic miRNAs with their host genes. RNA 2018. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.-Z.; Xu, S.-L.; Xin, Y.-H.; Yu, S.-C.; Ping, Y.-F.; Chen, L.; Xiao, H.-L.; Wang, B.; Yi, L.; Wang, Q.-L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Tomar, T.; Eggens-Meijer, E.; Bhat, K.; Copray, S.; Walenkamp, A.M.E.; Boddeke, E.; Balasubramanyian, V.; Wagemakers, M.; et al. TGF-β is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014, 5, e1443. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 2001, 53, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.-E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocaña, A.; Peñuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; Seoane, J. High TGFβ-Smad Activity Confers Poor Prognosis in Glioma Patients and Promotes Cell Proliferation Depending on the Methylation of the PDGF-B Gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Seystahl, K.; Tritschler, I.; Szabo, E.; Tabatabai, G.; Weller, M. Differential regulation of TGF-β-induced, ALK-5-mediated VEGF release by SMAD2/3 versus SMAD1/5/8 signaling in glioblastoma. Neuro. Oncol. 2015, 17, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.-Q.; Wang, J.-Y.; Xi, Y.; Wu, Z.-L.; Li, Y.-P.; Li, L. Nuclear Dvl, c-Jun, β-catenin, and TCF form a complex leading to stabiLization of β-catenin–TCF interaction. J. Cell Biol. 2008, 180, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tian, Y.; Du, J.; Hu, Z.; Yang, L.; Liu, J.; Gu, L. Dvl2-dependent activation of Daam1 and RhoA regulates Wnt5a-induced breast cancer cell migration. PLoS ONE 2012, 7, e37823. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Mulherkar, S.; Mukherjee, D.; Jana, N.R. Malin regulates Wnt signaling pathway through degradation of dishevelled2. J. Biol. Chem. 2012, 287, 6830–6839. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G. Others Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, K.; Shi, Z.; Zou, J.; Wang, Y.; Jia, Z.; Zhang, A.; Han, L.; Yue, X.; Liu, N.; et al. High β-catenin/Tcf-4 activity confers glioma progression via direct regulation of AKT2 gene expression. Neuro. Oncol. 2011, 13, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Truscott, M.; Islam, A.B.M.M.K.; Lightfoot, J.; López-Bigas, N.; Frolov, M.V. An intronic microRNA links Rb/E2F and EGFR signaling. PLoS Genet. 2014, 10, e1004493. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Terra, L.F.; Wailemann, R.A.; Labriola, L.; Sogayar, M.C. TGF-β1 modulates the homeostasis between MMPs and MMP inhibitors through p38 MAPK and ERK1/2 in highly invasive breast cancer cells. BMC Cancer 2012, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Dziembowska, M.; Danilkiewicz, M.; Wesolowska, A.; Zupanska, A.; Chouaib, S.; Kaminska, B. Cross-talk between Smad and p38 MAPK signalling in transforming growth factor β signal transduction in human glioblastoma cells. Biochem. Biophys. Res. Commun. 2007, 354, 1101–1106. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hübner, M.; Hinske, C.L.; Effinger, D.; Wu, T.; Thon, N.; Kreth, F.-W.; Kreth, S. Intronic miR-744 Inhibits Glioblastoma Migration by Functionally Antagonizing Its Host Gene MAP2K4. Cancers 2018, 10, 400. https://doi.org/10.3390/cancers10110400
Hübner M, Hinske CL, Effinger D, Wu T, Thon N, Kreth F-W, Kreth S. Intronic miR-744 Inhibits Glioblastoma Migration by Functionally Antagonizing Its Host Gene MAP2K4. Cancers. 2018; 10(11):400. https://doi.org/10.3390/cancers10110400
Chicago/Turabian StyleHübner, Max, Christian Ludwig Hinske, David Effinger, Tingting Wu, Niklas Thon, Friedrich-Wilhelm Kreth, and Simone Kreth. 2018. "Intronic miR-744 Inhibits Glioblastoma Migration by Functionally Antagonizing Its Host Gene MAP2K4" Cancers 10, no. 11: 400. https://doi.org/10.3390/cancers10110400
APA StyleHübner, M., Hinske, C. L., Effinger, D., Wu, T., Thon, N., Kreth, F.-W., & Kreth, S. (2018). Intronic miR-744 Inhibits Glioblastoma Migration by Functionally Antagonizing Its Host Gene MAP2K4. Cancers, 10(11), 400. https://doi.org/10.3390/cancers10110400