Aberrant RNA Splicing in Cancer and Drug Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
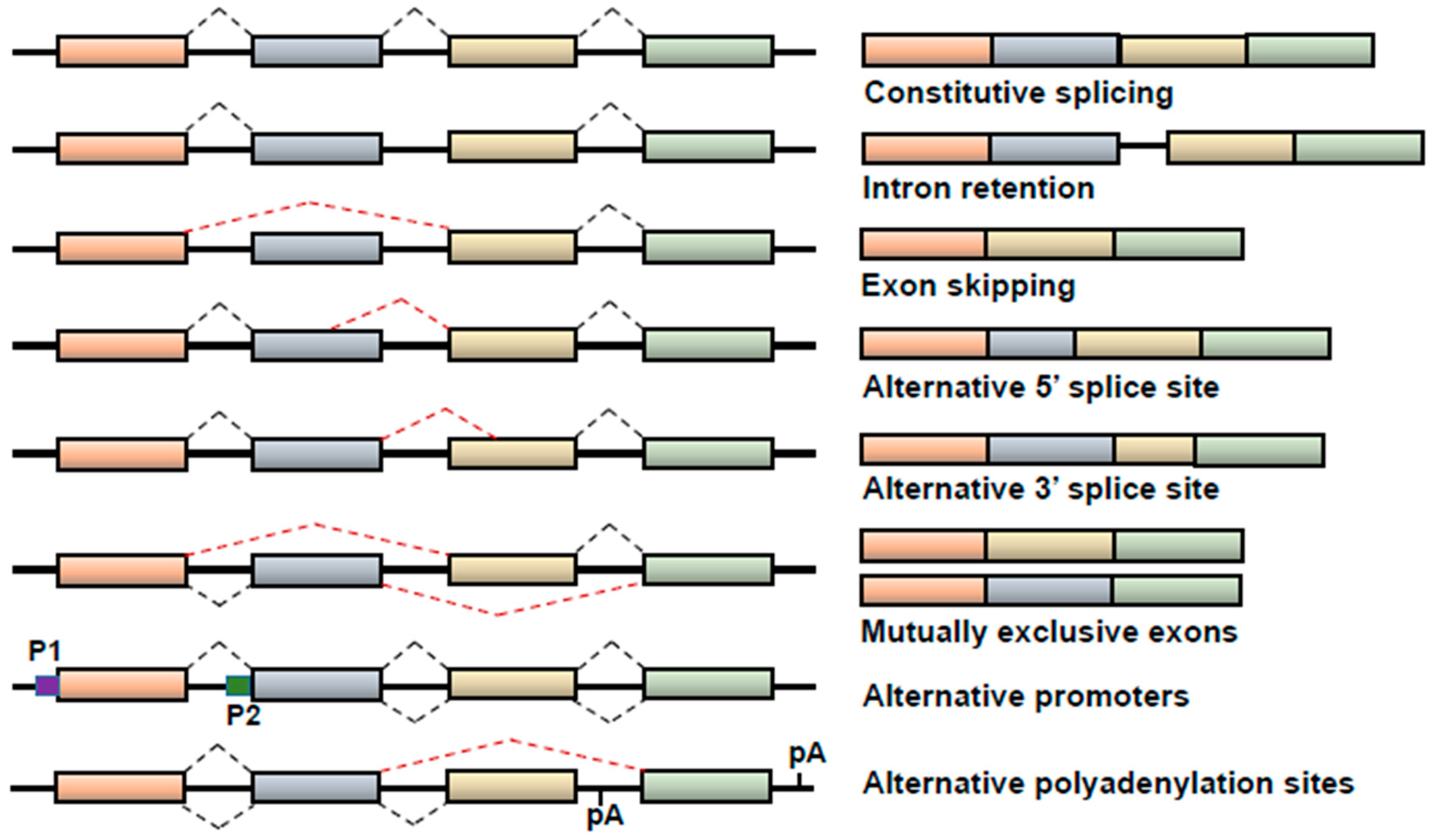
1. Alternative Splicing
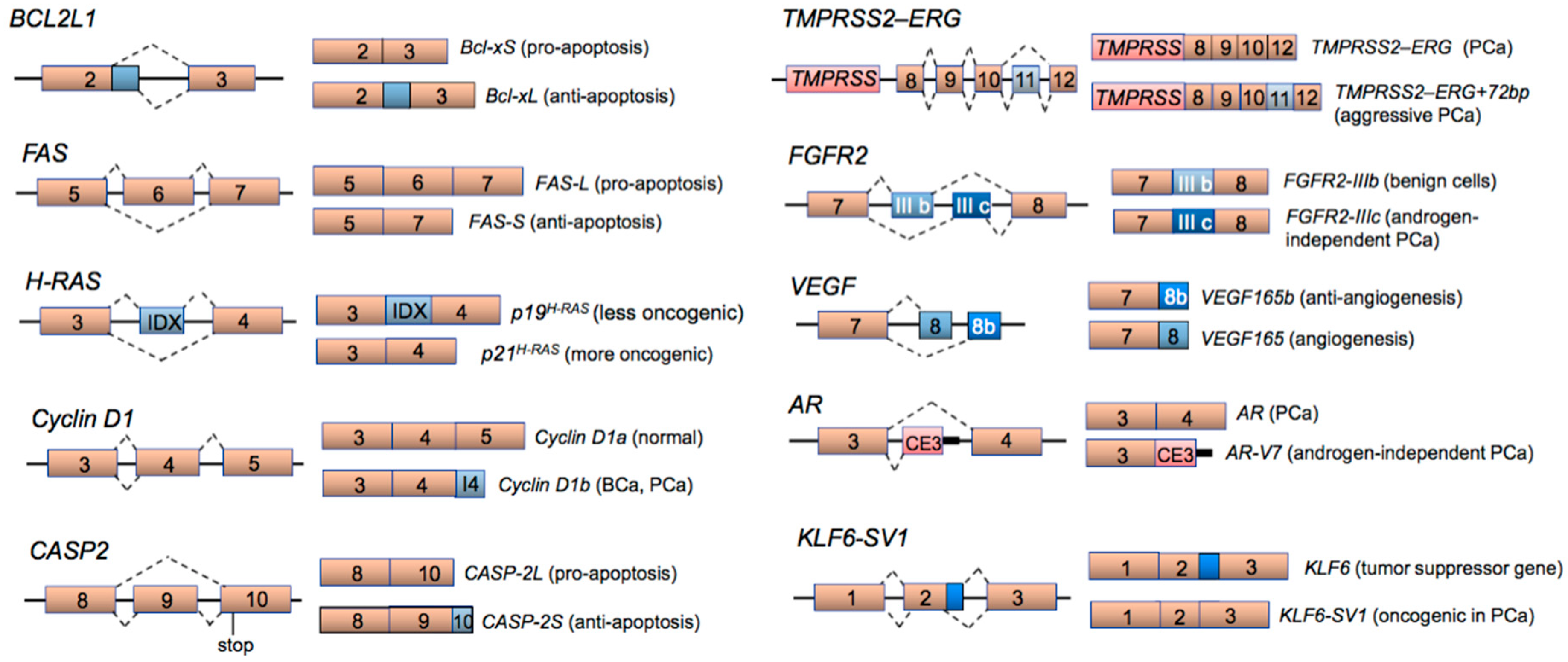
2. Aberrant Alternative Splicing in Cancer
3. Splicing Factors in Cancer Development and Progression
3.1. Aberrant Expression of Splicing Factors
3.2. Mutations in Splicing Factors
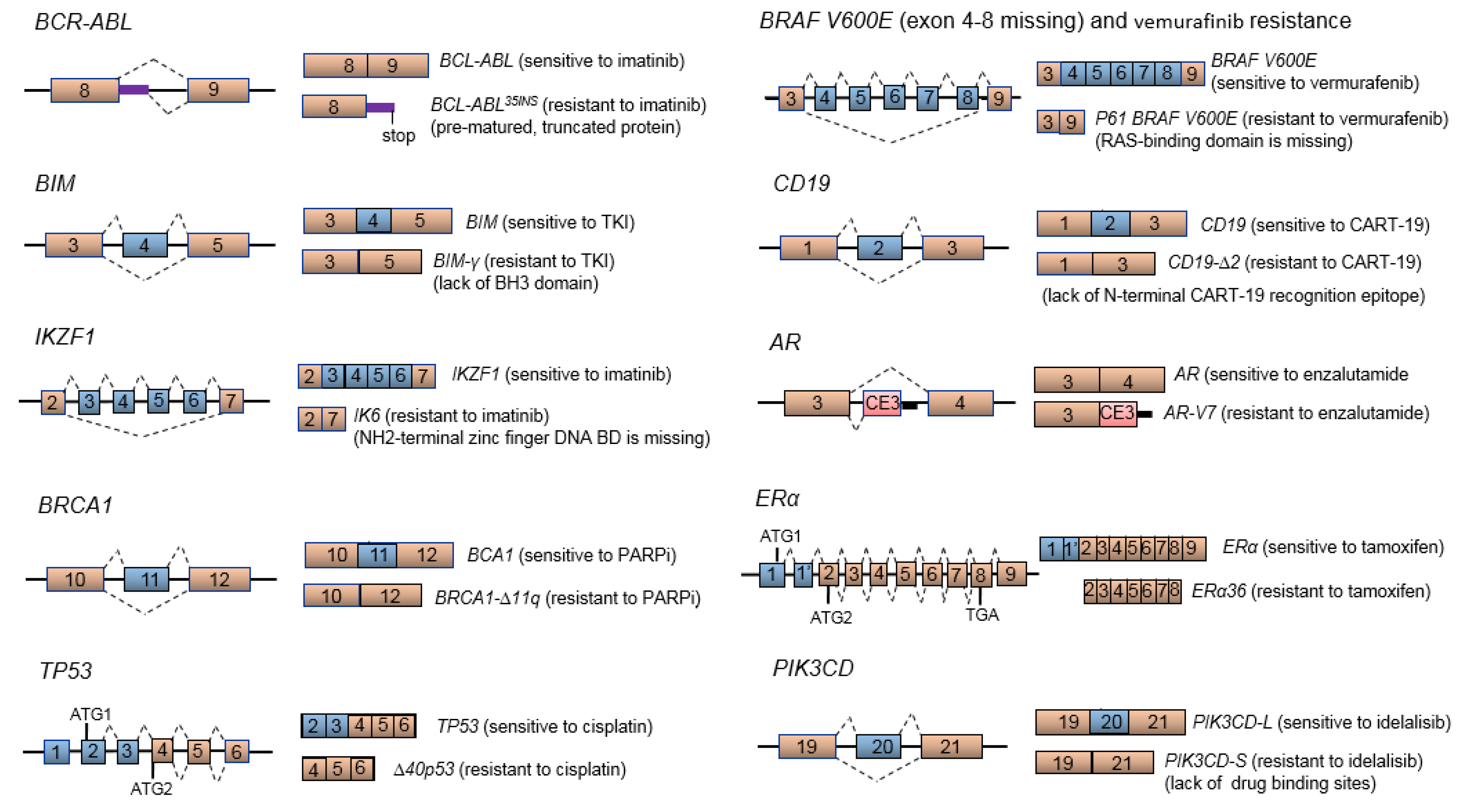
4. Aberrant mRNA Splicing and Cancer Drug Resistance
4.1. BCR-ABL Splice Variant and Imatinib Resistance
4.2. BCL2-Like 11 (BIM or BCL2L11) Splice Variant and TKI Resistance
4.3. BRCA Splice Variants Leading to PARP Inhibitor or Cytotoxic Drug Resistance
4.4. TP53 Splice Variants and Cisplatin Resistance
4.5. BRAF V600E Splice Variant and Vermurafenib Resistance
4.6. CD19 Splice Variant and CART-19 Immunotherapy Resistance in B-Cell Acute Lymphoblastic Leukemia
4.7. Truncated AR Variants and Androgen-Independent/Refractory Prostate Cancer Disease
4.8. ER Splice Variants and Tamoxifen Resistance
4.9. PIK3CD Splice Variant and Idelalisib Resistance
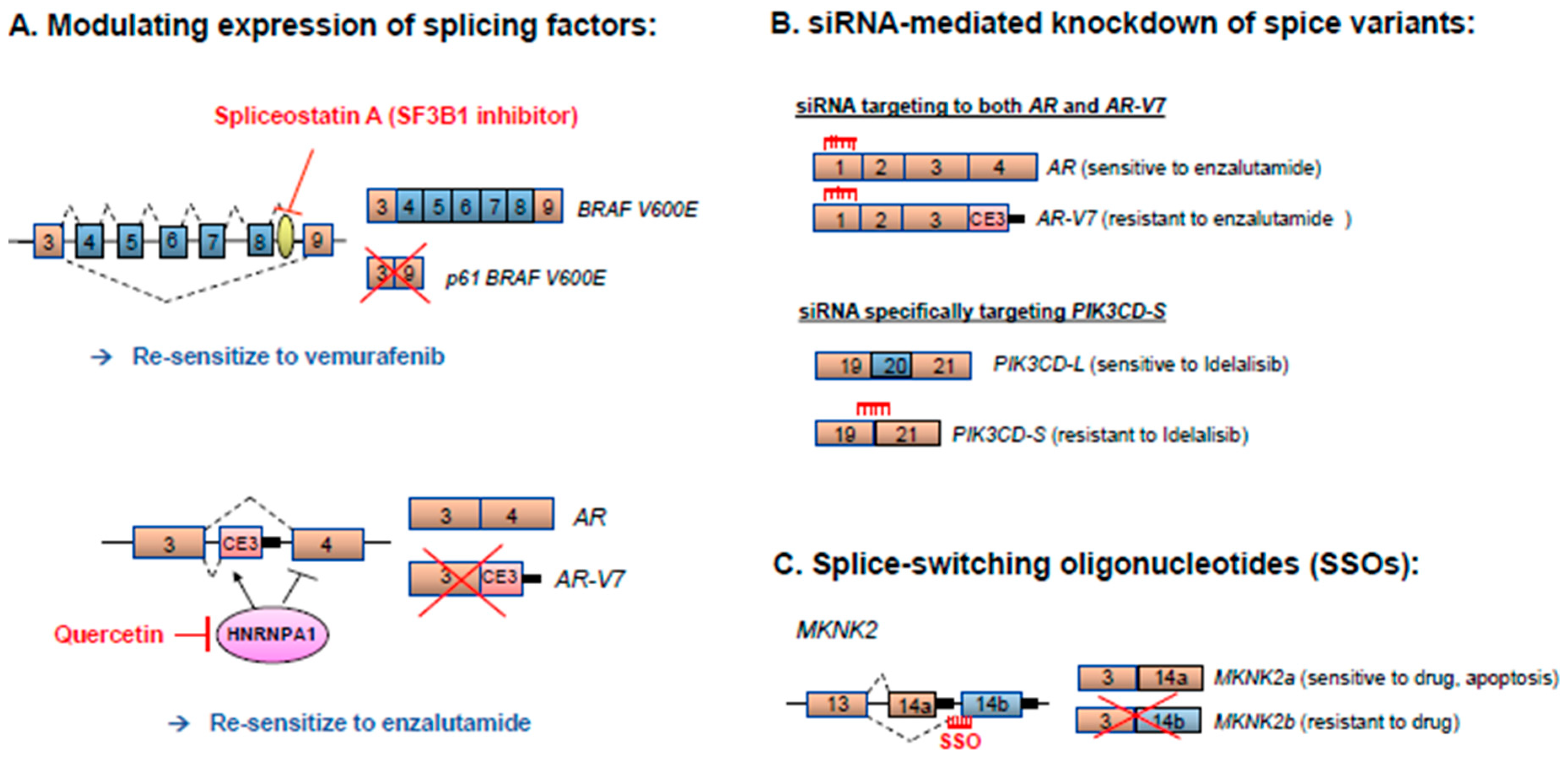
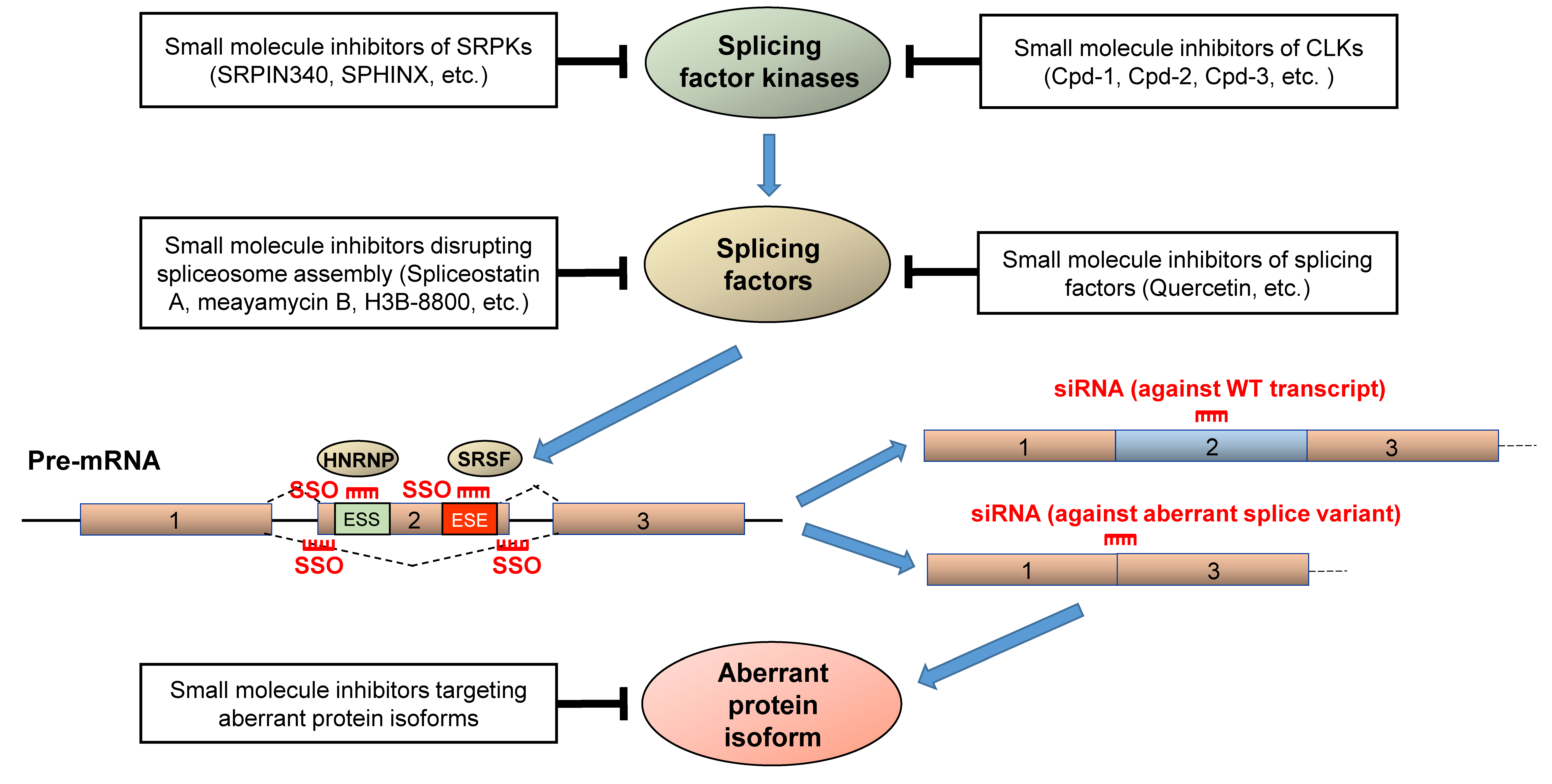
5. Therapeutic Strategies for Correcting Aberrant Splicing Errors
6. Concluding Remark and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Adams, M.D.; Kerlavage, A.R.; Fleischmann, R.D.; Fuldner, R.A.; Bult, C.J.; Lee, N.H.; Kirkness, E.F.; Weinstock, K.G.; Gocayne, J.D.; White, O.; et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature 1995, 377, 3–174. [Google Scholar] [PubMed]
- Hallegger, M.; Llorian, M.; Smith, C.W. Alternative splicing: Global insights. FEBS J. 2010, 277, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Sakharkar, M.K.; Chow, V.T.; Kangueane, P. Distributions of exons and introns in the human genome. In Silico Biol. 2004, 4, 387–393. [Google Scholar] [PubMed]
- Stamm, S.; Ben-Ari, S.; Rafalska, I.; Tang, Y.; Zhang, Z.; Toiber, D.; Thanaraj, T.A.; Soreq, H. Function of alternative splicing. Gene 2005, 344, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.A.; Schweitzer, A.C.; Chen, T.X.; Staples, M.K.; Lu, G.; Wang, H.; Williams, A.; Blume, J.E. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007, 8, R64. [Google Scholar] [CrossRef] [PubMed]
- De la Grange, P.; Gratadou, L.; Delord, M.; Dutertre, M.; Auboeuf, D. Splicing factor and exon profiling across human tissues. Nucleic Acids Res. 2010, 38, 2825–2838. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Benovoy, D.; Dias, C.; Gurd, S.; Provencher, C.; Beaulieu, P.; Hudson, T.J.; Sladek, R.; Majewski, J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008, 40, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Meth. 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.S.; Wang, E.T.; Vu, A.; David, M.P.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 2010, 38, 7651–7664. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.B.; Kawasawa, Y.I.; Mason, C.E.; Krsnik, Z.; Coppola, G.; Bogdanovic, D.; Geschwind, D.H.; Mane, S.M.; State, M.W.; Sestan, N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 2009, 62, 494–509. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R. Promoter usage and alternative splicing. Curr. Opin. Cell Biol. 2005, 17, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Rajan, P.; Elliott, D.J.; Robson, C.N.; Leung, H.Y. Alternative splicing and biological heterogeneity in prostate cancer. Nat. Rev. Urol. 2009, 6, 454–460. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P.; Klinck, R.; Koh, C.; Gervais-Bird, J.; Bramard, A.; Inkel, L.; Durand, M.; Couture, S.; Froehlich, U.; Lapointe, E.; et al. Cancer-associated regulation of alternative splicing. Nat. Struct. Mol. Biol. 2009, 16, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P. Unbalanced alternative splicing and its significance in cancer. Bioessays 2006, 28, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Skotheim, R.I.; Nees, M. Alternative splicing in cancer: Noise, functional, or systematic? Int. J. Biochem. Cell Biol. 2007, 39, 1432–1449. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Yocum, A.K.; Menon, R. Alternative splice variants, a new class of protein cancer biomarker candidates: Findings in pancreatic cancer and breast cancer with systems biology implications. Dis. Mark. 2010, 28, 241–251. [Google Scholar] [CrossRef]
- Srebrow, A.; Kornblihtt, A.R. The connection between splicing and cancer. J. Cell Sci. 2006, 119 Pt 13, 2635–2641. [Google Scholar] [CrossRef]
- Germann, S.; Gratadou, L.; Dutertre, M.; Auboeuf, D. Splicing programs and cancer. J. Nucleic Acids 2012, 2012, 269570. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Vagner, S.; Auboeuf, D. Alternative splicing and breast cancer. RNA Biol. 2010, 7, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P. Aberrant and alternative splicing in cancer. Cancer Res. 2004, 64, 7647–7654. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; Valacca, C.; Biamonti, G. Alternative splicing and tumor progression. Curr. Genom. 2008, 9, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; Gonzalez-Garcia, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nunez, G.; Thompson, C.B. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Akgul, C.; Moulding, D.A.; Edwards, S.W. Alternative splicing of Bcl-2-related genes: Functional consequences and potential therapeutic applications. Cell. Mol. Life Sci. 2004, 61, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Boise, L.H.; Thompson, C.B. Bcl-x(S) anatagonizes the protective effects of Bcl-x(L). J. Biol. Chem. 1996, 271, 6306–6312. [Google Scholar] [CrossRef] [PubMed]
- Olopade, O.I.; Adeyanju, M.O.; Safa, A.R.; Hagos, F.; Mick, R.; Thompson, C.B.; Recant, W.M. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J. Sci. Am. 1997, 3, 230–237. [Google Scholar] [PubMed]
- Takehara, T.; Liu, X.; Fujimoto, J.; Friedman, S.L.; Takahashi, H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001, 34, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 2002, 277, 49374–49382. [Google Scholar] [CrossRef] [PubMed]
- Bouillet, P.; O’Reilly, L.A. CD95, BIM and T cell homeostasis. Nat. Rev. Immunol. 2009, 9, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhou, T.; Liu, C.; Shapiro, J.P.; Brauer, M.J.; Kiefer, M.C.; Barr, P.J.; Mountz, J.D. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science 1994, 263, 1759–1762. [Google Scholar] [CrossRef] [PubMed]
- Cascino, I.; Fiucci, G.; Papoff, G.; Ruberti, G. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J. Immunol. 1995, 154, 2706–2713. [Google Scholar] [PubMed]
- Poh, T.W.; Pervaiz, S. LY294002 and LY303511 sensitize tumor cells to drug-induced apoptosis via intracellular hydrogen peroxide production independent of the phosphoinositide 3-kinase-Akt pathway. Cancer Res. 2005, 65, 6264–6274. [Google Scholar] [CrossRef] [PubMed]
- Tamm, C.; Zhivotovsky, B.; Ceccatelli, S. Caspase-2 activation in neural stem cells undergoing oxidative stress-induced apoptosis. Apoptosis 2008, 13, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Bonzon, C.; Bouchier-Hayes, L.; Pagliari, L.J.; Green, D.R.; Newmeyer, D.D. Caspase-2-induced apoptosis requires bid cleavage: A physiological role for bid in heat shock-induced death. Mol. Biol. Cell 2006, 17, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Logette, E.; Desoche, L.; Solary, E.; Corcos, L. Nonsense-mediated mRNA decay among human caspases: The caspase-2S putative protein is encoded by an extremely short-lived mRNA. Cell Death Differ. 2005, 12, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Droin, N.; Beauchemin, M.; Solary, E.; Bertrand, R. Identification of a caspase-2 isoform that behaves as an endogenous inhibitor of the caspase cascade. Cancer Res. 2000, 60, 7039–7047. [Google Scholar] [PubMed]
- Martinet, W.; Knaapen, M.W.; De Meyer, G.R.; Herman, A.G.; Kockx, M.M. Overexpression of the anti-apoptotic caspase-2 short isoform in macrophage-derived foam cells of human atherosclerotic plaques. Am. J. Pathol. 2003, 162, 731–736. [Google Scholar] [CrossRef]
- Parent, N.; Sane, A.T.; Droin, N.; Bertrand, R. Procaspase-2S inhibits procaspase-3 processing and activation, preventing ROCK-1-mediated apoptotic blebbing and body formation in human B lymphoma Namalwa cells. Apoptosis 2005, 10, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cai, Y.; Yu, W.; Ren, C.; Spencer, D.M.; Ittmann, M. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG fusion gene transcripts. Cancer Res. 2008, 68, 8516–8524. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Bjartell, A.; Chinnaiyan, A.M.; Jenster, G.; Nam, R.K.; Rubin, M.A.; Schalken, J.A. ETS gene fusions in prostate cancer: From discovery to daily clinical practice. Eur. Urol. 2009, 56, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.B.; Broz, S.D.; Levinson, A.D. Expression of the H-ras proto-oncogene is controlled by alternative splicing. Cell 1989, 58, 461–472. [Google Scholar] [CrossRef]
- Knudsen, K.E.; Diehl, J.A.; Haiman, C.A.; Knudsen, E.S. Cyclin D1: Polymorphism, aberrant splicing and cancer risk. Oncogene 2006, 25, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant expression of cyclin D1 in cancer. Sign. Transduct. Insights 2015, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, C.; Jiao, X.; Katiyar, S.; Casimiro, M.C.; Prendergast, G.C.; Powell, M.J.; Pestell, R.G. Alternate cyclin D1 mRNA splicing modulates p27KIP1 binding and cell migration. J. Biol. Chem. 2008, 283, 7007–7015. [Google Scholar] [CrossRef] [PubMed]
- Millar, E.K.; Dean, J.L.; McNeil, C.M.; O’Toole, S.A.; Henshall, S.M.; Tran, T.; Lin, J.; Quong, A.; Comstock, C.E.; Witkiewicz, A.; et al. Cyclin D1b protein expression in breast cancer is independent of cyclin D1a and associated with poor disease outcome. Oncogene 2009, 28, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dean, J.L.; Millar, E.K.; Tran, T.H.; McNeil, C.M.; Burd, C.J.; Henshall, S.M.; Utama, F.E.; Witkiewicz, A.; Rui, H.; et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res. 2008, 68, 5628–5638. [Google Scholar] [CrossRef] [PubMed]
- Gunthert, U.; Hofmann, M.; Rudy, W.; Reber, S.; Zoller, M.; Haussmann, I.; Matzku, S.; Wenzel, A.; Ponta, H.; Herrlich, P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 1991, 65, 13–24. [Google Scholar] [CrossRef]
- Sahadevan, K.; Darby, S.; Leung, H.Y.; Mathers, M.E.; Robson, C.N.; Gnanapragasam, V.J. Selective over-expression of fibroblast growth factor receptors 1 and 4 in clinical prostate cancer. J. Pathol. 2007, 213, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Gnanapragasam, V.J.; Robinson, M.C.; Marsh, C.; Robson, C.N.; Hamdy, F.C.; Leung, H.Y. FGF8 isoform b expression in human prostate cancer. Br. J. Cancer 2003, 88, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Woolard, J.; Wang, W.Y.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.G.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: Mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.J.; Ezponda, T.; Catena, R.; Calvo, A.; Pio, R.; Montuenga, L.M. Alternative splicing: An emerging topic in molecular and clinical oncology. Lancet Oncol. 2007, 8, 349–357. [Google Scholar] [CrossRef]
- Narla, G.; DiFeo, A.; Yao, S.; Banno, A.; Hod, E.; Reeves, H.L.; Qiao, R.F.; Camacho-Vanegas, O.; Levine, A.; Kirschenbaum, A.; et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005, 65, 5761–5768. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Delineation of prognostic biomarkers in prostate cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Narla, G.; DiFeo, A.; Fernandez, Y.; Dhanasekaran, S.; Huang, F.; Sangodkar, J.; Hod, E.; Leake, D.; Friedman, S.L.; Hall, S.J.; et al. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. J. Clin. Investig. 2008, 118, 2711–2721. [Google Scholar] [CrossRef] [PubMed]
- Maglic, D.; Stovall, D.B.; Cline, J.M.; Fry, E.A.; Mallakin, A.; Taneja, P.; Caudell, D.L.; Willingham, M.C.; Sui, G.; Inoue, K. DMP1beta, a splice isoform of the tumour suppressor DMP1 locus, induces proliferation and progression of breast cancer. J. Pathol. 2015, 236, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant splicing of the DMP1-ARF-MDM2-p53 pathway in cancer. Int. J. Cancer 2016, 139, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhu, Z.; Luo, J.; Fang, J.; Zhou, H.; Hu, M.; Maskey, N.; Yang, G. Impact of chemokine receptor CXCR3 on tumor-infiltrating lymphocyte recruitment associated with favorable prognosis in advanced gastric cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 14725–14732. [Google Scholar] [PubMed]
- Zhu, G.; Yan, H.H.; Pang, Y.; Jian, J.; Achyut, B.R.; Liang, X.; Weiss, J.M.; Wiltrout, R.H.; Hollander, M.C.; Yang, L. CXCR3 as a molecular target in breast cancer metastasis: Inhibition of tumor cell migration and promotion of host anti-tumor immunity. Oncotarget 2015, 6, 43408–43419. [Google Scholar] [CrossRef] [PubMed]
- Berchiche, Y.A.; Sakmar, T.P. CXC Chemokine Receptor 3 Alternative Splice Variants Selectively Activate Different Signaling Pathways. Mol. Pharmacol. 2016, 90, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Van der Feltz, C.; Anthony, K.; Brilot, A.; Pomeranz Krummel, D.A. Architecture of the spliceosome. Biochemistry 2012, 51, 3321–3333. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.A.; Yu, L.; Smith, P.G.; Buonamici, S. Targeting splicing abnormalities in cancer. Curr. Opin. Genet. Dev. 2018, 48, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 2001, 13, 290–301. [Google Scholar] [CrossRef]
- Graveley, B.R. Sorting out the complexity of SR protein functions. RNA 2000, 6, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.F.; Revil, T.; Chabot, B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007, 623, 123–147. [Google Scholar] [PubMed]
- Zhang, J.; Manley, J.L. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013, 3, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Grosso, A.R.; Martins, S.; Carmo-Fonseca, M. The emerging role of splicing factors in cancer. EMBO Rep. 2008, 9, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, H.; Piekielko-Witkowska, A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017, 396, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.C.; Noack, K.; Runnebaum, I.B.; Watermann, D.O.; Kieback, D.G.; Stamm, S.; Stickeler, E. Expression of splicing factors in human ovarian cancer. Oncol. Rep. 2004, 11, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Sun, Y.; Ding, J.H.; Lin, S.; Rose, D.W.; Rosenfeld, M.G.; Fu, X.D.; Li, X. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol. Cell. Biol. 2007, 27, 5393–5402. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Liao, W.; Zhou, X.; Wong, D.T.; Lichtenstein, A. Exon 11 skipping of E-cadherin RNA downregulates its expression in head and neck cancer cells. Mol. Cancer Ther. 2011, 10, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Li, C.; McCoy, J.P.; Deng, C.X.; Zheng, Z.M. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int. J. Biol. Sci. 2010, 6, 806–826. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.A.; Wilkinson, J.E.; Krainer, A.R. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat. Struct. Mol. Biol. 2014, 21, 189–197. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Ee, P.L.; Coon, J.S.; Beck, W.T. Alternative splicing of the multidrug resistance protein 1/ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20. Clin. Cancer Res. 2004, 10, 4652–4660. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Andersen, C.L.; Thorsen, K.; Orntoft, T.F.; Mu, D.; Karni, R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 2013, 229, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Akaike, Y.; Masuda, K.; Kuwano, Y.; Nishida, K.; Yamagishi, N.; Kajita, K.; Tanahashi, T.; Rokutan, K. Downregulation of serine/arginine-rich splicing factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells. Oncogene 2014, 33, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Patry, C.; Bouchard, L.; Labrecque, P.; Gendron, D.; Lemieux, B.; Toutant, J.; Lapointe, E.; Wellinger, R.; Chabot, B. Small interfering RNA-mediated reduction in heterogeneous nuclear ribonucleoparticule A1/A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res. 2003, 63, 7679–7688. [Google Scholar] [PubMed]
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.S.; Bakacs, A.; Coppola, L.; Karni, R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011, 71, 4464–4472. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Manley, J.L. Turning on a fuel switch of cancer: HnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010, 70, 8977–8980. [Google Scholar] [CrossRef] [PubMed]
- Balasubramani, M.; Day, B.W.; Schoen, R.E.; Getzenberg, R.H. Altered expression and localization of creatine kinase B, heterogeneous nuclear ribonucleoprotein F, and high mobility group box 1 protein in the nuclear matrix associated with colon cancer. Cancer Res. 2006, 66, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Minana, B.; Valcarcel, J.; Gabaldon, T.; Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Saez, B.; Walter, M.J.; Graubert, T.A. Splicing factor gene mutations in hematologic malignancies. Blood 2017, 129, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.A.; Cote, G.; Quintas-Cardama, A. The changing mutational landscape of acute myeloid leukemia and myelodysplastic syndrome. Mol. Cancer Res. 2013, 11, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J.; Dolatshad, H.; Varanasi, S.S.; Yip, B.H.; Pellagatti, A. The role of splicing factor mutations in the pathogenesis of the myelodysplastic syndromes. Adv. Biol. Regul. 2014, 54, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 2011, 118, 6239–6246. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kade, S.; Schlarmann, C.; Loffeld, P.; Morgan, M.; Krauter, J.; Wlodarski, M.W.; Kolking, B.; Wichmann, M.; Gorlich, K.; et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012, 119, 3578–3584. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhu, Y.; Liu, Z.J.; Ouyang, S. The emerging roles of the DDX41 protein in immunity and diseases. Protein Cell 2017, 8, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Lewinsohn, M.; Brown, A.L.; Weinel, L.M.; Phung, C.; Rafidi, G.; Lee, M.K.; Schreiber, A.W.; Feng, J.; Babic, M.; Chong, C.E.; et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood 2016, 127, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chng, W.J. Aberrant RNA splicing and mutations in spliceosome complex in acute myeloid leukemia. Stem Cell Investig. 2017, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Daubner, G.M.; Clery, A.; Jayne, S.; Stevenin, J.; Allain, F.H. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012, 31, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lieu, Y.K.; Ali, A.M.; Penson, A.; Reggio, K.S.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 2015, 112, E4726–E4734. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Hochhaus, A.; Branford, S.; Muller, M.C.; Kaeda, J.S.; Foroni, L.; Druker, B.J.; Guilhot, F.; Larson, R.A.; O’Brien, S.G.; et al. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: An analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood 2010, 116, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Itonaga, H.; Tsushima, H.; Imanishi, D.; Hata, T.; Doi, Y.; Mori, S.; Sasaki, D.; Hasegawa, H.; Matsuo, E.; Nakashima, J.; et al. Molecular analysis of the BCR-ABL1 kinase domain in chronic-phase chronic myelogenous leukemia treated with tyrosine kinase inhibitors in practice: Study by the Nagasaki CML Study Group. Leuk. Res. 2014, 38, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Berman, E.; Jhanwar, S.; Hedvat, C.; Arcila, M.E.; Wahab, O.A.; Levine, R.; Maloy, M.; Ma, W.; Albitar, M. Resistance to imatinib in patients with chronic myelogenous leukemia and the splice variant BCR-ABL1(35INS). Leuk. Res. 2016, 49, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, J.B.; Arnould, C.; Bravo, S.; Donadio, D.; Exbrayat, C.; Jourdan, E.; Reboul, D.; Chiesa, J.; Lavabre-Bertrand, T. Exon 7 deletion in the bcr-abl gene is frequent in chronic myeloid leukemia patients and is not correlated with resistance against imatinib. Mol. Cancer Ther. 2010, 9, 3083–3089. [Google Scholar] [CrossRef] [PubMed]
- Laudadio, J.; Deininger, M.W.; Mauro, M.J.; Druker, B.J.; Press, R.D. An intron-derived insertion/truncation mutation in the BCR-ABL kinase domain in chronic myeloid leukemia patients undergoing kinase inhibitor therapy. J. Mol. Diagn. 2008, 10, 177–180. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Zabriskie, M.S.; Eide, C.A.; Agarwal, A.; Adrian, L.T.; You, H.; Corbin, A.S.; Yang, F.; Press, R.D.; Rivera, V.M.; et al. The BCR-ABL35INS insertion/truncation mutant is kinase-inactive and does not contribute to tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Blood 2011, 118, 5250–5254. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Ma, W.; Zhang, X.; Giles, F.; Cortes, J.; Kantarjian, H.; Albitar, M. BCR-ABL alternative splicing as a common mechanism for imatinib resistance: Evidence from molecular dynamics simulations. Mol. Cancer Ther. 2008, 7, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Fedor, H.L.; Lotan, T.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, Z.; Karni, R. The role of alternative splicing in cancer drug resistance. Curr. Opin. Genet. Dev. 2018, 48, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.; Juan, W.C.; Ko, T.K.; Teo, A.S.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Puthalakath, H.; Cragg, M.S.; Kelly, P.N.; Bouillet, P.; Huang, D.C.; Kimura, S.; Ottmann, O.G.; Druker, B.J.; Villunger, A.; et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc. Natl. Acad. Sci. USA 2006, 103, 14907–14912. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.K.; Chin, H.S.; Chuah, C.T.; Huang, J.W.; Ng, K.P.; Khaw, S.L.; Huang, D.C.; Ong, S.T. The BIM deletion polymorphism: A paradigm of a permissive interaction between germline and acquired TKI resistance factors in chronic myeloid leukemia. Oncotarget 2016, 7, 2721–2733. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bhadra, M.; Sinnakannu, J.R.; Yue, W.L.; Tan, C.W.; Rigo, F.; Ong, S.T.; Roca, X. Overcoming imatinib resistance conferred by the BIM deletion polymorphism in chronic myeloid leukemia with splice-switching antisense oligonucleotides. Oncotarget 2017, 8, 77567–77585. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Cui, T.Y.; Jasin, M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001, 61, 4842–4850. [Google Scholar] [PubMed]
- Scully, R.; Chen, J.; Ochs, R.L.; Keegan, K.; Hoekstra, M.; Feunteun, J.; Livingston, D.M. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997, 90, 425–435. [Google Scholar] [CrossRef]
- Friedman, L.S.; Ostermeyer, E.A.; Szabo, C.I.; Dowd, P.; Lynch, E.D.; Rowell, S.E.; King, M.C. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat. Genet. 1994, 8, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.I.; King, M.C. Inherited breast and ovarian cancer. Hum. Mol. Genet. 1995, 4, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Srihari, S.; Cao, K.A.; Chenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine-scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, A.; Sharip, A.; Sharip, A.; Jiang, J.; Yang, Q.; Xie, Y. Reverse the Resistance to PARP Inhibitors. Int. J. Biol. Sci. 2017, 13, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Easton, D.; Breast Cancer Linkage Consortium. Variation in BRCA1 cancer risks by mutation position. Cancer Epidemiol. Biomark. Prev. 2002, 11, 329–336. [Google Scholar]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Fan, I.; Tang, J.; Li, S.; Zhang, S.; Shaw, P.A.; et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: A kin-cohort study in Ontario, Canada. J. Natl. Cancer Inst. 2006, 98, 1694–1706. [Google Scholar] [CrossRef] [PubMed]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Kwan, E.; Jack, E.; Vesprini, D.J.; Kuperstein, G.; Abrahamson, J.L.; et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am. J. Hum. Genet. 2001, 68, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Stevens, A.; Paredes, R.; Schneider, M.; Walker, M.J.; Williamson, A.J.K.; Gonzalez-Sanchez, M.B.; Smetsers, S.; Dalal, V.; Teng, H.Y.; et al. Acquired cross-linker resistance associated with a novel spliced BRCA2 protein variant for molecular phenotyping of BRCA2 disruption. Cell Death Dis. 2017, 8, e2875. [Google Scholar] [CrossRef] [PubMed]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco-Targets Ther. 2013, 7, 57–68. [Google Scholar] [PubMed]
- Bourdon, J.C.; Khoury, M.P.; Diot, A.; Baker, L.; Fernandes, K.; Aoubala, M.; Quinlan, P.; Purdie, C.A.; Jordan, L.B.; Prats, A.C.; et al. p53 mutant breast cancer patients expressing p53gamma have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011, 13, R7. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Zhang, X.D.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Huo, S.W.; Lu, J.J.; Liu, Z.; Fang, X.L.; Jin, X.B.; Yuan, M.Z. Expression of p53 isoforms in renal cell carcinoma. Chin. Med. J. 2009, 122, 921–926. [Google Scholar] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.K.; Slupsky, J.R.; Kalmes, H.A.; Rapp, U.R. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61, 3595–3598. [Google Scholar] [PubMed]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell. Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Kasprzak, W.K.; Voss, T.; Shapiro, B.A.; Poulikakos, P.I.; Misteli, T. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat. Commun. 2015, 6, 7103. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brooks, S.R.; Li, X.; Anzelon, A.N.; Rickert, R.C.; Carter, R.H. The physiologic role of CD19 cytoplasmic tyrosines. Immunity 2002, 17, 501–514. [Google Scholar] [CrossRef]
- Chung, E.Y.; Psathas, J.N.; Yu, D.; Li, Y.; Weiss, M.J.; Thomas-Tikhonenko, A. CD19 is a major B cell receptor-independent activator of MYC-driven B-lymphomagenesis. J. Clin. Investig. 2012, 122, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Rickert, R.C.; Rajewsky, K.; Roes, J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature 1995, 376, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Poe, J.C.; Minard-Colin, V.; Kountikov, E.I.; Haas, K.M.; Tedder, T.F. A c-Myc and surface CD19 signaling amplification loop promotes B cell lymphoma development and progression in mice. J. Immunol. 2012, 189, 2318–2325. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gokbuget, N.; Zugmaier, G.; Klappers, P.; Stelljes, M.; Neumann, S.; Viardot, A.; Marks, R.; Diedrich, H.; Faul, C.; et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J. Clin. Oncol. 2014, 32, 4134–4140. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.D.; Yang, Q.; Ceniccola, K.; Bianco, F.; Andrawis, R.; Jarrett, T.; Frazier, H.; Patierno, S.R.; Lee, N.H. Androgen receptor-target genes in African American prostate cancer disparities. Prostate Cancer 2013, 2013, 763569. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Okabe, S.; Seimiya, H. Pharmacological targeting of constitutively active truncated androgen receptor by nigericin and suppression of hormone-refractory prostate cancer cell growth. Mol. Pharmacol. 2010, 78, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Tummala, R.; Lou, W.; Gao, A.C.; Nadiminty, N. Quercetin Targets hnRNPA1 to Overcome Enzalutamide Resistance in Prostate Cancer Cells. Mol. Cancer Ther. 2017, 16, 2770–2779. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-kappaB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.C.; Chen, Y.J.; Chen, C.T.; Liu, Y.C.; Cheng, F.C.; Hsu, K.C.; Chow, L.P. Chemical proteomics identifies heterogeneous nuclear ribonucleoprotein (hnRNP) A1 as the molecular target of quercetin in its anti-cancer effects in PC-3 cells. J. Biol. Chem. 2014, 289, 22078–22089. [Google Scholar] [CrossRef] [PubMed]
- Marcinkiewicz, C.; Galasinski, W.; Gindzienski, A. EF-1 alpha is a target site for an inhibitory effect of quercetin in the peptide elongation process. Acta Biochim. Pol. 1995, 42, 347–350. [Google Scholar] [PubMed]
- Thomas, C.; Gustafsson, J.A. Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol. MeTable 2015, 26, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant Splicing of Estrogen Receptor, HER2, and CD44 Genes in Breast Cancer. Genet. Epigenet. 2015, 7, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Brusco, L.; Fuqua, S.A. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin. Cancer Res. 2010, 16, 2702–2708. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Xu, X.; Li, G.; Lin, B.; Cao, J.; Teng, L. ER-α36: A novel biomarker and potential therapeutic target in breast cancer. Onco-Targets Ther. 2014, 7, 1525–1533. [Google Scholar] [PubMed]
- Zou, Y.; Ding, L.; Coleman, M.; Wang, Z. Estrogen receptor-α (ER-α) suppresses expression of its variant ER-α 36. FEBS Lett. 2009, 583, 1368–1374. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Dong, B.; Li, Z.; Lu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B. Expression of ER-α36, a novel variant of estrogen receptor α, and resistance to tamoxifen treatment in breast cancer. J. Clin. Oncol. 2009, 27, 3423–3429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Yin, L. Estrogen receptor alpha-36 (ER-α36): A new player in human breast cancer. Mol. Cell. Endocrinol. 2015, 418 Pt 3, 193–206. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, J.; Ying, G.; Xie, X.Q.; Zhang, X.; Xu, W.; Zhang, X.; Song, E.; Bu, H.; Ping, Y.F.; et al. Tamoxifen enhances stemness and promotes metastasis of ERα36(+) breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. 2018, 28, 336–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Z.Y. Estrogen receptor-alpha variant, ER-α36, is involved in tamoxifen resistance and estrogen hypersensitivity. Endocrinology 2013, 154, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Berndt, A.; Miller, S.; Williams, O.; Le, D.D.; Houseman, B.T.; Pacold, J.I.; Gorrec, F.; Hon, W.C.; Liu, Y.; Rommel, C.; et al. The p110delta structure: Mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Modi, P.; Newcomb, T.; Queva, C.; Gandhi, V. Idelalisib: First-in-Class PI3K Delta Inhibitor for the Treatment of Chronic Lymphocytic Leukemia, Small Lymphocytic Leukemia, and Follicular Lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Byrd, J.C.; Coutre, S.E.; Benson, D.M.; Flinn, I.W.; Wagner-Johnston, N.D.; Spurgeon, S.E.; Kahl, B.S.; Bello, C.; Webb, H.K.; et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood 2014, 123, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Mangaonkar, A. Idelalisib: A Novel PI3Kdelta Inhibitor for Chronic Lymphocytic Leukemia. Ann. Pharmacother. 2015, 49, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulou, E.; Ladomery, M. Targeting Splicing in Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 1287. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, T.; Hosoya, T.; Shimizu, S.; Sumi, K.; Oshiro, T.; Yoshinaka, Y.; Suzuki, M.; Yamamoto, N.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. USA 2006, 103, 11329–11333. [Google Scholar] [CrossRef] [PubMed]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, R.P.; Barbosa Ede, A.; Poleto, M.D.; Righetto, G.L.; Seraphim, T.V.; Salgado, R.L.; Ferreira, J.G.; Barros, M.V.; de Oliveira, L.L.; Laranjeira, A.B.; et al. Potential Antileukemia Effect and Structural Analyses of SRPK Inhibition by N-(2-(Piperidin-1-yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (SRPIN340). PLoS ONE 2015, 10, e0134882. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Dairiki, R.; Nakayama, Y.; Murai, A.; Miyashita, R.; Iwatani, M.; Nomura, T.; Nakanishi, O. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS ONE 2015, 10, e0116929. [Google Scholar] [CrossRef] [PubMed]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.A.; Li, S.D.; Yang, A.; Huang, L.; Kole, R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010, 38, 8348–8356. [Google Scholar] [CrossRef] [PubMed]
- Galletti, G.; Matov, A.; Beltran, H.; Fontugne, J.; Miguel Mosquera, J.; Cheung, C.; MacDonald, T.Y.; Sung, M.; O’Toole, S.; Kench, J.G.; et al. ERG induces taxane resistance in castration-resistant prostate cancer. Nat. Commun. 2014, 5, 5548. [Google Scholar] [CrossRef] [PubMed]
- Hagen, R.M.; Adamo, P.; Karamat, S.; Oxley, J.; Aning, J.J.; Gillatt, D.; Persad, R.; Ladomery, M.R.; Rhodes, A. Quantitative analysis of ERG expression and its splice isoforms in formalin-fixed, paraffin-embedded prostate cancer samples: Association with seminal vesicle invasion and biochemical recurrence. Am. J. Clin. Pathol. 2014, 142, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Wood, M.J. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011, 27, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Takeda, J.; Suzuki, Y.; Sakate, R.; Sato, Y.; Seki, M.; Irie, T.; Takeuchi, N.; Ueda, T.; Nakao, M.; Sugano, S.; et al. Low conservation and species-specific evolution of alternative splicing in humans and mice: Comparative genomics analysis using well-annotated full-length cDNAs. Nucleic Acids Res. 2008, 36, 6386–6395. [Google Scholar] [CrossRef] [PubMed]
- Moroy, T.; Heyd, F. The impact of alternative splicing in vivo: Mouse models show the way. RNA 2007, 13, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, N.; Lu, Z.; Sun, S.; Huang, J.; Chen, Z.; He, J. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 2017, 393, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, Z.; Yong, L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol. Oncol. 2018, 148, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, S.S.; Panda, A.; Kumar, S.; Seiler, M.; Robinson, D.; Gheeya, J.; Yao, M.; Alnaes, G.I.G.; Toppmeyer, D.; Riis, M.; et al. Widespread alternative exon usage in clinically distinct subtypes of Invasive Ductal Carcinoma. Sci. Rep. 2017, 7, 5568. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Marcelino Meliso, F.; Hubert, C.G.; Favoretto Galante, P.A.; Penalva, L.O. RNA processing as an alternative route to attack glioblastoma. Hum. Genet. 2017, 136, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; The Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.-D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. https://doi.org/10.3390/cancers10110458
Wang B-D, Lee NH. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers. 2018; 10(11):458. https://doi.org/10.3390/cancers10110458
Chicago/Turabian StyleWang, Bi-Dar, and Norman H. Lee. 2018. "Aberrant RNA Splicing in Cancer and Drug Resistance" Cancers 10, no. 11: 458. https://doi.org/10.3390/cancers10110458
APA StyleWang, B.-D., & Lee, N. H. (2018). Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers, 10(11), 458. https://doi.org/10.3390/cancers10110458