Measurement of Fumonisins in Maize Using a Portable Mass Spectrometer

 ,
,  ,
,
Abstract
:
1. Introduction
2. Results
2.1. Sample Preparation and Collection of Spectra
2.2. Performance Criteria
2.3. Naturally Contaminated Samples
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. Samples
5.3. Extraction and Cleanup
5.4. Instrumentation
5.5. Data Analysis, Limits of Detection, Quantitation, and Cut-Off Value
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Disclaimer
References
- Ross, P.F.; Rice, L.G.; Osweiler, G.D.; Nelson, P.E.; Richard, J.L.; Wilson, T.M. A review and update of animal toxicoses associated with fumonisin-contaminated feeds and production of fumonisins by fusarium isolates. Mycopathologia 1992, 117, 109–114. [Google Scholar] [CrossRef]
- Marasas, W.F. Discovery and occurrence of the fumonisins: A historical perspective. Environ. Health Perspect. 2001, 109, 239–243. [Google Scholar] [PubMed]
- Stockmann-Juvala, H.; Savolainen, K. A review of the toxic effects and mechanisms of action of fumonisin b1. Hum. Exp. Toxicol. 2008, 27, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Gelineau-van Waes, J.; Voss, K.A.; Stevens, V.L.; Speer, M.C.; Riley, R.T. Chapter 5 maternal fumonisin exposure as a risk factor for neural tube defects. In Advances in Food and Nutrition Research; Academic Press: Cambridge, MA, USA, 2009; Volume 56, pp. 145–181. [Google Scholar]
- World Health Organization. Some traditional Herbal Medicines, Some Mycotoxins, Naphthalene and Styrene; International Agency for Research on Cancer: Lyon, France, 2002; Volume 82. [Google Scholar]
- Bacon, C.W.; Nelson, P.E. Fumonisin production in corn by toxigenic strains of Fusarium moniliforme and Fusarium proliferatum. J. Food Prot. 1994, 57, 514–521. [Google Scholar] [CrossRef]
- Shephard, G.S.; Van Der Westhuizen, L.; Thiel, P.G.; Gelderblom, W.C.A.; Marasas, W.F.O.; Van Schalkwyk, D.J. Disruption of sphingolipid metabolism in non-human primates consuming diets of fumonisin-containing Fusarium moniliforme culture material. Toxicon 1996, 34, 527–534. [Google Scholar] [CrossRef]
- Voss, K.A.; Riley, R.T.; Gardner, N.M.; Waes, J.G.-v. Chapter 47—Fumonisins. In Reproductive and developmental Toxicology, 2nd ed.; Gupta, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 925–943. [Google Scholar]
- United States Food and Drug Administration. Guidance for industry: Fumonisin levels in human foods and animal feeds. US Food and Drug Administration, C.F.S.A.N., Center for Veterinary Medicine, Ed. 2001. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-fumonisin-levels-human-foods-and-animal-feeds (accessed on 29 July 2022).
- Schneider, E.; Usleber, E.; Maertlbauer, E. Rapid detection of fumonisin B1 in corn-based food by competitive direct dipstick enzyme immunoassay/enzyme-linked immunofiltration assay with integrated negative control reaction. J. Agric. Food Chem. 1995, 43, 2548–2552. [Google Scholar] [CrossRef]
- Thompson, V.S.; Maragos, C.M. Fiber-optic immunosensor for the detection of fumonisin B1. J. Agric. Food Chem. 1996, 44, 1041–1046. [Google Scholar] [CrossRef]
- Molinelli, A.; Grossalber, K.; Krska, R. A rapid lateral flow test for the determination of total type b fumonisins in maize. Anal. Bioanal. Chem. 2009, 395, 1309–1316. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Y.; Lu, S.; Guo, D.; Ren, H.; Meng, X.; Zhi, B.; Lin, C.; Wang, Z.; Li, X.; et al. Development of a one-step test strip for rapid screening of fumonisins B1, B2 and B3 in maize. Food Control 2012, 24, 72–77. [Google Scholar] [CrossRef]
- Maragos, C.M. Multiplexed biosensors for mycotoxins. J. AOAC Int. 2016, 99, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Maragos, C.M.; Jolley, M.E.; Plattner, R.D.; Nasir, M.S. Fluorescence polarization as a means for determination of fumonisins in maize. J. Agric. Food Chem 2001, 49, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Sydenham, E.W.; Shephard, G.S.; Thiel, P.G.; Stockenström, S.; Snijman, P.W.; Van Schalkwyk, D.J.; Collaborators. Liquid chromatographic determination of fumonisins B1, B2, and B3 in corn: AOAC-IUPAC collaborative study. J. AOAC Int. 1996, 79, 688–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shephard, G.S. Chromatographic determination of the fumonisin mycotoxins. J. Chromatogr. A 1998, 815, 31–39. [Google Scholar] [CrossRef]
- Visconti, A.; Solfrizzo, M.; Girolamo, A.D.; Collaborators. Determination of fumonisins B1 and B2 in corn and corn flakes by liquid chromatography with immunoaffinity column cleanup: Collaborative study. J. AOAC Int. 2001, 84, 1828–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscarella, M.; Lo Magro, S.; Nardiello, D.; Palermo, C.; Centonze, D. Determination of fumonisins B1 and B2 in maize food products by a new analytical method based on high-performance liquid chromatography and fluorimetric detection with post-column derivatization. In Microbial Toxins: Methods and Protocols, Methods in Molecular Biology; Humana: Totowa, NJ, USA, 2011; pp. 187–194. [Google Scholar]
- Ndube, N.; van der Westhuizen, L.; Green, I.R.; Shephard, G.S. HPLC determination of fumonisin mycotoxins in maize: A comparative study of naphthalene-2,3-dicarboxaldehyde and o-phthaldialdehyde derivatization reagents for fluorescence and diode array detection. J. Chromatogr. B 2011, 879, 2239–2243. [Google Scholar] [CrossRef]
- Solfrizzo, M.; De Girolamo, A.; Gambacorta, L.; Visconti, A.; Stroka, J.; van Egmond, H.P. Determination of fumonisins B1 and B2 in corn-based foods for infants and young children by LC with immunoaffinity column cleanup: Interlaboratory validation study. J. AOAC Int. 2011, 94, 900–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, M.; Herderich, M.; Humpf, H.U. Rapid determination of fumonisin FB1 in corn products by high-performance liquid chromatography-electrospray mass spectrometry. Eur. Food Res. Technol. 1999, 209, 348–351. [Google Scholar] [CrossRef]
- Bartok, T.; Szekeres, A.; Szecsi, A.; Bartok, M.; Mesterhazy, A. Detection of new fumonisin mycotoxins and fumonisin-like compounds by reverse-phase high-performance liquid chromatography/electrospray ionization ion trap mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 20, 2447–2462. [Google Scholar] [CrossRef]
- Senyuva, H.; Ozcan, S.; Cimen, D.; Gilbert, J. Determination of fumonisins B1 and B2 in corn by liquid chromatography/mass spectrometry with immunoaffinity column cleanup: Single-laboratory validation. J. AOAC Int. 2008, 91, 598–606. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Zhang, Y.; Han, S.; Han, Z.; Wu, Y. Simultaneous determination of fumonisins B1, B2 and B3 contaminants in maize by ultra high-performance liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2011, 692, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, Y.; Yang, T.; Huang-Fu, W. Rapid determination of fumonisins B1 and B2 in corn by liquid chromatography-tandem mass spectrometry with ultrasonic extraction. J. Chromatogr. Sci. 2012, 50, 57–63. [Google Scholar] [CrossRef]
- Soleimany, F.; Jinap, S.; Faridah, A.; Khatib, A. A UPLC-MS/MS for simultaneous determination of aflatoxins, ochratoxin A, zearalenone, DON, fumonisins, T-2 toxin and HT-2 toxin, in cereals. Food Control 2012, 25, 647–653. [Google Scholar] [CrossRef]
- Renaud, J.B.; Kelman, M.J.; Qi, T.F.; Seifert, K.A.; Sumarah, M.W. Product ion filtering with rapid polarity switching for the detection of all fumonisins and AAL-toxins. Rapid Commun. Mass Spectrom. RCM 2015, 29, 2131–2139. [Google Scholar] [CrossRef]
- Vega, V.A. A quick assay for the quantitation of fumonisin B1 and B2 in maize samples by liquid chromatography and mass spectrometry. J. AOAC Int. 2016, 99, 717–720. [Google Scholar] [CrossRef] [PubMed]
- De Girolamo, A.; Lippolis, V.; Pascale, M. Overview of recent liquid chromatography mass spectrometry-based methods for natural toxins detection in food products. Toxins 2022, 14, 328. [Google Scholar] [CrossRef]
- Tittlemier, S.A.; Cramer, B.; Dall’Asta, C.; DeRosa, M.C.; Lattanzio, V.M.T.; Malone, R.; Maragos, C.; Stranska, M.; Sumarah, M.W. Developments in mycotoxin analysis: An update for 2020–2021. World Mycotoxin J. 2022, 15, 3–25. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, D. Application of stable isotope dilution and liquid chromatography tandem mass spectrometry for multi-mycotoxin analysis in edible oils. J. AOAC Int. 2019, 102, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Andrade, P.D.; Dantas, R.R.; Moura-Alves, T.L.d.S.d.; Caldas, E.D. Determination of multi-mycotoxins in cereals and of total fumonisins in maize products using isotope labeled internal standard and liquid chromatography/tandem mass spectrometry with positive ionization. J. Chromatogr. A 2017, 1490, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Malachová, A.; Stránská, M.; Václavíková, M.; Elliott, C.T.; Black, C.; Meneely, J.; Hajšlová, J.; Ezekiel, C.N.; Schuhmacher, R.; Krska, R. Advanced LC-MS-based methods to study the co-occurrence and metabolization of multiple mycotoxins in cereals and cereal-based food. Anal. Bioanal. Chem. 2018, 410, 801–825. [Google Scholar] [CrossRef] [Green Version]
- Hickert, S.; Cramer, B.; Letzel, M.C.; Humpf, H.-U. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry imaging of ochratoxin A and fumonisins in mold-infected food. Rapid Commun. Mass Spectrom. 2016, 30, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Kecskemeti, A.; Nagy, C.; Biro, P.; Szabo, Z.; Pocsi, I.; Bartok, T.; Gaspar, A. Analysis of fumonisin mycotoxins with capillary electrophoresis-mass spectrometry. Food Addit. Contam. Part A 2020, 37, 1553–1563. [Google Scholar] [CrossRef]
- Ramalho, R.R.F.; Pereira, I.; Lima, G.d.S.; Santos, G.F.d.; Maciel, L.I.L.; Simas, R.C.; Vaz, B.G. Fumonisin B1 analysis in maize by molecularly imprinted polymer paper spray ionization mass spectrometry (MIP-PSI-MS). J. Food Compos. Anal. 2022, 107, 104362. [Google Scholar] [CrossRef]
- Mulligan, C.C.; Talaty, N.; Cooks, R.G. Desorption electrospray ionization with a portable mass spectrometer: In situ analysis of ambient surfaces. Chem. Commun. 2006, 28, 1709–1711. [Google Scholar] [CrossRef]
- Lawton, Z.E.; Traub, A.; Fatigante, W.L.; Mancias, J.; O’Leary, A.E.; Hall, S.E.; Wieland, J.R.; Oberacher, H.; Gizzi, M.C.; Mulligan, C.C. Analytical validation of a portable mass spectrometer featuring interchangeable, ambient ionization sources for high throughput forensic evidence screening. J. Am. Soc. Mass Spectrom. 2017, 28, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Noll, R.J.; Cooks, R.G. Handheld miniature ion trap mass spectrometers. Anal. Chem. 2009, 81, 2421–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Commission. Comission regulation (EU) no 519/2014 of 16 may 2014 amending regulation (EC) no 401/2006 as regards methods of sampling large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis. Off. J. Eur. Union 2014, 147, 29–43. [Google Scholar]
- Illinois Department of Agriculture. Fumonisin in Corn, Illinois Department of Agriculture County Survey—2021. Bureau of Agricultural Products Inspection. Available online: https://www2.illinois.gov/sites/agr/Plants/Mycotoxin/Pages/Survey.aspx (accessed on 27 June 2022).
- Renaud, J.B.; Miller, J.D.; Sumarah, M.W. Mycotoxin testing paradigm: Challenges and opportunities for the future. J. AOAC Int. 2019, 102, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Vaclavik, L.; Zachariasova, M.; Hrbek, V.; Hajslova, J. Analysis of multiple mycotoxins in cereals under ambient conditions using direct analysis in real time (DART) ionization coupled to high resolution mass spectrometry. Talanta 2010, 82, 1950–1957. [Google Scholar] [CrossRef]
- Tsagkaris, A.S.; Hrbek, V.; Dzuman, Z.; Hajslova, J. Critical comparison of direct analysis in real time orbitrap mass spectrometry (DART-orbitrap MS) towards liquid chromatography mass spectrometry (LC-MS) for mycotoxin detection in cereal matrices. Food Control 2022, 132, 108548. [Google Scholar] [CrossRef]
- United States Department of Agriculture, Agricultural Marketing Service, Federal Grain Inspection Service. Design Criteria and test Performance Specifications for Quantitative Fumonisin Test Kits. Available online: https://www.ams.usda.gov/services/fgis/standardization/tke (accessed on 27 July 2022).
- Poling, S.M.; Plattner, R.D. Rapid purification of fumonisins and their hydrolysis products with solid phase extraction columns. J. Agric. Food Chem. 1999, 47, 2344–2349. [Google Scholar] [CrossRef]
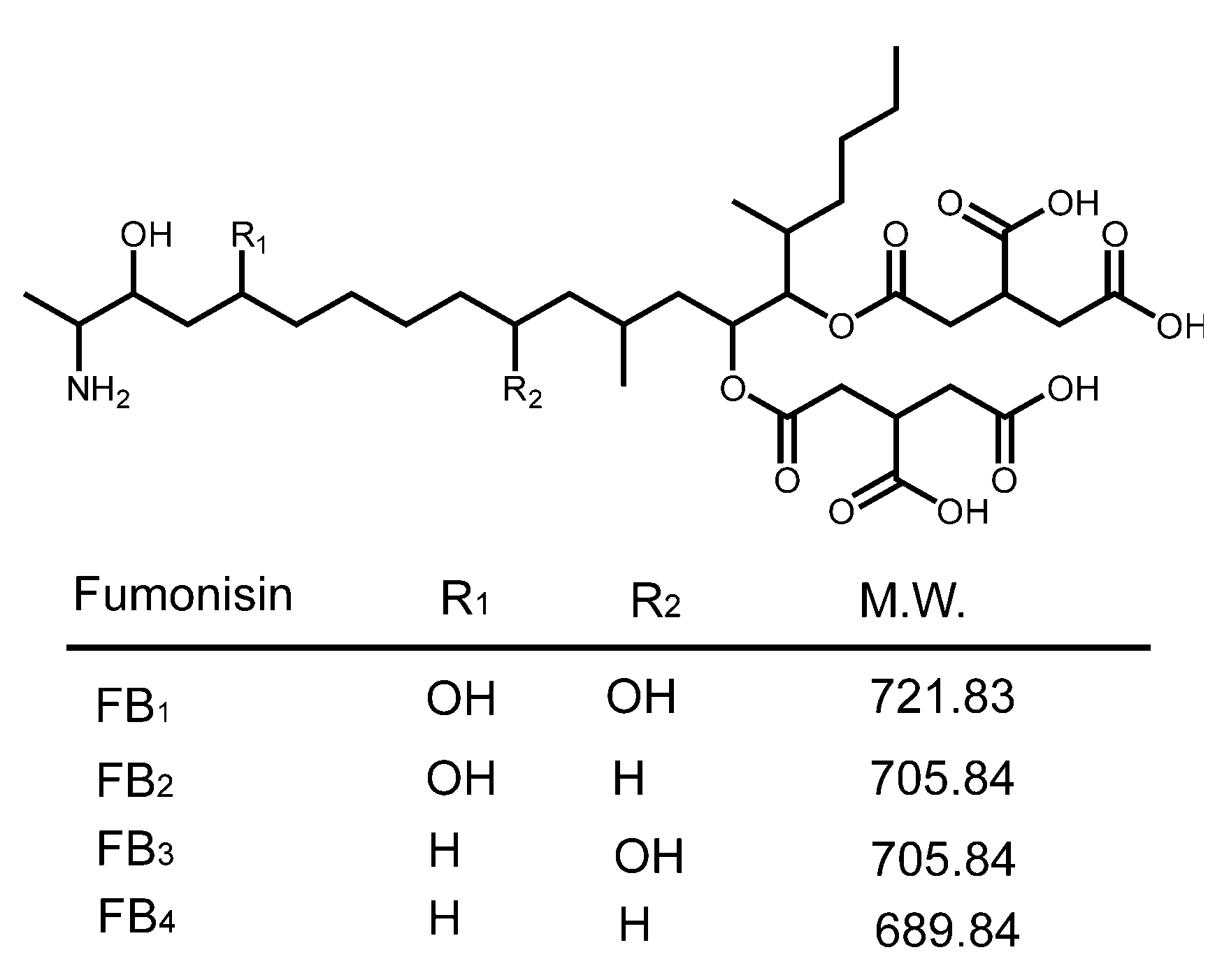



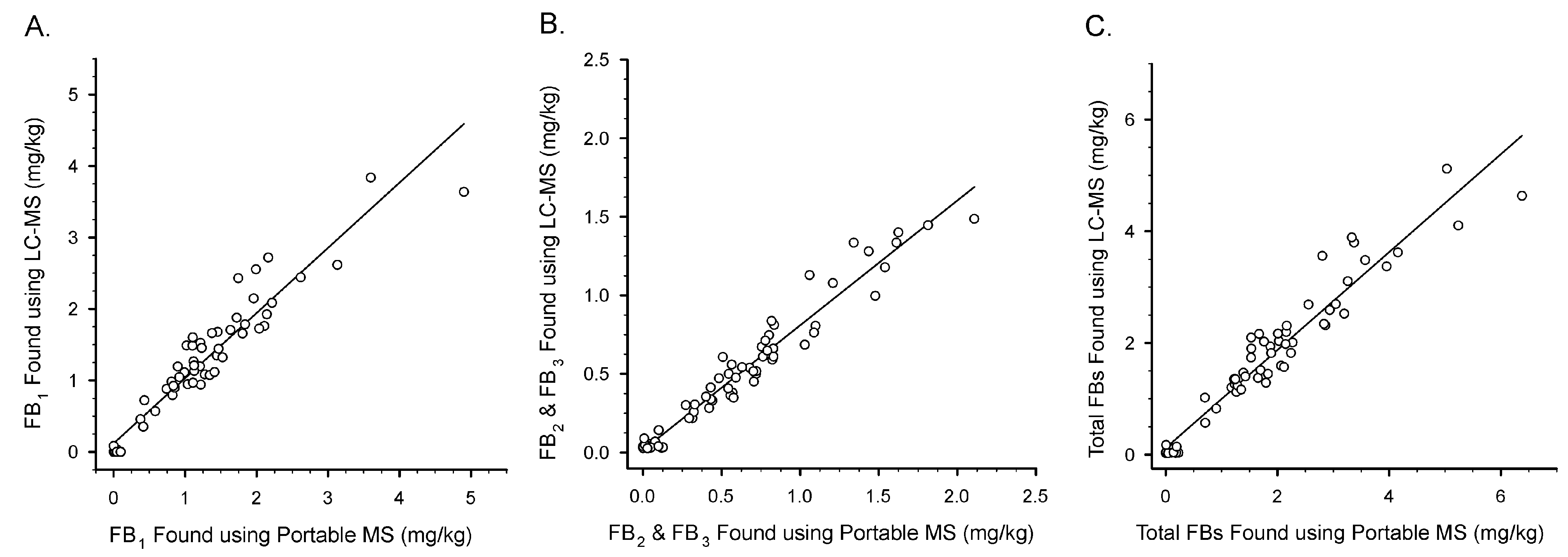

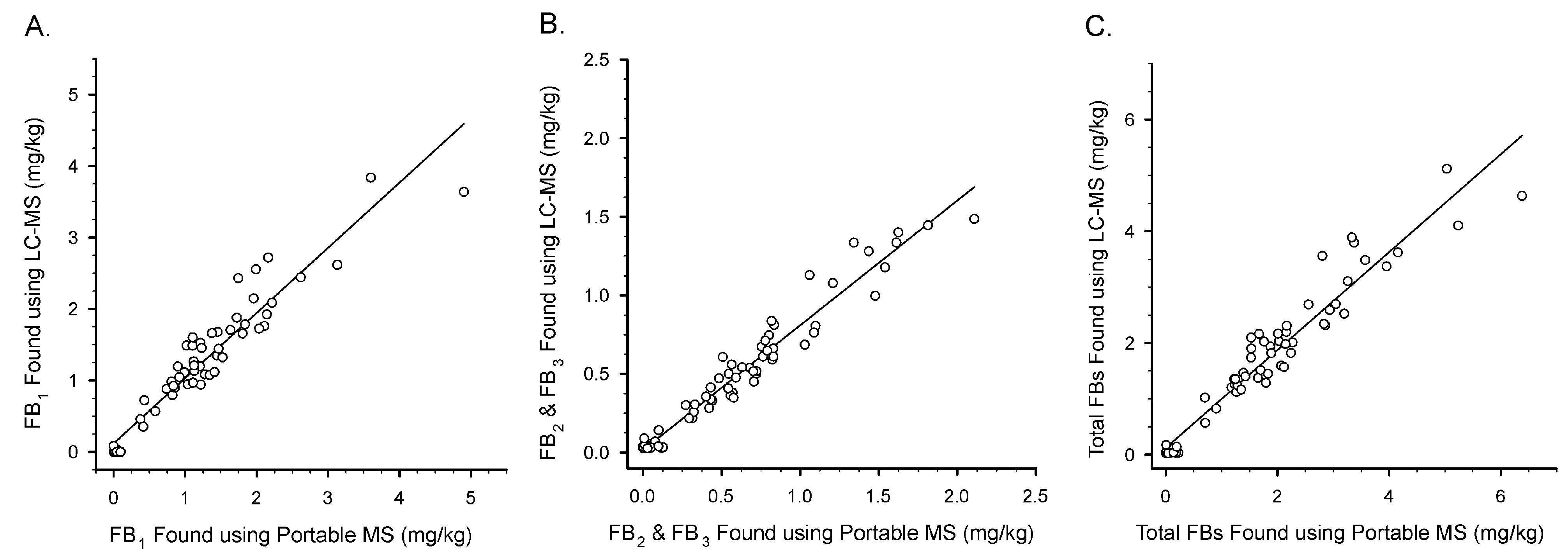
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
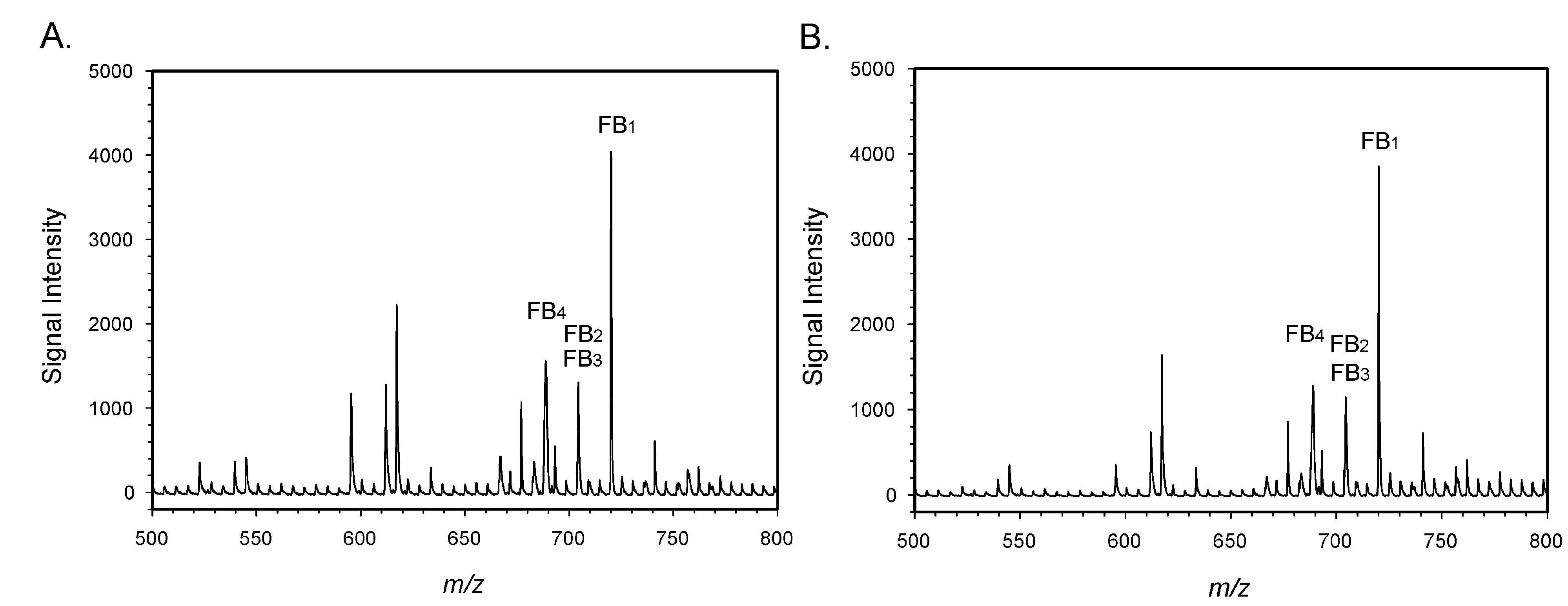
{kind=link}
{kind=link}
| Toxin | Spiking Level (mg/kg) | Recovery (% ± SD) 1 | RSD (%) | n |
|---|---|---|---|---|
| FB1 | 0.38 | 101.6 ± 19.2 | 18.9 | 24 |
| 0.76 | 101.7 ± 15.7 | 15.4 | 7 | |
| 1.91 | 93.6 ± 11.6 | 12.4 | 7 | |
| 3.81 | 96.2 ± 28.7 | 29.8 | 7 | |
| FB2 & FB3 | 0.19 | 108.6 ± 29.0 | 26.7 | 24 |
| 0.37 | 107.0 ± 17.5 | 16.4 | 7 | |
| 0.92 | 99.2 ± 11.9 | 12.0 | 7 | |
| 1.85 | 105.8 ± 23.4 | 22.1 | 7 | |
| Total FB | 0.57 | 106.7 ± 18.8 | 17.6 | 24 |
| 1.13 | 104.4 ± 14.8 | 14.2 | 7 | |
| 2.83 | 95.8 ± 11.6 | 12.1 | 7 | |
| 5.66 | 99.5 ± 27.0 | 27.1 | 7 |
| Reference Material | Toxin | Assigned Value (mg/kg) | Reported SD 1 | Value Found with the Portable MS (mg/kg) | SD (n = 3) |
|---|---|---|---|---|---|
| M1811-100 | FB1 | 2.2 | 0.2 | 2.0 | 0.1 |
| FB2 | 0.6 | 0.1 | 1.1 | 0.04 | |
| FB3 | 0.3 | 0.1 | |||
| Total FB | 3.1 | NR | 3.1 | 0.12 | |
| MT-100 | FB1 | 28.3 | NR | 21.1 | 3.4 |
| FB2 | 7.1 | NR | 11.3 | 1.0 | |
| FB3 | 1.7 | NR | |||
| Total FB | 37.1 | 4.2 | 32.3 | 4.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maragos, C.M.; Barnett, K.; Morgan, L.; Vaughan, M.M.; Sieve, K.K. Measurement of Fumonisins in Maize Using a Portable Mass Spectrometer. Toxins 2022, 14, 523. https://doi.org/10.3390/toxins14080523
Maragos CM, Barnett K, Morgan L, Vaughan MM, Sieve KK. Measurement of Fumonisins in Maize Using a Portable Mass Spectrometer. Toxins. 2022; 14(8):523. https://doi.org/10.3390/toxins14080523
Chicago/Turabian StyleMaragos, Chris M., Kristin Barnett, Luke Morgan, Martha M. Vaughan, and Kristal K. Sieve. 2022. "Measurement of Fumonisins in Maize Using a Portable Mass Spectrometer" Toxins 14, no. 8: 523. https://doi.org/10.3390/toxins14080523
APA StyleMaragos, C. M., Barnett, K., Morgan, L., Vaughan, M. M., & Sieve, K. K. (2022). Measurement of Fumonisins in Maize Using a Portable Mass Spectrometer. Toxins, 14(8), 523. https://doi.org/10.3390/toxins14080523