2.1. Identification of Core Gene TASs
The TASmania database [
23] was queried to identify the putative TASs in the 352
Lm genomes that are currently in the database (list of
L. monocytogenes genomes in TASmania,
Table S1, list of TASs,
Table S2). These 352 genomes were typed using the scheme cgMLST1748 at the Pasteur BIGSdb-Lm web site [
24] to obtain the core gene alleles for all strains. By comparing with the TASmania hits we identified n = 14 core gene TASs (cgTASs) (
Table 1) and their respective alleles (list of alleles in the 14 cgTASs,
Table S3). The current knowledge on TASs in
L. monocytogenes is rather scarce and our list included the two cgTASs already identified by TADB2 (
lmo0113-0114 and
lmo0887-0888). TASmania extended the number of TAS candidates by one order of magnitude in all
L. monocytogenes strains, but only a subset of them consists of core genes according to cgMLST1748.
Out of these 14 cgTASs, two cgTASs appear as orphans (
lmo0168 and
lmo0887). Their genetic environment was studied in the annotations of the EGD-e strain [
25]. In the case of
lmo0168, no possible partner was found, as the gene is surrounded by genes located on the other strand, confirming a probable orphan antitoxin. The potential partner of
lmo0887 is
lmo0888, which likely is a toxin mRNA interferase containing a pemK-like domain and a plasmid_toxin domain (according to TASmania), is a mazEF (according to TADB2); however, it is not identified as a core gene according to the cgMLST1748 scheme and thus must be ignored in our co-evolution analysis below.
In a second step, we added our own collection of
L. monocytogenes strains [
26] (n = 227) with their cgMLST alleles (
Table S4) and metadata annotation (
Table S5). We extracted their non-redundant gene alleles corresponding to the previously identified 14 cgTASs (
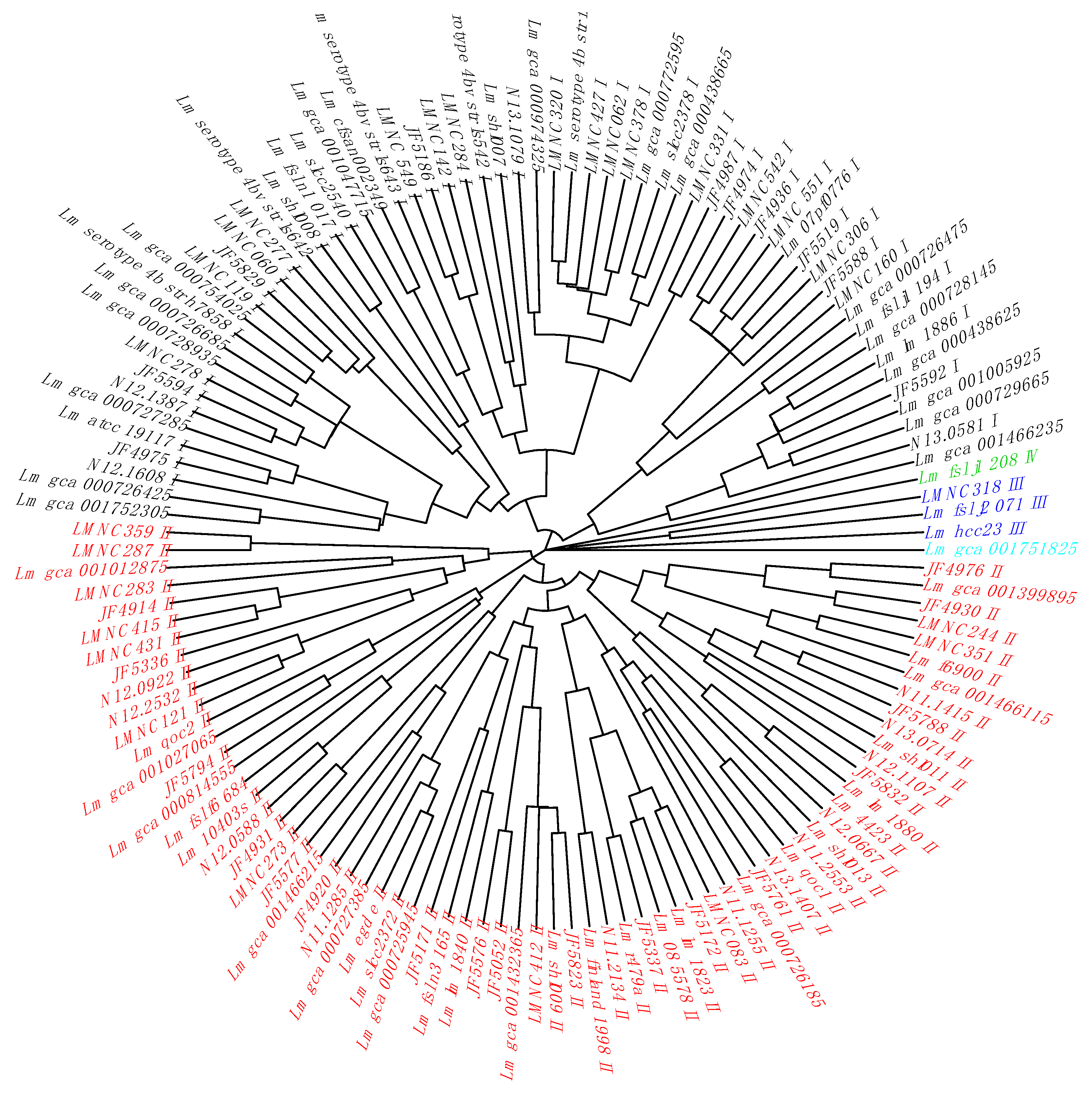
Table S6). By clustering the non-redundant cgTAS alleles patterns with nominal hierarchical clustering, we obtained the dendrogram (
Figure 1) showing that the 14 cgTASs are sufficient to separate the
L. monocytogenes lineages (I, II, III, and IV). These results are in agreement with previous MLST analysis using seven housekeeping genes [
14]. This clustering allows for characterizing the lineage membership of strains that did not have metadata information available (e.g., Lm_gca_000729665 as Lineage I and Lm_gca_001466115 as Lineage II). However, one cannot infer any causal role for the cgTASs in the lineage separation, as clustering the alleles of some subsets of core genes among the 1748 core genes would potentially show the same lineage split.
2.2. Co-Evolution Analysis
We analyzed the six complete cgTAS pairs for co-evolutionary residues and excluded the orphan cgTASs (
lmo0168 and
lmo0887). We grouped the sequences by gene, translated them to proteins and fused the toxin (T) alleles with the corresponding antitoxin (A) alleles per genome in a multiple sequence alignment (MSA) using our own Perl script and MAFFT [
27]. With this MSA, the BIS2Analyzer server [
28] was able to identify potential co-evolutionary residues, i.e. a mutated amino acid in a toxin associated to another mutated residue in the cognate antitoxin of the same TAS (e.g., red arrows in
Figure 2 and
Table 2). Only two cgTAS pairs (
lmo1466-1467 and
lmo1309-1310) revealed co-evolving residues between the two partners. For instance, the residue at location 435 (within the first partner
lmo1466) co-evolves with the residue at location 828 (within the second partner
lmo1467) (
Figure 2a and
Table 2). More than one residue per partner can be co-evolving as shown with the pair
lmo1309-1310 (
Figure 2b and
Table 2). One cgTAS pair (
lmo2793-2794) had co-evolving residues visible on the MSA using Jalview (
Figure 2c red arrows), but it failed to reach a significant p-value (<0.05) in BIS2Analyzer.
2.3. Accessory Gene TAS Analysis
By subtracting the cgTAS hits from the
L. monocytogenes strains in TASmania, all other TASs are classified as accessory TASs (acTASs) (
Table S7,
Figure A1). In order to obtain a better understanding of the role of accessory TASs, we performed a gene analysis with a set of 227
L. monocytogenes strains [
26] for which we have the corresponding metadata (
Table S5). These isolates are not part of the TASmania database, so we processed them by first predicting protein genes with Prodigal [
30] and annotating them with the TASmania HMMs [
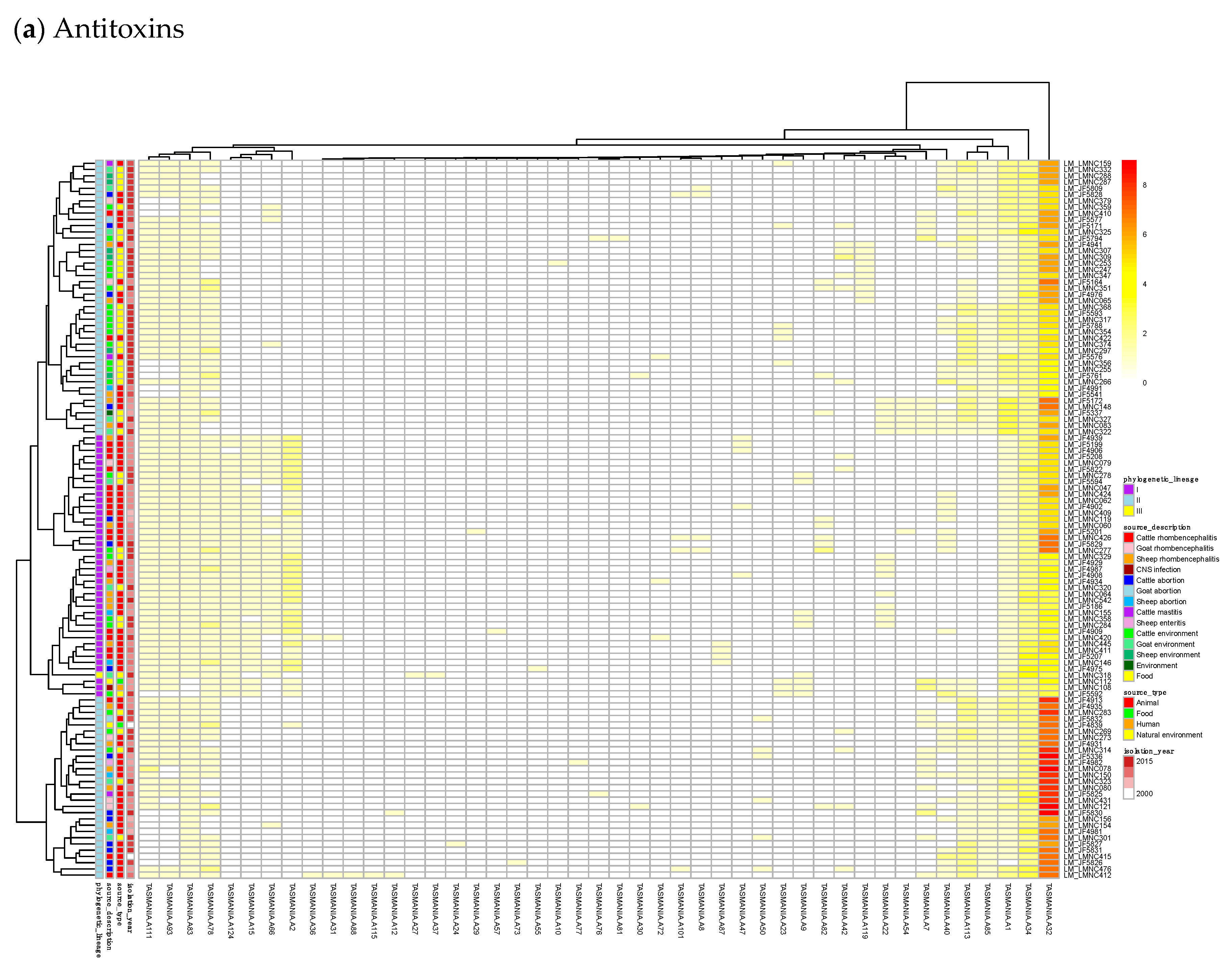
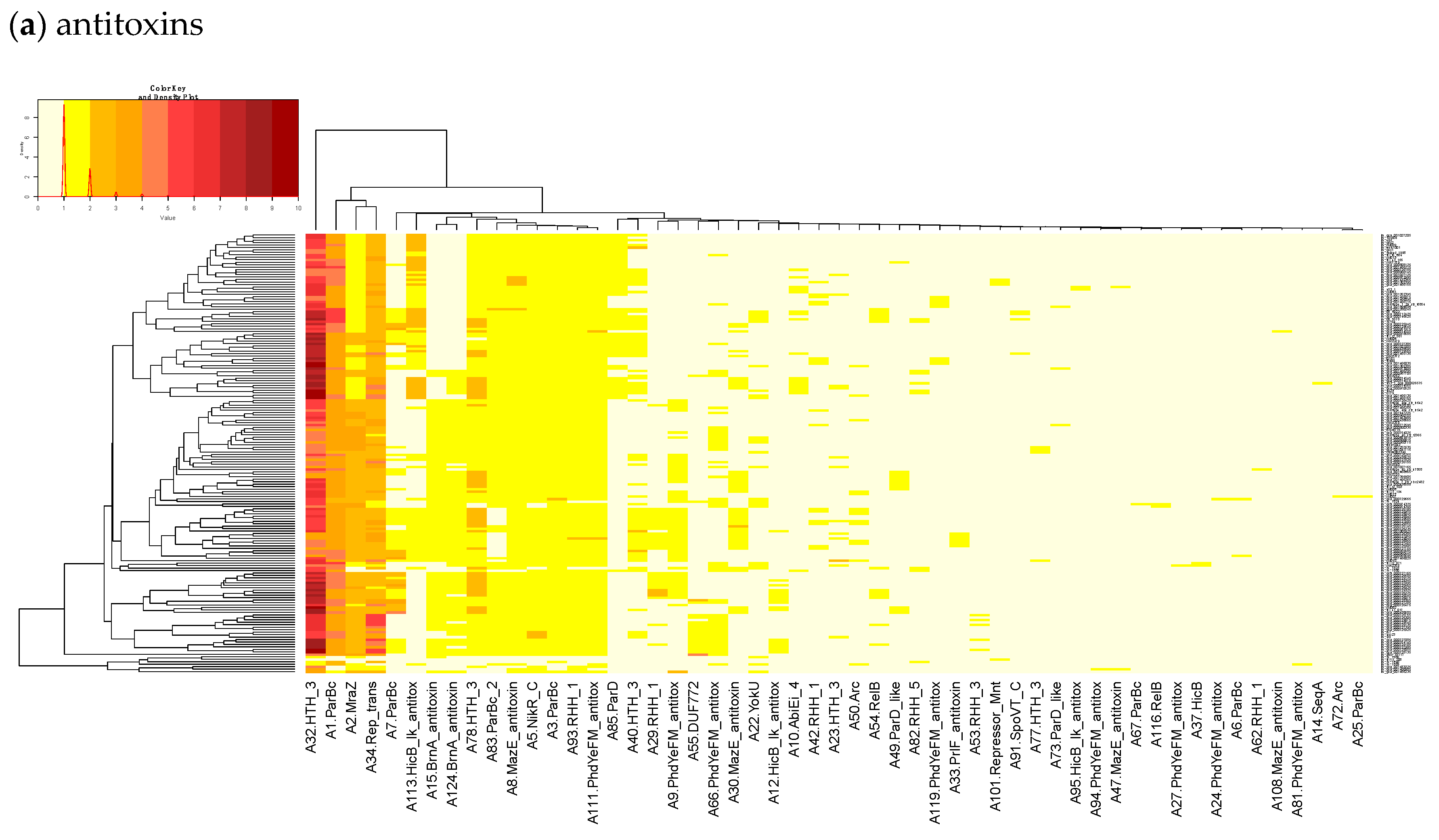
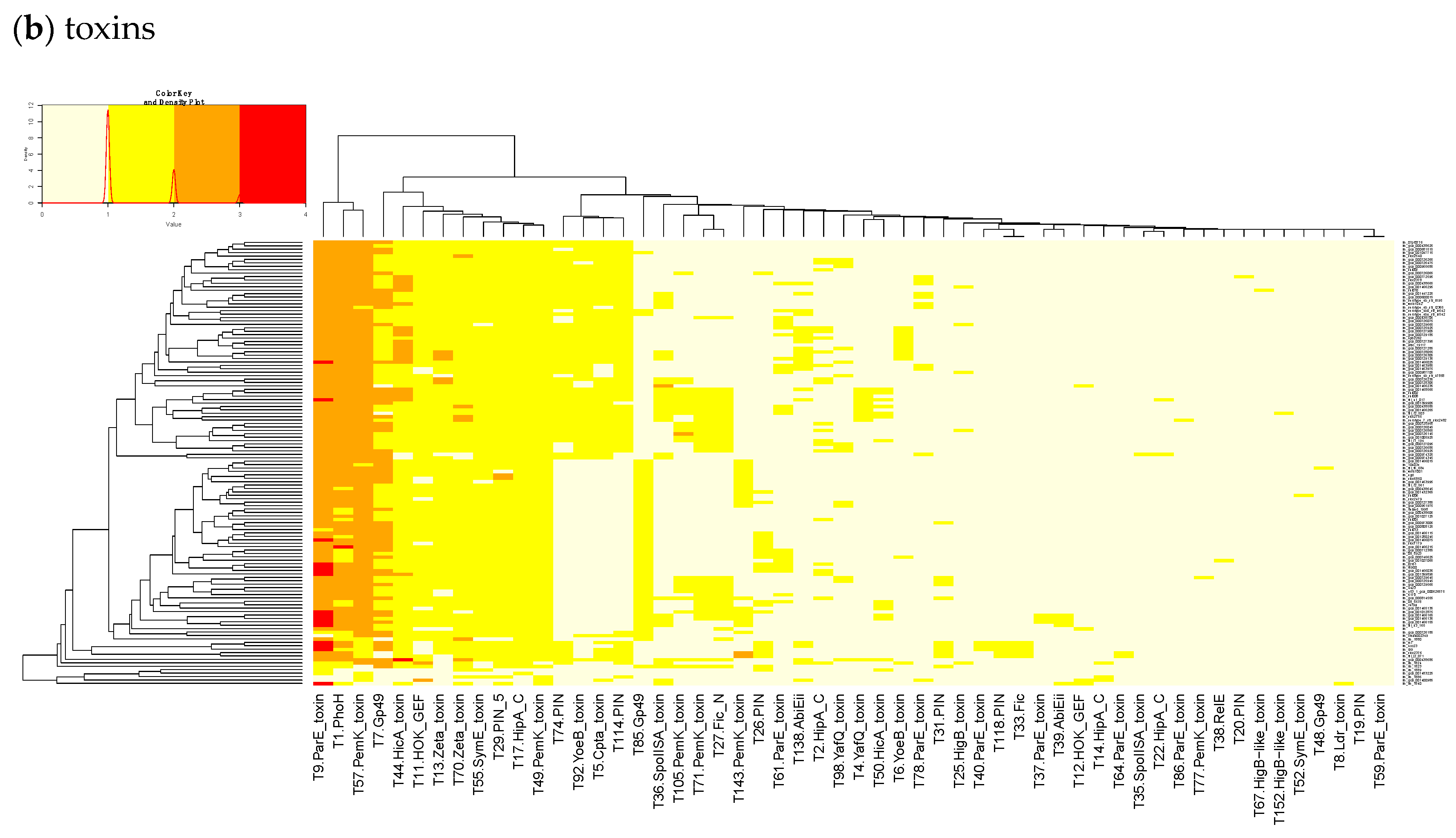
23]. Finally, we built a heatmap of those acTASs (
Table S8) for antitoxins (
Figure 3a) and toxins (
Figure 3b) after removing the core genes and the redundancy as described above.
Figure 3a shows on the rightmost column that the antitoxin cluster A32 (HTH_3) is the most abundant and is more frequently found in animal cases of Lineage II. In TASmania, this A32 antitoxin cluster is observed as being paired with at least six different toxin clusters (T2, T13, T14, T38, T48, and T85, whose nearest Pfam identifiers are HipA_C, Zeta_toxin, HipA_C, RelE, Gp49 and Gp49, respectively). All of these pairs were analyzed for their co-occurrence in the
L. monocytogenes strains above, but no candidate pair seems to exist in Lineage I, while only A32.T2 (nearest Pfam HTH_3.HipA_C) can be found rarely in Lineage II (
Table A2).
In
Figure 3b, a small, interesting group of isolates carry one or two genes having a hit to the T138 (nearest Pfam AbiEii) toxin cluster. It is mainly found in Lineage I isolates causing rhombencephalitis, 12 out of 15 (9 in cattle, 2 in goat, and 1 in sheep). The three remaining isolates are not rhombencephalitis, but could be related by their proximity to animals: 1 sheep abortion and 2 environmental isolates. In addition, one case carrying T138 in Lineage II, a cattle rhombencephalitis, is reported. This renders this toxin quite interesting regarding rhombencephalitis. However, in the
L. monocytogenes strains of TASmania, no antitoxin partner is known for this toxin that seems to be often encoded in a prophage region.
For the 352
L. monocytogenes strains described in TASmania unfortunately, no metadata is available, but the list of acTASs and their abundance is similar to our annotated dataset, with A32, A1, A2, and A34 (nearest Pfam HTH_3, ParBc, MraZ, and Omega_Repress/Rep_trans, respectively) being the most prevalent antitoxin clusters, and T9, T1, T57, T7, and T44 (nearest Pfam ParE, PhoH, PemK_toxin, Gp49, and HicA_toxin, respectively) being the main toxin clusters, as shown in
Figure A1.
Using TASmania, a closer look at the Pfam annotation of the toxin clusters highlights the diversity of the TA systems uncovered in
L. monocytogenes. Indeed, ParE targets the DNA gyrase and, along with Gp49, belongs to the Pfam clan called “plasmid_antitox CL0136”, whose members are originally described as plasmid-encoded TASs involved in plasmid maintenance. The PhOH domain is found in cytoplasmic proteins predicted as ATPase and which are induced by phosphate starvation [
31]. PemK toxins belong to the Pfam superfamily of CcdB/PemK (CL0624) known as growth inhibitors that can bind to their own promoter and act also as endonucleases. HicA is an mRNA interferase that binds to target mRNA potentially in a translation-independent manner. The
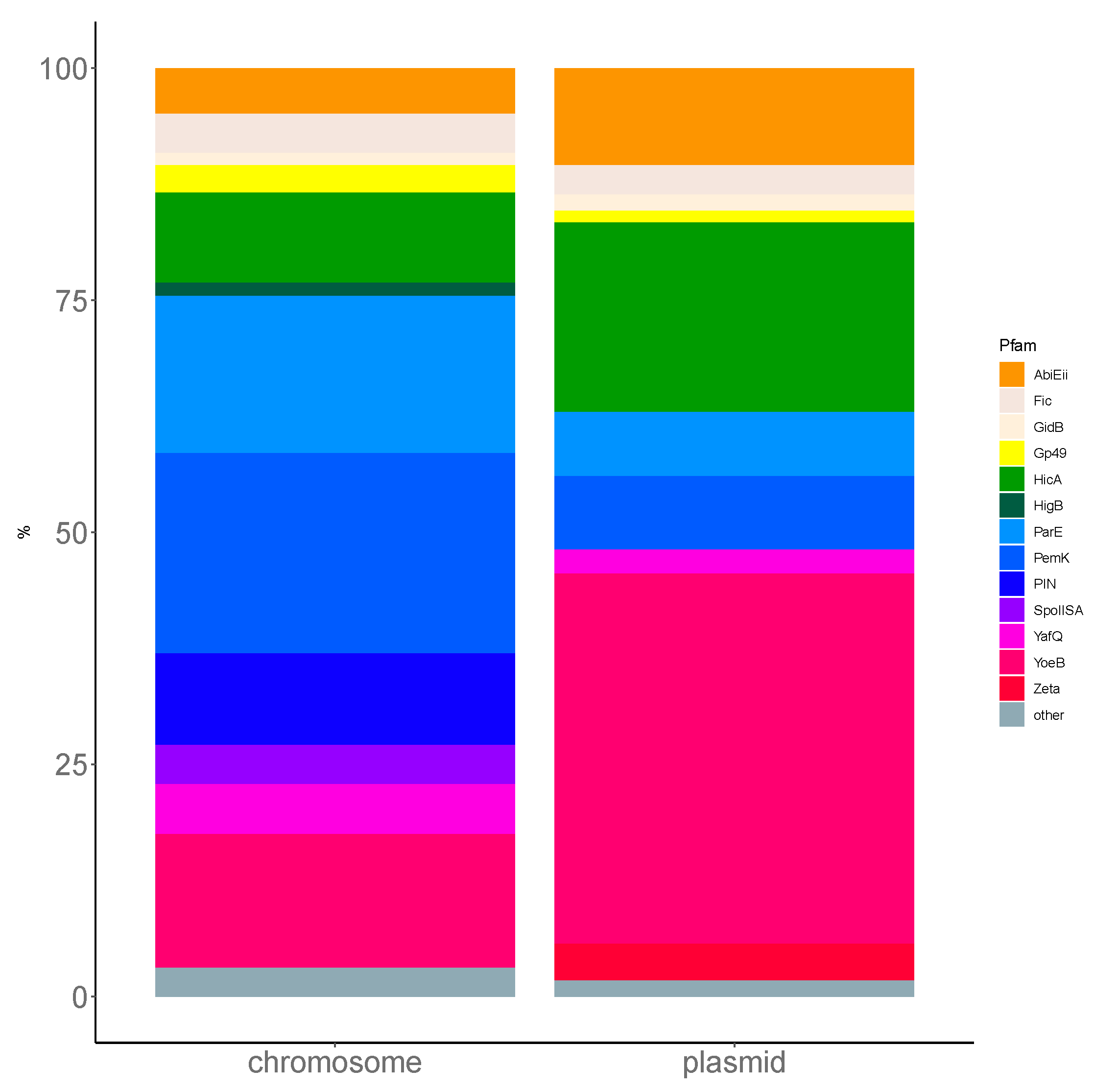
Firmicutes phylum has a prevalence of ParE and PemK like toxins in chromosomal TASs, which correspond to the prevalence in
L. monocytogenes (
Figure A2). More experimental investigation is required to confirm these putative TASs and to understand the conditions that regulate their expression in
L. monocytogenes of various pathogenicity.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



