Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
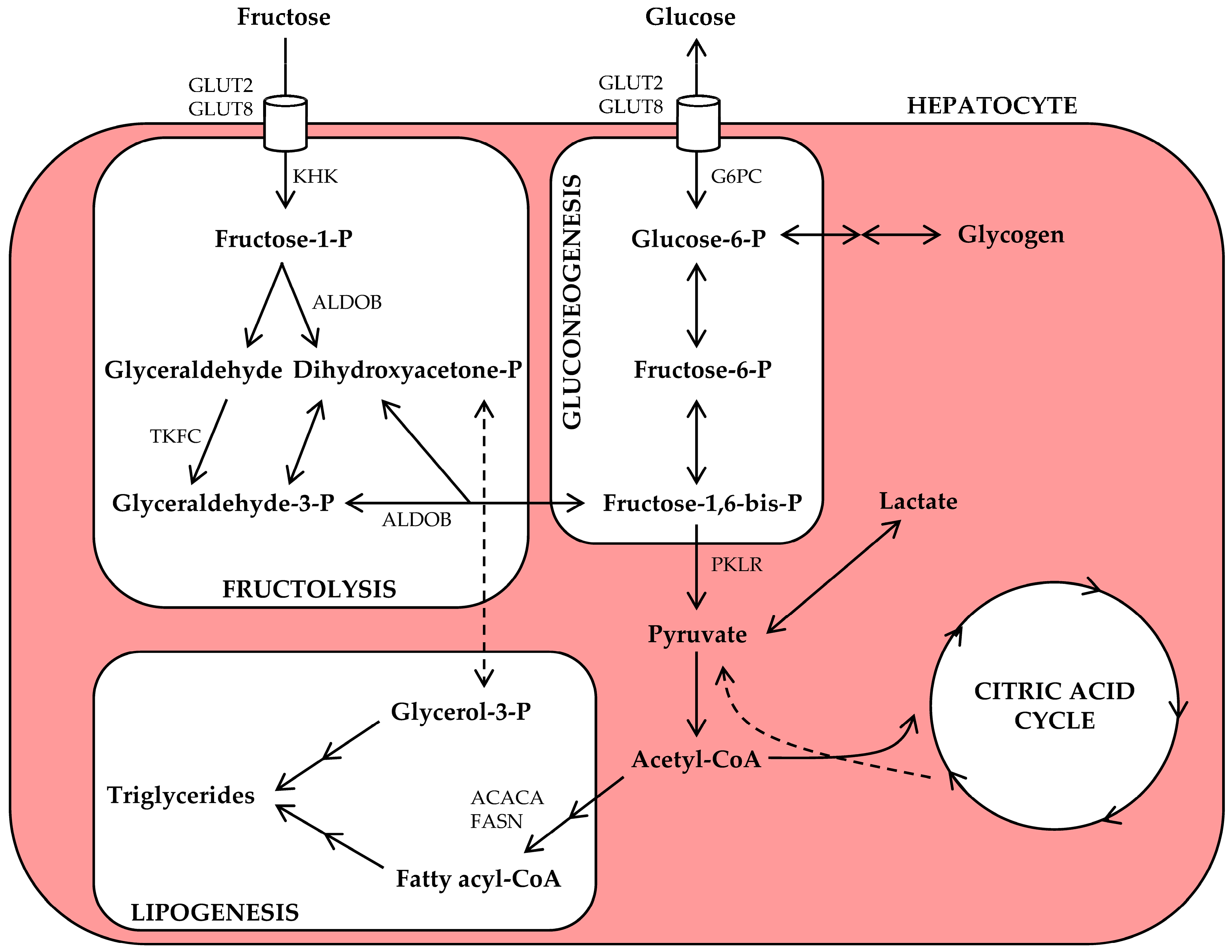
2. Fructose Metabolism in Physiology
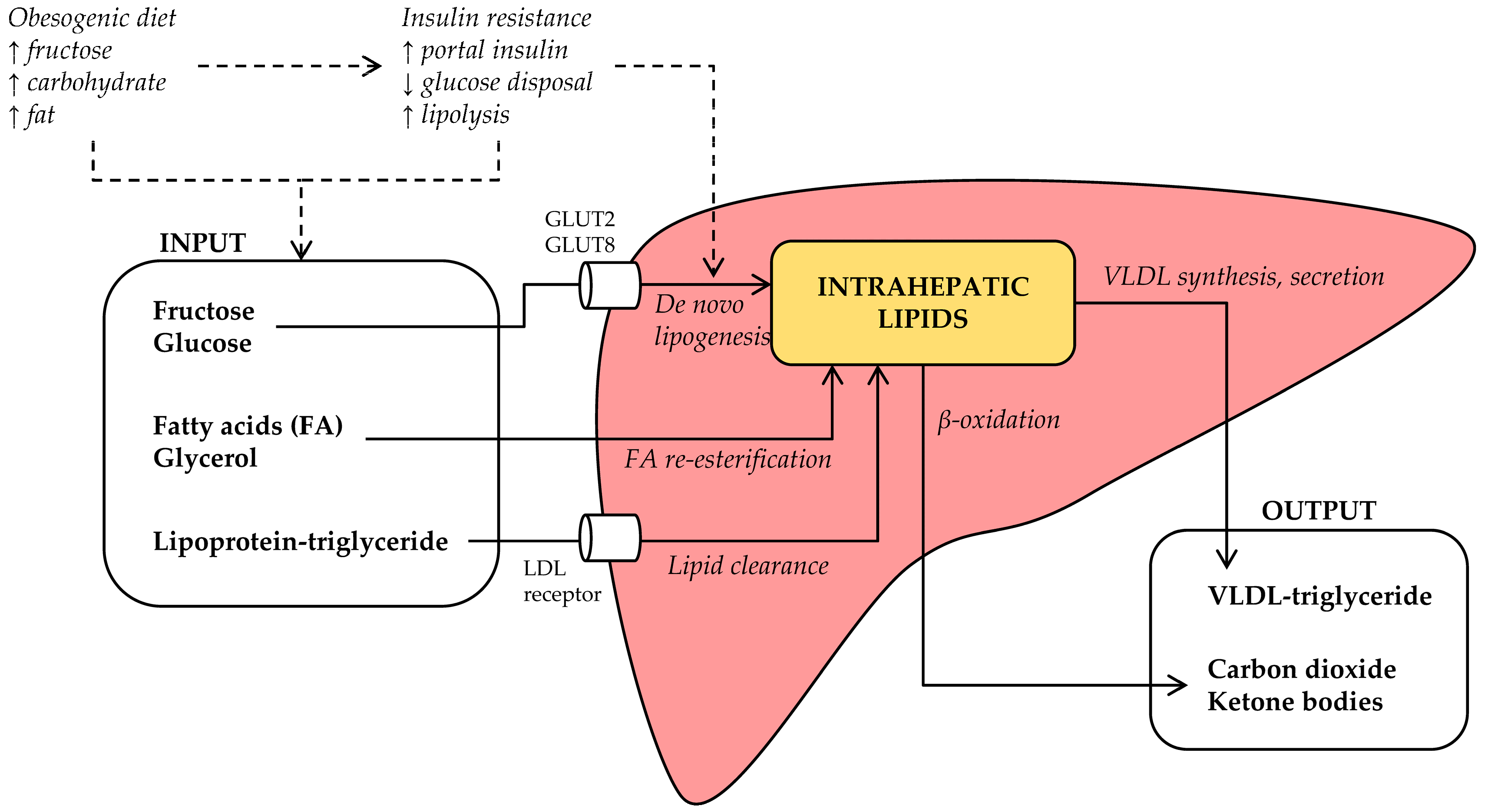
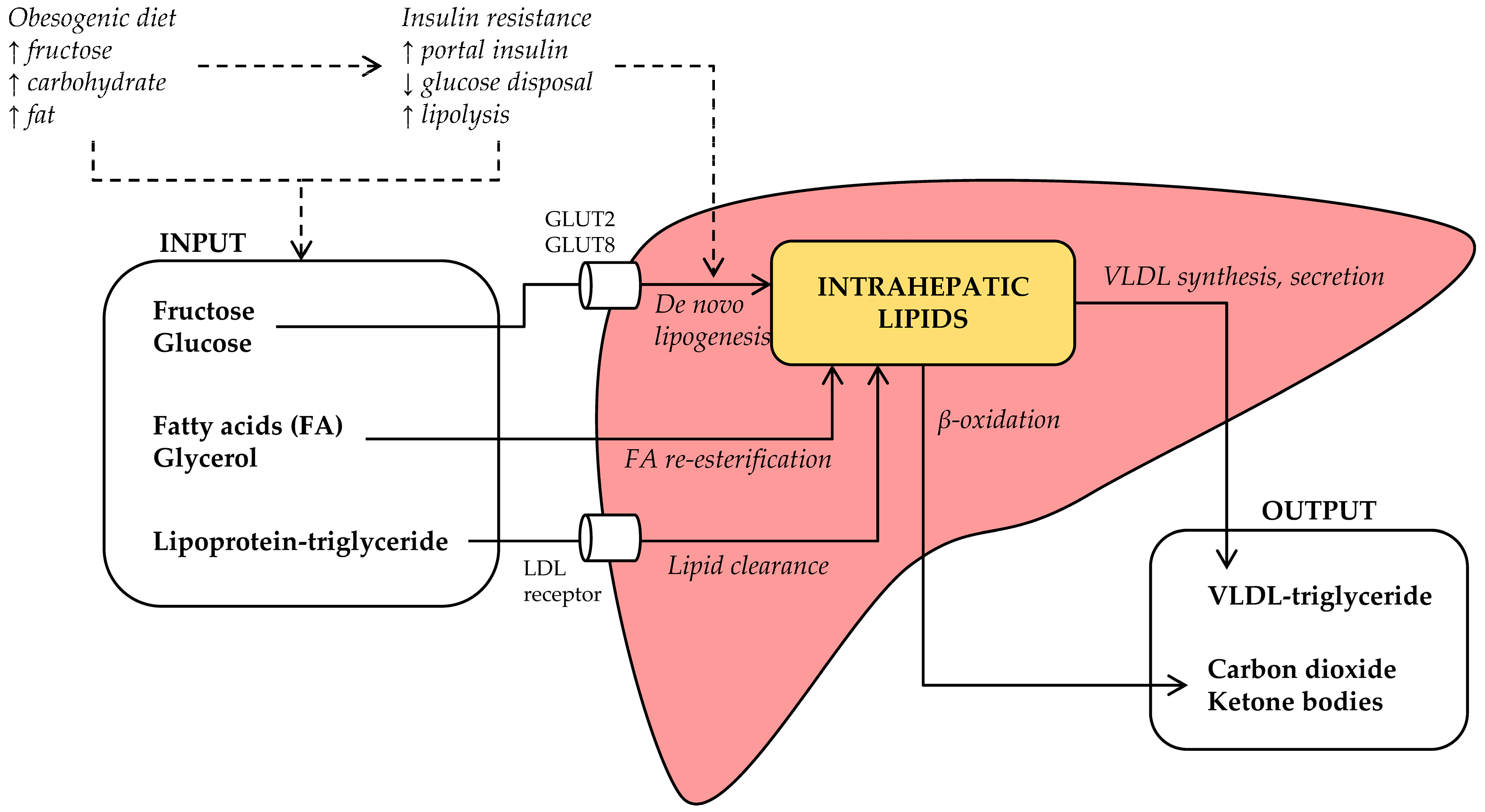
3. What Are the Mechanisms Linking Fructose to NAFLD?
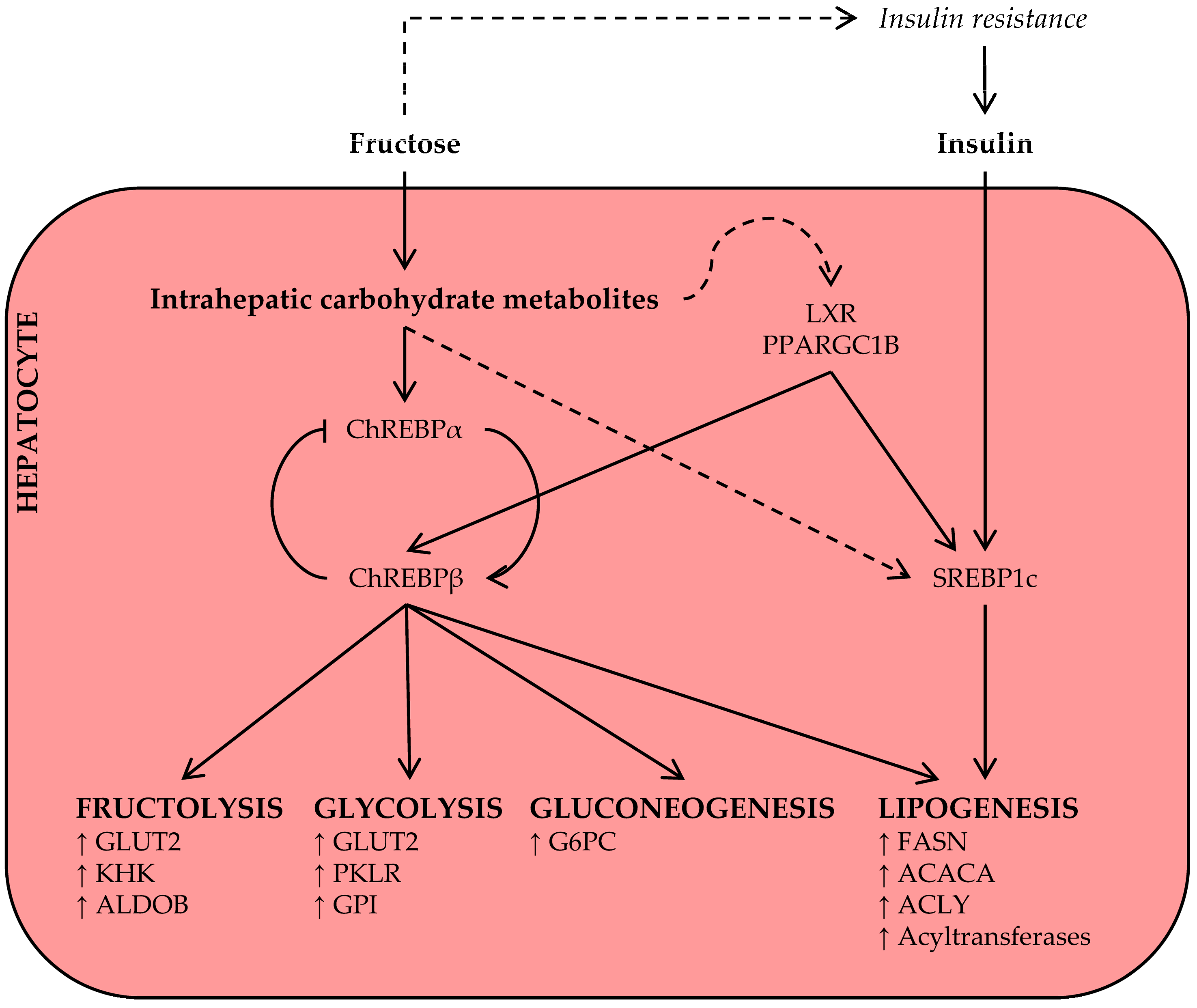
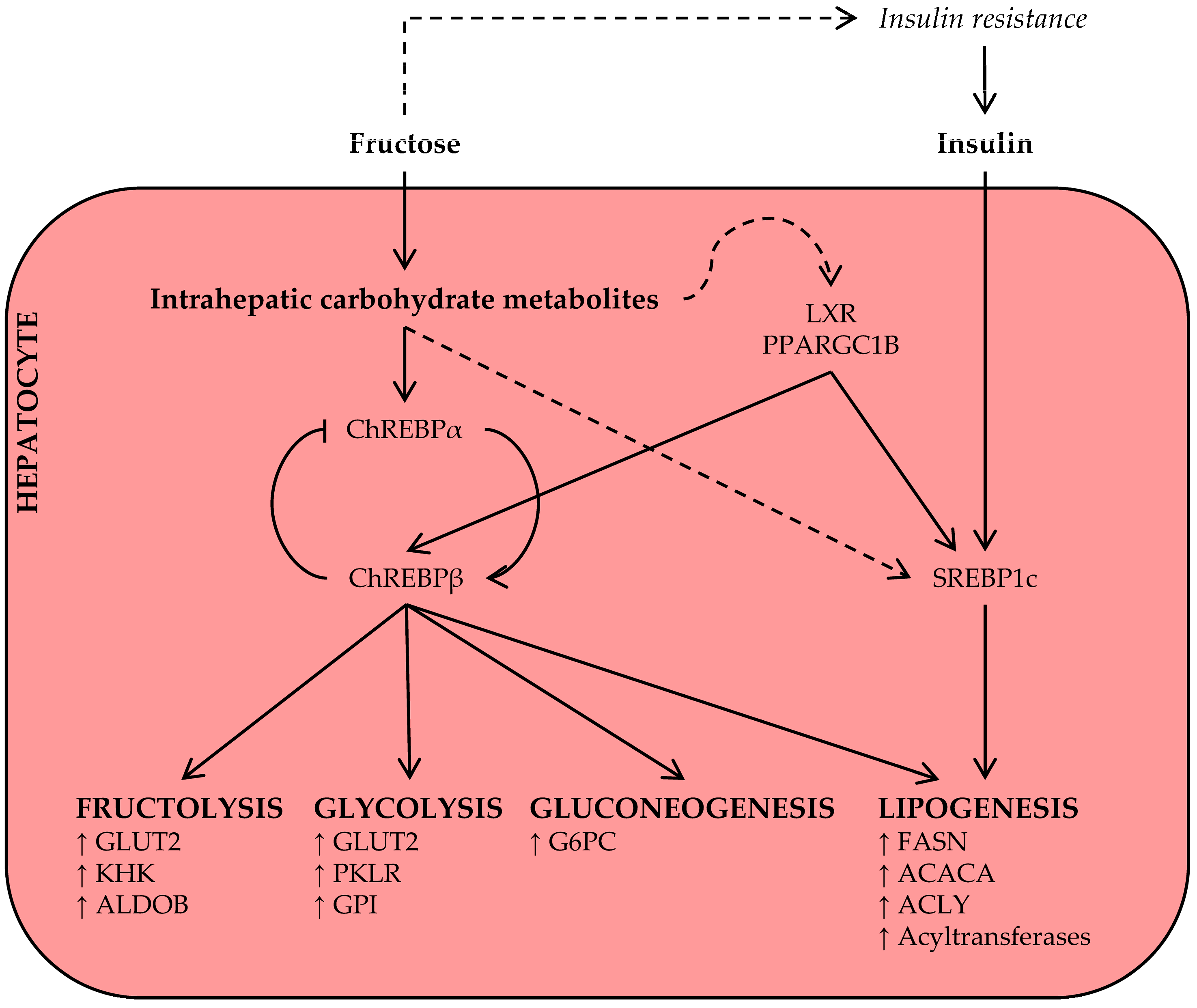
3.1. Fructose and Lipid Synthesis
3.2. Fructose and Lipolysis
3.3. Fructose and Hepatic Lipoprotein-Triglyceride Uptake
3.4. Fructose and β-Oxidation
3.5. Fructose and Hepatic VLDL Secretion
4. Does Fructose Consumption Cause Hepatic Steatosis in Humans?
4.1. Epidemiological Evidence
4.2. Controlled Diet-Intervention Studies
4.3. Fructose Reduction as a Therapeutic Strategy
5. Does Fructose Consumption Cause Disease Progression in Humans?
5.1. Fructose and Hepatic Insulin Resistance
5.2. Fructose and NASH
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Literature Search and Selection Strategy
Appendix B. Recommended Further Reading
References
- Zimmet, P.; Alberti, K.G.; Shaw, J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Malnick, S.D.; Knobler, H. The medical complications of obesity. QJM 2006, 99, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Global BMI Mortality Collaboration. Body-mass index and all-cause mortality: Individual-participant-data meta-analysis of 239 prospective studies in four continents. Lancet 2016, 388, 776–786. [Google Scholar]
- Yu, E.; Ley, S.H.; Manson, J.E.; Willet, W.; Satija, A.; Hu, F.B.; Stokes, A. Weight history and all-cause and cause-specific mortality in three prospective cohort studies. Ann. Intern. Med. 2017, 166, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E462–E468. [Google Scholar] [CrossRef] [PubMed]
- Donhoffer, H. Quantitative estimation of lipids in needle biopsy sized specimens of cadaver liver. Acta Med. Acad. Sci. Hung. 1974, 31, 47–49. [Google Scholar] [PubMed]
- Petäjä, E.M.; Yki-Järvinen, H. Definitions of normal liver fat and the association of insulin sensitivity with acquired and genetic nafld—A systematic review. Int. J. Mol. Sci. 2016, 17, 633. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Li, M.K.; Chan, H.Y.; Chim, A.M.; Yu, J.; Sung, J.J.; Chan, H.L. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Zezos, P.; Renner, E.L. Liver transplantation and non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15532–15538. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Soft drink consumption and obesity: It is all about fructose. Curr. Opin. Lipidol. 2010, 21, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [PubMed]
- Bray, G.A.; Popkin, B.M. Dietary sugar and body weight: Have we reached a crisis in the epidemic of obesity and diabetes? Health be damned! Pour on the sugar. Diabetes Care 2014, 37, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Després, J.P.; Hu, F.B. Sugar sweetened beverages, obesity, type 2 diabetes and cardiovascular disease risk. Circulation 2010, 121, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, K.W.; Schene, M.R.; Holman, R.; Romijn, J.A.; Serlie, M.J. Effect of fructose consumption on insulin sensitivity in nondiabetic subjects: A systematic review and meta-analysis of diet-intervention trials. Am. J. Clin. Nutr. 2016, 104, 1562–1576. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.-M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Diraison, F.; Yankah, V.; Letexier, D.; Dusserre, E.; Jones, P.; Beylot, M. Differences in the regulation of adipose tissue and liver lipogenesis by carbohydrates in humans. J. Lipid Res. 2003, 44, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.J. Tracing lipogenesis in humans using deuterated water. Can. J. Physiol. Pharmacol. 1996, 74, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Samuel, V.T. The sweet path to metabolic demise: Fructose and lipid synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, 754s–765s. [Google Scholar] [PubMed]
- Samuel, V.T. Fructose induced lipogenesis: From sugar to fat to insulin resistance. Trends Endocrinol. Metab. 2011, 22, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Drozdowski, L.A.; Thomson, A.B.R. Intestinal sugar transport. World J. Gastroenterol. 2006, 12, 1657–1670. [Google Scholar] [CrossRef] [PubMed]
- Ebert, K.; Witt, H. Fructose malabsorption. Mol. Cell. Pediatr. 2016, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter Glut5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef] [PubMed]
- Leturque, A.; Brot-Laroche, E.; Le Gall, M.; Stolarczyk, E.; Tobin, V. The role of Glut2 in dietary sugar handling. J. Physiol. Biochem. 2005, 61, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Debosch, B.J.; Chen, Z.; Saben, J.L.; Finck, B.N.; Moley, K.H. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J. Biol. Chem. 2014, 289, 10989–10998. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Adams, D.H.; Lalor, P.F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 2012, 18, 6771–6781. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—What isotopic tracer studies tell us. Nutr. Metab. 2012, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K. The role of carbohydrate response element binding protein in intestinal and hepatic fructose metabolism. Nutrients 2017, 9, 181. [Google Scholar] [CrossRef] [PubMed]
- Dushay, J.; Toschi, E.; Mitten, E.K.; Fisher, F.M.; Herman, M.A.; Maratos-Flier, E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol. Metab. 2014, 4, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Theytaz, F.; de Giorgi, S.; Hodson, L.; Stefanoni, N.; Rey, V.; Schneiter, P.; Giusti, V.; Tappy, L. Metabolic fate of fructose ingested with and without glucose in a mixed meal. Nutrients 2014, 6, 2632–2649. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [PubMed]
- Parks, E.J.; Skokan, L.E.; Timlin, M.T.; Dingfelder, C.S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 2008, 138, 1039–1046. [Google Scholar] [PubMed]
- Teff, K.L.; Grudziak, J.; Townsend, R.R.; Dunn, T.N.; Grant, R.W.; Adams, S.H.; Keim, N.L.; Cummings, B.P.; Stanhope, K.L.; Havel, P.J. Endocrine and metabolic effects of consuming fructose- and glucose-sweetened beverages with meals in obese men and women: Influence of insulin resistance on plasma triglyceride responses. J. Clin. Endocrinol. Metab. 2009, 94, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Seppala-Lindroos, A.; Vehkavaara, S.; Bergholm, R.; Frayn, K.N.; Fielding, B.A.; Yki-Jarvinen, H. Liver fat and lipid oxidation in humans. Liver Int. 2009, 29, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Cassader, M.; Gambino, R.; Musso, G.; Depetris, N.; Mecca, F.; Cavallo-Perin, P.; Pacini, G.; Rizzetto, M.; Pagano, G. Postprandial triglyceride-rich lipoprotein metabolism and insulin sensitivity in nonalcoholic steatohepatitis patients. Lipids 2001, 36, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Watts, G.F.; Adiels, M.; Soderlund, S.; Chan, D.C.; Hakkarainen, A.; Lundbom, N.; Matikainen, N.; Kahri, J.; Verges, B.; et al. Kinetic and related determinants of plasma triglyceride concentration in abdominal obesity: Multicenter tracer kinetic study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Hakkinen, A.; Olofsson, S.O.; Yki-Jarvinen, H.; et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Sreekumar, R.; Rasmussen, D.; Lindor, K.; Nair, K.S. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology 2002, 35, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Jacot-Descombes, D.; Lecoultre, V.; Fielding, B.A.; Carrel, G.; Le, K.A.; Schneiter, P.; Bortolotti, M.; Frayn, K.N.; Tappy, L. Sex differences in lipid and glucose kinetics after ingestion of an acute oral fructose load. Br. J. Nutr. 2010, 104, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Faeh, D.; Minehira, K.; Schwarz, J.M.; Periasamy, R.; Park, S.; Tappy, L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005, 54, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Horikawa, Y. ChREBP: A glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr. J. 2008, 55, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [PubMed]
- Li, M.V.; Chang, B.; Imamura, M.; Poungvarin, N.; Chan, L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes 2006, 55, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizuka, K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim. Biophys. Acta 2017, 1863, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose-2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose-5-phosphate mediates glucose-induced lipogenesis by xylulose-5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; Miyashita, M.; Cho, B.H.; Nakamura, M.T. Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochem. Biophys. Res. Commun. 2009, 390, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar] [CrossRef] [PubMed]
- ter Horst, K.W.; Gilijamse, P.W.; Demirkiran, A.; van Wagensveld, B.A.; Ackermans, M.T.; Verheij, J.; Romijn, J.A.; Nieuwdorp, M.; Maratos-Flier, E.; Herman, M.A.; et al. The FGF21 response to fructose predicts metabolic health and persists after bariatric surgery in obese humans. Mol. Metab. 2017. [Google Scholar] [CrossRef]
- Zhang, D.; Tong, X.; VanDommelen, K.; Gupta, N.; Stamper, K.; Brady, G.F.; Meng, Z.; Lin, J.; Rui, L.; Omary, M.B.; et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J. Clin. Investig. 2017, 127, 2855–2867. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Finck, B.N. ChREBP refines the hepatic response to fructose to protect the liver from injury. J. Clin. Investig. 2017, 127, 2533–2535. [Google Scholar] [CrossRef] [PubMed]
- Eissing, L.; Scherer, T.; Tödter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef] [PubMed]
- Aragno, M.; Tomasinelli, C.E.; Vercellinatto, I.; Catalano, M.G.; Collino, M.; Fantozzi, R.; Danni, O.; Boccuzzi, G. SREBP-1c in nonalcoholic fatty liver disease induced by western-type high-fat diet plus fructose in rats. Free Radic. Biol. Med. 2009, 47, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, R.M.; Ueno, M.; Silva, M.S.; Tavares, D.Q.; Carvalho, C.R.; Saad, M.J.; Gontijo, J.A. A high-fructose diet induces insulin resistance but not blood pressure changes in normotensive rats. Braz. J. Med. Biol. Res. 2001, 34, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Rizza, R.A.; Romero, J.C. High-fructose feeding elicits insulin resistance, hyperinsulinism, and hypertension in normal mongrel dogs. Hypertension 1994, 23, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.T.; Yuen, V.G.; McNeill, J.H. The fructose-fed rat: A review on the mechanisms of fructose-induced insulin resistance and hypertension. Mol. Cell. Biochem. 2009, 332, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; Tomita, S.; Sekiya, M.; et al. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes 2004, 53, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Dobrzyn, A.; Man, W.C.; Chu, K.; Sampath, H.; Kim, H.J.; Ntambi, J.M. Stearoyl-CoA desaturase 1 gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J. Biol. Chem. 2004, 279, 25164–25171. [Google Scholar] [CrossRef] [PubMed]
- Bindesboll, C.; Fan, Q.; Norgaard, R.C.; MacPherson, L.; Ruan, H.B.; Wu, J.; Pedersen, T.A.; Steffensen, K.R.; Yang, X.; Matthews, J.; et al. Liver X receptor regulates hepatic nuclear o-glcnac signaling and carbohydrate responsive element-binding protein activity. J. Lipid Res. 2015, 56, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Loffler, M.G.; et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Randin, J.P.; Felber, J.P.; Chiolero, R.; Simonson, D.C.; Jequier, E.; DeFronzo, R.A. Comparison of thermogenic effect of fructose and glucose in normal humans. Am. J. Physiol. 1986, 250, E718–E724. [Google Scholar] [PubMed]
- Rizkalla, S.W.; Luo, J.; Guilhem, I.; Boillot, J.; Bruzzo, F.; Chevalier, A.; Slama, G. Comparative effects of 6 week fructose, dextrose and starch feeding on fat-cell lipolysis in normal rats: Effects of isoproterenol, theophylline and insulin. Mol. Cell. Biochem. 1992, 109, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Sayed, A.; Binnert, C.; Le, K.A.; Bortolotti, M.; Schneiter, P.; Tappy, L. A high-fructose diet impairs basal and stress-mediated lipid metabolism in healthy male subjects. Br. J. Nutr. 2008, 100, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Lafontan, M.; Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res. 2009, 48, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Camporez, J.P.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, E.; Jensen, M.D. Quantification of adipose tissue insulin sensitivity. J. Investig. Med. 2016, 64, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Taskinen, M.R.; Olofsson, S.O.; Levin, M. Ectopic lipid storage and insulin resistance: A harmful relationship. J. Int. Med. 2013, 274, 25–40. [Google Scholar] [CrossRef] [PubMed]
- ter Horst, K.W.; van Galen, K.A.; Gilijamse, P.W.; Hartstra, A.V.; de Groot, P.F.; van der Valk, F.M.; Ackermans, M.T.; Nieuwdorp, M.; Romijn, J.A.; Serlie, M.J. Methods for quantifying adipose tissue insulin resistance in overweight/obese humans. Int. J. Obes. 2017, 41, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Haymond, M.W.; Rizza, R.A.; Cryer, P.E.; Miles, J.M. Influence of body fat distribution on free fatty acid metabolism in obesity. J. Clin. Investig. 1989, 83, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Baena, M.; Sangüesa, G.; Dávalos, A.; Latasa, M.-J.; Sala-Vila, A.; Sánchez, R.M.; Roglans, N.; Laguna, J.C.; Alegret, M. Fructose, but not glucose, impairs insulin signaling in the three major insulin-sensitive tissues. Sci. Rep. 2016, 6, 26149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrana, A.; Fabry, P.; Slabochova, Z.; Kazdova, L. Effect of dietary fructose on free fatty acid release from adipose tissue and serum free fatty acid concentration in the rat. Nutr. Metab. 1974, 17, 74–83. [Google Scholar] [PubMed]
- Ma, J.; Sloan, M.; Fox, C.S.; Hoffmann, U.; Smith, C.E.; Saltzman, E.; Rogers, G.T.; Jacques, P.F.; McKeown, N.M. Sugar-sweetened beverage consumption is associated with abdominal fat partitioning in healthy adults. J. Nutr. 2014, 144, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Fox, C.; Speliotes, E.; Hoffmann, U.; Smith, C.; Saltzman, E.; Jacques, P.; McKeown, N. Sugar-sweetened beverage intake is associated with fatty liver in the Framingham offspring study. J. Hepatol. 2015, 63, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Sievenpiper, J.L.; de Souza, R.J.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; Beyene, J.; Chiavaroli, L.; Di Buono, M.; Jenkins, A.L.; Leiter, L.A.; et al. Effect of fructose on body weight in controlled feeding trials: A systematic review and meta-analysis. Ann. Intern. Med. 2012, 156, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, S. Implications of diet on nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 2010, 26, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Bjornson, E.; Adiels, M.; Taskinen, M.R.; Boren, J. Kinetics of plasma triglycerides in abdominal obesity. Curr. Opin. Lipidol. 2017, 28, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.J.; Hellerstein, M.K. Thematic review series: Patient-oriented research. Recent advances in liver triacylglycerol and fatty acid metabolism using stable isotope labeling techniques. J. Lipid Res. 2006, 47, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Barrows, B.R.; Parks, E.J. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Nemes, K.; Aberg, F. Interpreting lipoproteins in nonalcoholic fatty liver disease. Curr. Opin. Lipidol. 2017, 28, 355–360. [Google Scholar] [CrossRef] [PubMed]
- David Wang, D.; Sievenpiper, J.L.; de Souza, R.J.; Cozma, A.I.; Chiavaroli, L.; Ha, V.; Mirrahimi, A.; Carleton, A.J.; Di Buono, M.; Jenkins, A.L.; et al. Effect of fructose on postprandial triglycerides: A systematic review and meta-analysis of controlled feeding trials. Atherosclerosis 2014, 232, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Sievenpiper, J.L.; Carleton, A.J.; Chatha, S.; Jiang, H.Y.; de Souza, R.J.; Beyene, J.; Kendall, C.W.; Jenkins, D.J. Heterogeneous effects of fructose on blood lipids in individuals with type 2 diabetes: Systematic review and meta-analysis of experimental trials in humans. Diabetes Care 2009, 32, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Havel, P.J. Fructose consumption: Potential mechanisms for its effects to increase visceral adiposity and induce dyslipidemia and insulin resistance. Curr. Opin. Lipidol. 2008, 19, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Singh, A.B.; Azhar, S.; Seidah, N.G.; Liu, J. High-fructose feeding promotes accelerated degradation of hepatic LDL receptor and hypercholesterolemia in hamsters via elevated circulating Pcsk9 levels. Atherosclerosis 2015, 239, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Campos, V.C.; Tappy, L. Physiological handling of dietary fructose-containing sugars: Implications for health. Int. J. Obes. 2016, 40 (Suppl. 1), S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Topping, D.L.; Mayes, P.A. The immediate effects of insulin and fructose on the metabolism of the perfused liver. Changes in lipoprotein secretion, fatty acid oxidation and esterification, lipogenesis and carbohydrate metabolism. Biochem. J. 1972, 126, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Prager, G.N.; Ontko, J.A. Direct effects of fructose metabolism on fatty acid oxidation in a recombined rat liver mitochondria-hish speed supernatant system. Biochim. Biophys. Acta 1976, 424, 386–395. [Google Scholar] [CrossRef]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Havel, P.J. Fructose consumption: Recent results and their potential implications. Ann. N. Y. Acad. Sci. 2010, 1190, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Cederbaum, A.I.; Zhang, Y.L.; Ginsberg, H.N.; Williams, K.J.; Fisher, E.A. Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J. Clin. Investig. 2004, 113, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Marriott, B.P.; Cole, N.; Lee, E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr. 2009, 139, 1228s–1235s. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, N.; Sandboge, S.; Kaartinen, N.E.; Mannisto, S.; Eriksson, J.G. Higher fructose intake is inversely associated with risk of nonalcoholic fatty liver disease in older Finnish adults. Am. J. Clin. Nutr. 2014, 100, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with NAFLD. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Ma, J.; Patel, K.; Berger, S.; Lau, J.; Lichtenstein, A.H. Fructose, high-fructose corn syrup, sucrose, and nonalcoholic fatty liver disease or indexes of liver health: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2014, 100, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Te Morenga, L.; Mallard, S.; Mann, J. Dietary sugars and body weight: Systematic review and meta-analyses of randomised controlled trials and cohort studies. BMJ 2012, 346, e7492. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Health effects of fructose and fructose-containing caloric sweeteners: Where do we stand 10 years after the initial whistle blowings? Curr. Diabetes Rep. 2015, 15, 627. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.; Sievenpiper, J.L.; de Souza, R.J.; Cozma, A.I.; Mirrahimi, A.; Carleton, A.J.; Ha, V.; Di Buono, M.; Jenkins, A.L.; Leiter, L.A.; et al. Effect of fructose on markers of non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of controlled feeding trials. Eur. J. Clin. Nutr. 2014, 68, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Prior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a high-fructose weight-maintaining diet on lipogenesis and liver fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Agebratt, C.; Strom, E.; Romu, T.; Dahlqvist-Leinhard, O.; Borga, M.; Leandersson, P.; Nystrom, F.H. A randomized study of the effects of additional fruit and nuts consumption on hepatic fat content, cardiovascular risk factors and basal metabolic rate. PLoS ONE 2016, 11, e0147149. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, M.A.; Nagy, G.A.; Mircea, P.A. Current management of NAFLD. Clujul Med. 2016, 89, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly, Y.M.; Carling, D. Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Choi, S.E.; Yi, S.A.; Jung, J.G.; Jung, I.R.; Shin, M.; Kang, S.; Oh, H.; Kim, H.J.; Kim, D.J.; et al. Glutamate dehydrogenase activator BCH stimulating reductive amination prevents high fat/high fructose diet-induced steatohepatitis and hyperglycemia in C57Bl/6J mice. Sci. Rep. 2016, 5, 37468. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The multifaceted aspects of fructose metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Machann, J.; Kuper, M.A.; Maier, I.B.; Spruss, A.; Konigsrainer, A.; Bischoff, S.C.; Bergheim, I. A moderate weight reduction through dietary intervention decreases hepatic fat content in patients with non-alcoholic fatty liver disease (NAFLD): A pilot study. Eur. J. Nutr. 2013, 52, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K.; et al. Effects of dietary fructose restriction on liver fat, de novo lipogenesis, and insulin kinetics in children with obesity. Gastroenterology 2017. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Weber, M.B.; Welsh, J.; Khatoon, F.; Jones, D.P.; Whitington, P.F.; McClain, C.J. Fructose and oxidized low-density lipoprotein in pediatric nonalcoholic fatty liver disease: A pilot study. Arch. Pediatr. Adolesc. Med. 2009, 163, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Haghighatdoost, F.; Salehi-Abargouei, A.; Surkan, P.J.; Azadbakht, L. The effects of low carbohydrate diets on liver function tests in nonalcoholic fatty liver disease: A systematic review and meta-analysis of clinical trials. J. Res. Med. Sci. 2016, 21, 53. [Google Scholar] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Seppala-Lindroos, A.; Vehkavaara, S.; Hakkinen, A.M.; Goto, T.; Westerbacka, J.; Sovijarvi, A.; Halavaara, J.; Yki-Jarvinen, H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocrinol. Metab. 2002, 87, 3023–3028. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Oral, E.A.; Dufour, S.; Befroy, D.; Ariyan, C.; Yu, C.; Cline, G.W.; DePaoli, A.M.; Taylor, S.I.; Gorden, P.; et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Investig. 2002, 109, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Lehrke, M.; Hendler, R.E.; Shulman, G.I. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005, 54, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Deivanayagam, S.; Mohammed, B.S.; Vitola, B.E.; Naguib, G.H.; Keshen, T.H.; Kirk, E.P.; Klein, S. Nonalcoholic fatty liver disease is associated with hepatic and skeletal muscle insulin resistance in overweight adolescents. Am. J. Clin. Nutr. 2008, 88, 257–262. [Google Scholar] [PubMed]
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Yki-Jarvinen, H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 2008, 135, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Birkenfeld, A.L.; Jurczak, M.J.; Kanda, S.; Guigni, B.A.; Jiang, D.C.; Zhang, D.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc. Natl. Acad. Sci. USA 2011, 108, 5748–5752. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Madiraju, A.K.; Gassaway, B.M.; Marcel, M.; Nasiri, A.R.; Butrico, G.; Marcucci, M.J.; Zhang, D.; Abulizi, A.; Zhang, X.M.; et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Investig. 2016, 126, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- ter Horst, K.W.; Gilijamse, P.W.; Versteeg, R.I.; Ackermans, M.T.; Nederveen, A.J.; la Fleur, S.E.; Romijn, J.A.; Nieuwdorp, M.; Zhang, D.; Samuel, V.T.; et al. Hepatic diacylglycerol-associated protein kinase cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017, 19, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Sun, R.Q.; Zeng, X.Y.; Choong, Z.H.; Wang, H.; Watt, M.J.; Ye, J.M. Activation of PPARalpha ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress. Diabetes 2013, 62, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, M.J.; Lee, A.H.; Jornayvaz, F.R.; Lee, H.Y.; Birkenfeld, A.L.; Guigni, B.A.; Kahn, M.; Samuel, V.T.; Glimcher, L.H.; Shulman, G.I. Dissociation of inositol-requiring enzyme (IRE1alpha)-mediated C-jun n-terminal kinase activation from hepatic insulin resistance in conditional x-box-binding protein-1 (XPB1) knock-out mice. J. Biol. Chem. 2012, 287, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Kopf, T.; Schaefer, H.L.; Troetzmueller, M.; Koefeler, H.; Broenstrup, M.; Konovalova, T.; Schmitz, G. Influence of fenofibrate treatment on triacylglycerides, diacylglycerides and fatty acids in fructose fed rats. PLoS ONE 2014, 9, e106849. [Google Scholar] [CrossRef] [PubMed]
- Mosca, A.; Nobili, V.; De Vito, R.; Crudele, A.; Scorletti, E.; Villani, A.; Alisi, A.; Byrne, C.D. Serum uric acid concentrations and fructose consumption are independently associated with nash in children and adolescents. J. Hepatol. 2017, 66, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, C.; Calabria, D.; Roda, A.; Cicero, A.F.G. Fructose intake, serum uric acid, and cardiometabolic disorders: A critical review. Nutrients 2017, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Sautin, Y.Y.; Oliver, W.J.; Roncal, C.; Mu, W.; Gabriela Sanchez-Lozada, L.; Rodriguez-Iturbe, B.; Nakagawa, T.; Benner, S.A. Lessons from comparative physiology: Could uric acid represent a physiologic alarm signal gone awry in western society? J. Comp. Physiol. B 2009, 179, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Park, S.K.; Lee, I.K.; Johnson, R.J. Uric acid-induced C-reactive protein expression: Implication on cell proliferation and nitric oxide production of human vascular cells. J. Am. Soc. Nephrol. 2005, 16, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Sievenpiper, J.L.; de Souza, R.J.; Chiavaroli, L.; Ha, V.; Cozma, A.I.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; Di Buono, M.; et al. The effects of fructose intake on serum uric acid vary among controlled dietary trials. J. Nutr. 2012, 142, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Madero, M.; Arriaga, J.C.; Jalal, D.; Rivard, C.; McFann, K.; Perez-Mendez, O.; Vazquez, A.; Ruiz, A.; Lanaspa, M.A.; Jimenez, C.R.; et al. The effect of two energy-restricted diets, a low-fructose diet versus a moderate natural fructose diet, on weight loss and metabolic syndrome parameters: A randomized controlled trial. Metabolism 2011, 60, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Zamudio-Cuevas, Y.; Hernandez-Diaz, C.; Pineda, C.; Reginato, A.M.; Cerna-Cortes, J.F.; Ventura-Rios, L.; Lopez-Reyes, A. Molecular basis of oxidative stress in gouty arthropathy. Clin. Rheumatol. 2015, 34, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [PubMed]
- Stack, A.G.; Hanley, A.; Casserly, L.F.; Cronin, C.J.; Abdalla, A.A.; Kiernan, T.J.; Murthy, B.V.; Hegarty, A.; Hannigan, A.; Nguyen, H.T. Independent and conjoint associations of gout and hyperuricaemia with total and cardiovascular mortality. QJM 2013, 106, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Bu, H.; Ma, X.; Zhao, S.; Li, X.; Lu, S. The relation between serum uric acid and HbA1c is dependent upon hyperinsulinemia in patients with newly diagnosed type 2 diabetes mellitus. J. Diabetes Res. 2016, 2016, 7184123. [Google Scholar] [CrossRef] [PubMed]
- Moulin, S.; Seematter, G.; Seyssel, K. Fructose use in clinical nutrition: Metabolic effects and potential consequences. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Mosca, A.; Della Corte, C.; Sartorelli, M.R.; Ferretti, F.; Nicita, F.; Vania, A.; Nobili, V. Beverage consumption and paediatric NAFLD. Eat. Weight Disord. 2016, 21, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Rippe, J.M.; Angelopoulos, T.J. Sugars, obesity, and cardiovascular disease: Results from recent randomized control trials. Eur. J. Nutr. 2016, 55, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2017, 91, 1545–1563. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.; Sievenpiper, J.L. Dietary sugar and body weight: Have we reached a crisis in the epidemic of obesity and diabetes? We have, but the pox on sugar is overwrought and overworked. Diabetes Care 2014, 37, 957–962. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. https://doi.org/10.3390/nu9090981
Ter Horst KW, Serlie MJ. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients. 2017; 9(9):981. https://doi.org/10.3390/nu9090981
Chicago/Turabian StyleTer Horst, Kasper W., and Mireille J. Serlie. 2017. "Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease" Nutrients 9, no. 9: 981. https://doi.org/10.3390/nu9090981
APA StyleTer Horst, K. W., & Serlie, M. J. (2017). Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients, 9(9), 981. https://doi.org/10.3390/nu9090981