Can Skin Exposure to Sunlight Prevent Liver Inflammation?




Abstract
:1. What Is Non-Alcoholic Fatty Liver Disease?
2. Sunlight-Derived Ultraviolet Radiation—A Systemic Modulator of Inflammation
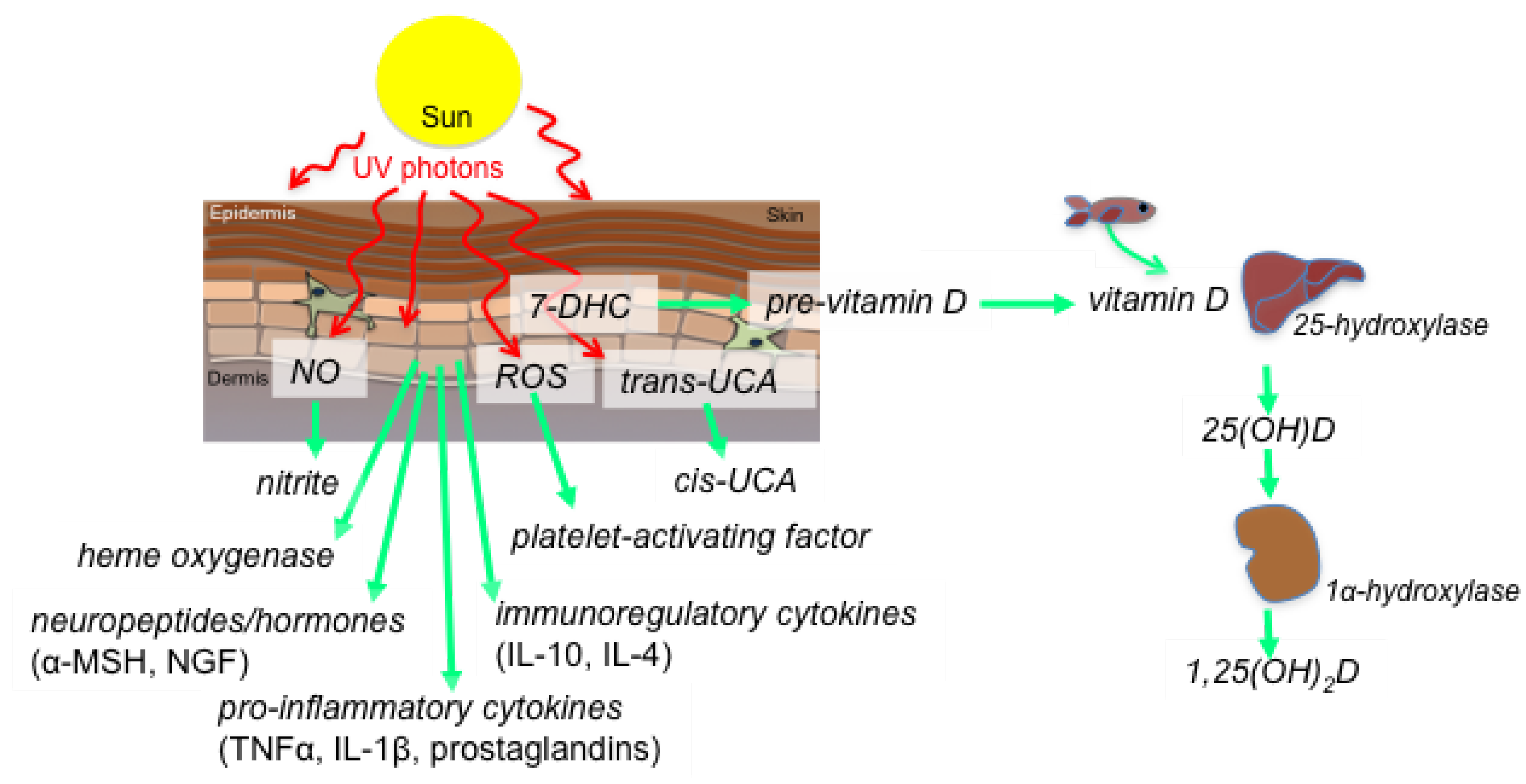
3. Hepatic Inflammation Linked with NAFLD
3.1. What Are the Roles of Resident and Infiltrating Immune Cells in Modulating Liver Inflammation?
3.2. What Are the Central Effector Molecules that Drive Liver Inflammation?
4. Evidence for the Control of Hepatic Inflammation by Ultraviolet Radiation: Vitamin D-Dependent and Independent Pathways
4.1. Effects of Phototherapy on Severity of NAFLD
4.2. Associations between Vitamin D and NAFLD

{kind=link}
{kind=link}
| Reference | Location | Sample Size | Percent Male | Age | NAFLD Detection | Assay Method | Significant Association | Summary of Findings |
|---|---|---|---|---|---|---|---|---|
| Barchetta et al., 2011 [31] | Italy | 262 | 53 | Adults | Ultrasound | DiaSorin LIAISON | Inverse | Low 25(OH)D was associated with NAFLD independent of metabolic syndrome, diabetes and insulin-resistance profile |
| Bril et al., 2014 [33] | USA | 239 | 85 | 18–70 year | Biopsy | DiaSorin LIAISON | None | Plasma 25(OH)D was not associated with insulin resistance, liver fat accumulation or severity of NASH |
| Dasarathy et al., 2014 [34] | USA | 187 | 27 | Adults | Biopsy | Standard automatic colorimetric methods | Inverse | Plasma 25(OHD was an independent predictor of NAFLD activity score |
| Hao et al., 2014 [35] | China | 514 | 100 | Adults | Ultrasound | Electrochemiluminescence immunoassay (Roche) | Inverse | Serum 25(OH)D was inversely associated with NAFLD, even in subjects with normal total body fat |
| Jablonski et al., 2013 [36] | USA | 1214 | 26 | ≥18 year | Ultrasound | RIA | Inverse | Compared with matched controls, NAFLD patients had significantly lower serum 25(OH)D |
| Kasapoglu et al., 2013 [37] | Turkey | 613 | 21 | Adults | Ultrasound | Not reported | Inverse | Low 25(OH)D was associated with NAFLD among non-obese subjects |
| Kucukazman et al., 2014 [38] | Turkey | 211 | 44 | Adults | Ultrasound | RIA | Inverse | Serum 25(OH)D was lower in patients with NAFLD compared to those without NAFLD |
| Li et al., 2013 [39] | China | 1248 | 55 | ≥20 year | Ultrasound | DiaSorin RIA | None | Serum 25(OH)D was not significantly associated with the presence of NAFLD |
| Liangpunsakul et al., 2011 [40] | USA | 1287 | 50 | ≥20 year | Alanine transaminase | RIA | Inverse | Significant inverse association between serum 25(OH)D and unexplained elevation in alanine aminotransaminase |
| Rhee et al., 2013 [41] | Korea | 6567 | 100 | 24–75 year | Ultrasound | Electrochemiluminescence immunoassay (Roche) | Inverse | Reduced risk for NAFLD with higher serum 25(OH)D, independent of obesity and metabolic syndrome |
| Seo et al., 2013 [42] | Korea | 1081 | 32 | 40–69 year | Computed tomography | DiaSorin LIAISON | Inverse | In subjects with diabetes or insulin resistance, low vitamin D status was associated with NAFLD, independent of visceral obesity |
| Targher et al., 2007 [43] | Italy | 120 | 67 | Adults | Biopsy | DiaSorin LIAISON | Inverse | Compared with matched controls, NAFLD patients had lower serum 25(OH)D |
| Bhatt et al., 2013 [32] | India | 335 | 71 | Adults | Ultrasound | DiaSorin RIA | Inverse | Low serum 25(OH)D was independently associated with NAFLD |
| Black et al., 2014 [44] | Australia | 994 | 52 | 17 year | Ultrasound | LC-MS/MS | Inverse | Lower serum 25(OH)D was significantly associated with NAFLD, independent of adiposity and insulin resistance |
| Katz et al., 2010 [45] | USA | 1630 | 52 | 12–19 year | Alanine transaminase | DiaSorin RIA | None | No independent association between vitamin D status and NAFLD after adjusting for obesity |
| Malespin et al., 2014 [46] | USA (Chinese American) | 407 | 51 | 6–18 year | Alanine transaminase | Immunochemiluminometric assay | Inverse | Suspected NAFLD was associated with lower 25(OH)D after adjusting for BMI, sex, triglycerides, total cholesterol, LDL, HDL |
| Nobili et al., 2014 [47] | Italy | 73 | 64 | 10–15 year | Ultrasound | HPLC | Inverse | 25(OH)D was inversely associated with NASH and fibrosis in overweight and obese children with NAFLD |
| Pirgon et al., 2013 [48] | Turkey | 87 | 48 | 11–15 year | Ultrasound | DiaSorin LIAISON | Inverse | In obese adolescents, those with NAFLD had significantly lower 25(OH)D than those without NAFLD |
4.3. Vitamin D Supplementation and NAFLD
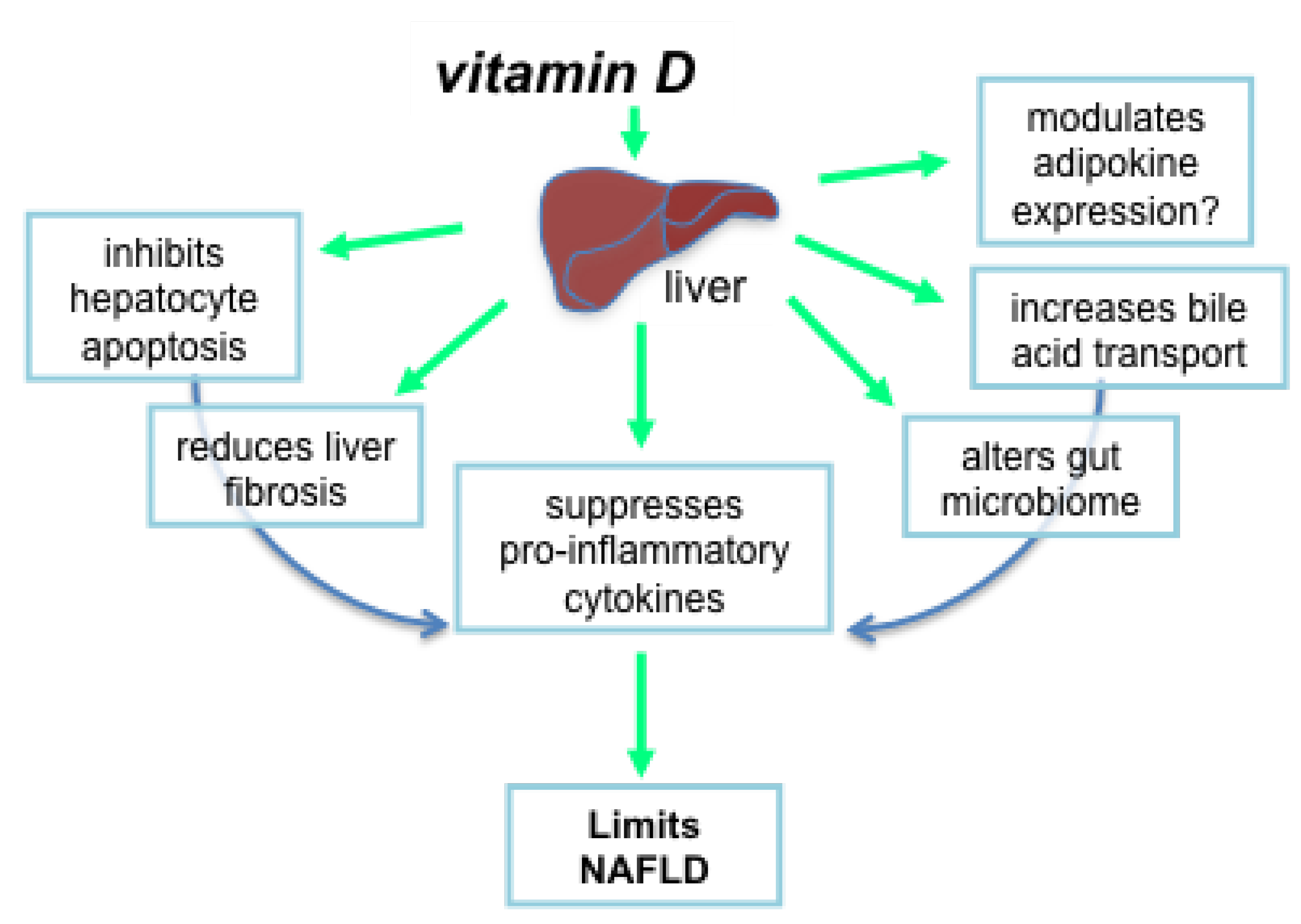
4.4. Mechanisms by Which Vitamin D Could Suppress Liver Pathology
4.4.1. Vitamin D Inhibits Hepatocyte Apoptosis
4.4.2. Vitamin D Reduces Liver Fibrosis
4.4.3. Does Vitamin D Modulate Adipokine Expression?
4.4.4. Vitamin D Alters the Gut Microbiome and Production of Bile Acids
4.4.5. Vitamin D Suppresses Proinflammatory Cytokines and Mediators of Oxidative Stress
4.5. Other UVR-Induced Mediators That Could Suppress Liver Inflammation
4.5.1. Activation of Dermal Nitric Oxide (NO) by UVR
4.5.2. NO-Mediated Upregulation of Heme Oxygenase Expression
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- LaBrecque, D.; Anania, F.; Ferenci, P.; Ghafoor Khan, A.; Goh, K.-L.; Hamid, S.S.; Isakov, V.; Lizarzabal, M.; Mojica Pernaranda, M.; Rivera Ramos, J.F.; et al. Nonalcoholic Fatty Liver Disease and Nonalcholic Steatohepatitis; World Gastroenterology Organisation: Milwaukee, WI, USA, 2012. [Google Scholar]
- Berlanga, A.; Guiu-Jurado, E.; Porras, J.A.; Auguet, T. Molecular pathways in non-alcoholic fatty liver disease. Clin. Exp. Gastroent. 2014, 7, 221–239. [Google Scholar]
- Lade, A.; Noon, L.A.; Friedman, S.L. Contributions of metabolic dysregulation and inflammation to nonalcoholic steatohepatitis, hepatic fibrosis, and cancer. Curr. Opin. Oncol. 2014, 26, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating tnf-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in nash. Hepatol. Inter. 2013, 7, 771–781. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Gorman, S.; Finlay-Jones, J.J. Modulation of the immune system by UV radiation: More than just the effects of vitamin D? Nat. Rev. Immunol. 2011, 11, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.M.; Gorman, S.; Geldenhuys, S.; Hart, P.H. Vitamin D and immunity. F1000prime Reports 2014, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Gorman, S. Exposure to UV wavelengths in sunlight suppresses immunity. To what extent is uv-induced vitamin D3 the mediator responsible? Clini. Biochem. 2013, 34, 3–13. [Google Scholar]
- Kundu, R.; Chain, B.M.; Coussens, A.K.; Khoo, B.; Noursadeghi, M. Regulation of CYP27B1 and CYP24A1 hydroxylases limits cell-autonomous activation of vitamin D in dendritic cells. Eur. J. Immunol. 2014, 44, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin d receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef] [PubMed]
- Gascon-Barre, M.; Demers, C.; Mirshahi, A.; Neron, S.; Zalzal, S.; Nanci, A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology 2003, 37, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.; Schwentker, A.; Billiar, T.R.; Vodovotz, Y. Autologous nitric oxide protects mouse and human keratinocytes from ultraviolet B radiation-induced apoptosis. Am. J. Physiol. 2003, 284, 1140–1148. [Google Scholar] [CrossRef]
- Yuen, K.S.; Nearn, M.R.; Halliday, G.M. Nitric oxide-mediated depletion of langerhans cells from the epidermis may be involved in uva radiation-induced immunosuppression. Nitric Oxide 2002, 6, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Grochot-Przeczek, A.; Dulak, J.; Jozkowicz, A. Haem oxygenase-1: Non-canonical roles in physiology and pathology. Clin. Sci. 2012, 122, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, R.M.; Reeve, V.E. Potential protection of skin by acute UVA irradiation—From cellular to animal models. Prog. Biophysics Mol. Biol. 2006, 92, 86–91. [Google Scholar] [CrossRef]
- Reeve, V.E.; Tyrrell, R.M.; Allanson, M.; Domanski, D.; Blyth, L. The role of interleukin-6 in UVA protection against UVB-induced immunosuppression. J. Invest. Dermatol. 2009, 129, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Reeve, V.E.; Allanson, M.; Cho, J.L.; Arun, S.J.; Domanski, D. Interdependence between heme oxygenase-1 induction and estrogen-receptor-beta signaling mediates photoimmune protection by UVA radiation in mice. J. Invest. Dermatol. 2009, 129, 2702–2710. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroent. Hepatol. 2012, 27, 65–68. [Google Scholar] [CrossRef]
- Martel, C.; Allouche, M.; Esposti, D.D.; Fanelli, E.; Boursier, C.; Henry, C.; Chopineau, J.; Calamita, G.; Kroemer, G.; Lemoine, A.; et al. Glycogen synthase kinase 3-mediated voltage-dependent anion channel phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation. Hepatology 2013, 57, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, N.; Reeve, V.E.; Nishimura, H.; Satoh, M.; Tohyama, C. Cutaneous metallothionein induction by ultraviolet B irradiation in interleukin-6 null mice. J. Invest. Dermatol. 2000, 114, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, N.; Tohyama, C.; Satoh, M.; Nishimura, H.; Reeve, V.E. Defective immune response and severe skin damage following UVB irradiation in interleukin-6-deficient mice. Immunology 1999, 97, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Shreedhar, V.; Giese, T.; Sung, V.W.; Ullrich, S.E. A cytokine cascade including prostaglandin e2, il-4, and il-10 is responsible for UV-induced systemic immune suppression. J. Immunol. 1998, 160, 3783–3789. [Google Scholar] [PubMed]
- Mowbray, M.; McLintock, S.; Weerakoon, R.; Lomatschinsky, N.; Jones, S.; Rossi, A.G.; Weller, R.B. Enzyme-independent no stores in human skin: Quantification and influence of uv radiation. J. Invest. Dermatol. 2009, 129, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Cheng, Y.F.; Lai, C.Y.; Hsu, L.W.; Chang, Y.C.; Deng, J.Y.; Huang, Y.Z.; Honda, H.; Chen, K.D.; Wang, C.C.; et al. Impact of artificial sunlight therapy on the progress of non-alcoholic fatty liver disease in rats. J. Hepatol. 2011, 55, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, S.; Hart, P.H.; Endersby, R.; Jacoby, P.; Feelisch, M.; Weller, R.B.; Matthews, V.; Gorman, S. Ultraviolet radiation suppresses obesity and symptoms of metabolic syndrome independently of vitamin D in mice fed a high-fat diet. Diabetes 2014, 63, 3759–3769. [Google Scholar] [CrossRef] [PubMed]
- Gorman, S.; Scott, N.M.; Tan, D.H.; Weeden, C.E.; Tuckey, R.C.; Bisley, J.L.; Grimbaldeston, M.A.; Hart, P.H. Acute erythemal ultraviolet radiation causes systemic immunosuppression in the absence of increased 25-hydroxyvitamin D3 levels in male mice. PLoS ONE 2012, 7, e46006. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Angelico, F.; Del Ben, M.; Baroni, M.G.; Pozzilli, P.; Morini, S.; Cavallo, M.G. Strong association between non alcoholic fatty liver disease (NAFLD) and low 25(OH) vitamin D levels in an adult population with normal serum liver enzymes. BMC Med. 2011, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.P.; Nigam, P.; Misra, A.; Guleria, R.; Qadar Pasha, M.A. Independent associations of low 25 hydroxy vitamin D and high parathyroid hormonal levels with nonalcoholic fatty liver disease in Asian Indians residing in north India. Atherosclerosis 2013, 230, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Maximos, M.; Portillo-Sanchez, P.; Biernacki, D.; Lomonaco, R.; Subbarayan, S.; Correa, M.; Lo, M.; Suman, A.; Cusi, K. Relationship of vitamin D with insulin resistance and disease severity in non-alcoholic steatohepatitis. J. Hepatol. 2015, 62, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, J.; Periyalwar, P.; Allampati, S.; Bhinder, V.; Hawkins, C.; Brandt, P.; Khiyami, A.; McCullough, A.J.; Dasarathy, S. Hypovitaminosis D is associated with increased whole body fat mass and greater severity of non-alcoholic fatty liver disease. Liver Inter. 2014, 34, 118–127. [Google Scholar] [CrossRef]
- Hao, Y.P.; Ma, X.J.; Luo, Y.Q.; Ni, J.; Dou, J.X.; Hu, Y.Q.; Zhu, J.A.; Bao, Y.Q.; Jia, W.P. Serum vitamin D is associated with non-alcoholic fatty liver disease in chinese males with normal weight and liver enzymes. Acta Pharmacol. Sinica 2014, 35, 1150–1156. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Jovanovich, A.; Holmen, J.; Targher, G.; McFann, K.; Kendrick, J.; Chonchol, M. Low 25-hydroxyvitamin D level is independently associated with non-alcoholic fatty liver disease. NMCD 2013, 23, 792–798. [Google Scholar] [PubMed]
- Kasapoglu, B.; Turkay, C.; Yalcin, K.S.; Carlioglu, A.; Sozen, M.; Koktener, A. Low vitamin D levels are associated with increased risk for fatty liver disease among non-obese adults. Clin. Med. 2013, 13, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Kucukazman, M.; Ata, N.; Dal, K.; Yeniova, A.O.; Kefeli, A.; Basyigit, S.; Aktas, B.; Akin, K.O.; Agladioglu, K.; Ure, O.S.; et al. The association of vitamin D deficiency with non-alcoholic fatty liver disease. Clinics 2014, 69, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, L.; Pan, S.; Wu, X.; Yin, X. No significant association between vitamin D and nonalcoholic fatty liver disease in a chinese population. Dig. Dis. Sci. 2013, 58, 2376–2382. [Google Scholar] [CrossRef] [PubMed]
- Liangpunsakul, S.; Chalasani, N. Serum vitamin D concentrations and unexplained elevation in alt among us adults. Dig. Dis. Sci. 2011, 56, 2124–2129. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.J.; Kim, M.K.; Park, S.E.; Park, C.Y.; Baek, K.H.; Lee, W.Y.; Kang, M.I.; Park, S.W.; Kim, S.W.; Oh, K.W. High serum vitamin D levels reduce the risk for nonalcoholic fatty liver disease in healthy men independent of metabolic syndrome. Endocr. J. 2013, 60, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.A.; Eun, C.R.; Cho, H.; Lee, S.K.; Yoo, H.J.; Kim, S.G.; Choi, K.M.; Baik, S.H.; Choi, D.S.; Yim, H.J.; et al. Low vitamin D status is associated with nonalcoholic fatty liver disease independent of visceral obesity in korean adults. PLoS ONE 2013, 8, e75197. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Scala, L.; Cigolini, M.; Zenari, L.; Falezza, G.; Arcaro, G. Associations between serum 25-hydroxyvitamin D3 concentrations and liver histology in patients with non-alcoholic fatty liver disease. NMCD 2007, 17, 517–524. [Google Scholar] [PubMed]
- Black, L.J.; Jacoby, P.; She Ping-Delfos, W.C.; Mori, T.A.; Beilin, L.J.; Olynyk, J.K.; Ayonrinde, O.T.; Huang, R.C.; Holt, P.G.; Hart, P.H.; et al. Low serum 25-hydroxyvitamin D concentrations associate with non-alcoholic fatty liver disease in adolescents independent of adiposity. J. Gastroent. Hepatol. 2014, 29, 1215–1222. [Google Scholar] [CrossRef]
- Katz, K.; Brar, P.C.; Parekh, N.; Liu, Y.H.; Weitzman, M. Suspected nonalcoholic fatty liver disease is not associated with vitamin D status in adolescents after adjustment for obesity. J. Obes. 2010, 2010, 496829. [Google Scholar] [CrossRef] [PubMed]
- Malespin, M.; Sleesman, B.; Lau, A.; Wong, S.S.; Cotler, S.J. Prevalence and correlates of suspected nonalcoholic fatty liver disease in chinese american children. J. Clin. Gastroent. 2015, 49, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Giorgio, V.; Liccardo, D.; Bedogni, G.; Morino, G.; Alisi, A.; Cianfarani, S. Vitamin D levels and liver histological alterations in children with nonalcoholic fatty liver disease. Eur. J. Endocr. 2014, 170, 547–553. [Google Scholar] [CrossRef]
- Pirgon, O.; Cekmez, F.; Bilgin, H.; Eren, E.; Dundar, B. Low 25-hydroxyvitamin D level is associated with insulin sensitivity in obese adolescents with non-alcoholic fatty liver disease. Obes. Res. Clin. Pract. 2013, 7, e275–e283. [Google Scholar] [CrossRef] [PubMed]
- Eliades, M.; Spyrou, E.; Agrawal, N.; Lazo, M.; Brancati, F.L.; Potter, J.J.; Koteish, A.A.; Clark, J.M.; Guallar, E.; Hernaez, R. Meta-analysis: Vitamin D and non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2013, 38, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, R.; Lazo, M.; Bonekamp, S.; Kamel, I.; Brancati, F.L.; Guallar, E.; Clark, J.M. Diagnostic accuracy and reliability of ultrasonography for the detection of fatty liver: A meta-analysis. Hepatology 2011, 54, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.S.; Barlow, S.E.; Dietz, W.H. Prevalence of abnormal serum aminotransferase values in overweight and obese adolescents. J. Pediatr. 2000, 136, 727–733. [Google Scholar] [PubMed]
- Carter, G.D. Accuracy of 25-hydroxyvitamin D assays: Confronting the issues. Curr. Drug Targets 2011, 12, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.K.; Lucas, R.M.; Banks, E.; Ponsonby, A.L. Variability in vitamin D assays impairs clinical assessment of vitamin d status. Intern. Med. J. 2011, 42, 43–50. [Google Scholar] [CrossRef]
- Binkley, N.; Sempos, C.T. Standardizing vitamin D assays: The way forward. J. Bone Miner. Res. 2014, 29, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Feelisch, M.; Gorman, S.; Weller, R.B. Vitamin D status and ill health. Lancet Diab. Endocrin. 2014, 2, e8. [Google Scholar] [CrossRef]
- Bolt, M.J.; Sitrin, M.D.; Favus, M.J.; Rosenberg, I.H. Hepatic vitamin D 25-hydroxylase: Inhibition by bile duct ligation or bile salts. Hepatology 1981, 1, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver vitamin D receptor, CYP2R1, and CYP27A1 expression: Relationship with liver histology and vitamin D3 levels in patients with nonalcoholic steatohepatitis or hepatitis C virus. Hepatology 2012, 56, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Grunhage, F.; Hochrath, K.; Krawczyk, M.; Hoblinger, A.; Obermayer-Pietsch, B.; Geisel, J.; Trauner, M.; Sauerbruch, T.; Lammert, F. Common genetic variation in vitamin D metabolism is associated with liver stiffness. Hepatology 2012, 56, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; White, S.W.; Marsh, J.A.; Lye, S.J.; Connor, K.L.; Maganga, R.; Ayonrinde, O.T.; Olynyk, J.K.; Mori, T.A.; Beilin, L.J.; et al. Association between liver-specific gene polymorphisms and their expression levels with nonalcoholic fatty liver disease. Hepatology 2013, 57, 590–600. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Institutes of Health. Clinicaltrials.Gov. Avaiable online: https://clinicaltrials.gov/ (accessed on 22 April 2015).
- Sharifi, N.; Amani, R.; Hajiani, E.; Cheraghian, B. Does vitamin D improve liver enzymes, oxidative stress, and inflammatory biomarkers in adults with non-alcoholic fatty liver disease? A randomized clinical trial. Endocrine 2014, 47, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Cortez-Pinto, H. Exercise and improvements of nafld: Practical recommendations. J. Hepatol. 2015. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight loss via lifestyle modification significantly reduces features of nonalcoholi steatohepatitis. Gastroenterology 2015. [Google Scholar] [CrossRef]
- Keating, S.E.; Hackett, D.A.; George, J.; Johnson, N.A. Exercise and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 57, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, Y.; Xie, H.; Zheng, S. Calcitriol inhibits hepatocyte apoptosis in rat allograft by regulating apoptosis-associated genes. Int. Immunopharmacol. 2007, 7, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.B.; Zheng, S.S.; Jia, C.K.; Wang, Y. Role of 1,25-dihydroxyvitamin D3 in preventing acute rejection of allograft following rat orthotopic liver transplantation. Chin. Med. J. 2004, 117, 408–412. [Google Scholar] [PubMed]
- Potter, J.J.; Liu, X.; Koteish, A.; Mezey, E. 1,25-dihydroxyvitamin D3 and its nuclear receptor repress human alpha1 (i) collagen expression and type i collagen formation. Liver Internat. 2013, 33, 677–686. [Google Scholar] [CrossRef]
- Beilfuss, A.; Sowa, J.P.; Sydor, S.; Beste, M.; Bechmann, L.P.; Schlattjan, M.; Syn, W.K.; Wedemeyer, I.; Mathe, Z.; Jochum, C.; et al. Vitamin D counteracts fibrogenic TGF-beta signalling in human hepatic stellate cells both receptor-dependently and independently. Gut 2015, 64, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.M.; Torres, D.M.; Harrison, S.A. Vitamin D and nonalcoholic fatty liver disease (nafld): Is it more than just an association? Hepatology 2013, 58, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Giby, V.G.; Ajith, T.A. Role of adipokines and peroxisome proliferator-activated receptors in nonalcoholic fatty liver disease. World J. Hepatol. 2014, 6, 570–579. [Google Scholar] [PubMed]
- Husemoen, L.L.; Skaaby, T.; Martinussen, T.; Jorgensen, T.; Thuesen, B.H.; Kistorp, C.; Jeppesen, J.; Thyssen, J.P.; Meldgaard, M.; Szecsi, P.B.; et al. Investigating the causal effect of vitamin D on serum adiponectin using a mendelian randomization approach. Eur. J. Clin. Nutr. 2014, 68, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and toll-like receptor activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Chen, Y.; Zhu, G.; Zhao, Q.; Li, Y.C. 1,25-dihydroxyvitamin D3 upregulates leptin expression in mouse adipose tissue. J. Endocrinol. 2013, 216, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Edita, S.; Aleksandar, K.; Dragana, T.N.; Dragana, S.; Branka, K.Z.; Biljana, S.G.; Sanja, S.; Esma, I.R. Vitamin d and dysfunctional adipose tissue in obesity. Angiology 2014. [Google Scholar] [CrossRef]
- Roth, C.L.; Elfers, C.; Kratz, M.; Hoofnagle, A.N. Vitamin d deficiency in obese children and its relationship to insulin resistance and adipokines. J. Obesity 2011, 2011, 495101. [Google Scholar] [CrossRef]
- Vaidya, A.; Underwood, P.C.; Annes, J.P.; Sun, B.; Williams, G.H.; Forman, J.P.; Williams, J.S. The influence of sodium- and calcium-regulatory hormone interventions on adipocytokines in obesity and diabetes. Metabol. Clin. Exp. 2013, 62, 539–547. [Google Scholar] [CrossRef]
- Vilarrasa, N.; Vendrell, J.; Maravall, J.; Elio, I.; Solano, E.; San Jose, P.; Garcia, I.; Virgili, N.; Soler, J.; Gomez, J.M. Is plasma 25(OH)D related to adipokines, inflammatory cytokines and insulin resistance in both a healthy and morbidly obese population? Endocrine 2010, 38, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.P.; Kong, M.; Zheng, S.; Ren, Y.; Zhu, L.; Shi, H.; Duan, Z. Vitamin d in liver diseases: From mechanisms to clinical trials. J. Gastroent. Hepatol. 2013, 28, 49–55. [Google Scholar] [CrossRef]
- Bouillon, R.; Carmeliet, G.; Lieben, L.; Watanabe, M.; Perino, A.; Auwerx, J.; Schoonjans, K.; Verstuyf, A. Vitamin D and energy homeostasis: Of mice and men. Nature Rev. Endocrinol. 2014, 10, 79–87. [Google Scholar] [CrossRef]
- George, N.; Kumar, T.P.; Antony, S.; Jayanarayanan, S.; Paulose, C.S. Effect of vitamin D3 in reducing metabolic and oxidative stress in the liver of streptozotocin-induced diabetic rats. Br. J. Nutr. 2012, 108, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Zhu, L.; Bai, L.; Zhang, X.; Chen, Y.; Liu, S.; Zheng, S.; Pandol, S.J.; Han, Y.P.; Duan, Z. Vitamin D deficiency promotes nonalcoholic steatohepatitis through impaired enterohepatic circulation in animal model. Am. J. Physiol. Gastroint. Liver Physiol. 2014, 307, G883–G893. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Gu, Q.; Cui, Z. Responses to: Nitric oxide as a protector from NAFLD. Hepatology 2015. [Google Scholar] [CrossRef]
- Vinciguerra, M. Nitric oxide as a protector from NAFLD. Hepatology 2014. [Google Scholar] [CrossRef]
- Kim, P.K.; Zuckerbraun, B.S.; Otterbein, L.E.; Vodovotz, Y.; Billiar, T.R. 'Til cell death do us part: Nitric oxide and mechanisms of hepatotoxicity. Biol. Chem. 2004, 385, 11–15. [Google Scholar] [PubMed]
- Suzuki, H.; Menegazzi, M.; Carcereri de Prati, A.; Mariotto, S.; Armato, U. Nitric oxide in the liver: Physiopathological roles. Adv. Nuroimmunol. 1995, 5, 379–410. [Google Scholar] [CrossRef]
- Molinari, C.; Uberti, F.; Grossini, E.; Vacca, G.; Carda, S.; Invernizzi, M.; Cisari, C. 1alpha,25-dihydroxycholecalciferol induces nitric oxide production in cultured endothelial cells. Cell. Physiol. Biochem. 2011, 27, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.M.; Kuo, M.C.; Kuo, H.T.; Hwang, S.J.; Tsai, J.C.; Chen, H.C.; Lai, Y.H. 1-alpha,25-dihydroxyvitamin D3 regulates inducible nitric oxide synthase messenger rna expression and nitric oxide release in macrophage-like raw 264.7 cells. J. Lab. Clin. Med. 2004, 143, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Aparna, R.; Subhashini, J.; Roy, K.R.; Reddy, G.S.; Robinson, M.; Uskokovic, M.R.; Venkateswara Reddy, G.; Reddanna, P. Selective inhibition of cyclooxygenase-2 (COX-2) by 1alpha,25-dihydroxy-16-ene-23-yne-vitamin D3, a less calcemic vitamin d analog. J. Cell. Biol. 2008, 104, 1832–1842. [Google Scholar]
- Zhou, Y.; Zhou, X.; Wang, X. 1,25-dihydroxyvitamin D3 prevented allergic asthma in a rat model by suppressing the expression of inducible nitric oxide synthase. Allergy Asthma Proc. 2008, 29, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Weitzberg, E.; Hezel, M.; Lundberg, J.O. Nitrate-nitrite-nitric oxide pathway: Implications for anesthesiology and intensive care. Anesthesiology 2010, 113, 1460–1475. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Fernandez, B.O.; Hamilton, M.B.; Lang, N.N.; Gallagher, J.M.C.; Newby, D.E.; Feelisch, M.; Weller, R.B. UVA irradiation of human skin vasodilates arterial vasculature and lowers blood pressure independently of nitric oxide synthase. J. Invest. Dermatol. 2014, 134, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Miranda, K.M.; Espey, M.G.; Pluta, R.M.; Hewett, S.J.; Colton, C.; Vitek, M.; Feelisch, M.; Grisham, M.B. Mechanisms of the antioxidant effects of nitric oxide. Antioxid. Redox. Signal. 2001, 3, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Bossy-Wetzel, E. Impact of nitric oxide on metabolism in health and age-related disease. Diabetes Obes. Metabol. 2010, 12, 126–133. [Google Scholar] [CrossRef]
- Malaguarnera, L.; Madeddu, R.; Palio, E.; Arena, N.; Malaguarnera, M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J. Hepatol. 2005, 42, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Depner, C.M.; Torres-Gonzalez, M.; Tripathy, S.; Milne, G.; Jump, D.B. Menhaden oil decreases high-fat diet-induced markers of hepatic damage, steatosis, inflammation, and fibrosis in obese LDLR-/- mice. J. Nutr. 2012, 142, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Kathirvel, E.; Chen, P.; Morgan, K.; French, S.W.; Morgan, T.R. Oxidative stress and regulation of anti-oxidant enzymes in cytochrome p4502e1 transgenic mouse model of non-alcoholic fatty liver. J. Gastroent. Hepatol. 2010, 25, 1136–1143. [Google Scholar] [CrossRef]
- Ahmed, U.; Redgrave, T.G.; Oates, P.S. Effect of dietary fat to produce non-alcoholic fatty liver in the rat. J. Gastroent. Hepatol. 2009, 24, 1463–1471. [Google Scholar] [CrossRef]
- Jung, T.S.; Kim, S.K.; Shin, H.J.; Jeon, B.T.; Hahm, J.R.; Roh, G.S. Alpha-lipoic acid prevents non-alcoholic fatty liver disease in oletf rats. Liver Internat. 2012, 32, 1565–1573. [Google Scholar] [CrossRef]
- Panchal, S.K.; Poudyal, H.; Brown, L. Quercetin ameliorates cardiovascular, hepatic, and metabolic changes in diet-induced metabolic syndrome in rats. J. Nutr. 2012, 142, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Widyarini, S.; Domanski, D.; Painter, N.; Reeve, V.E. Photoimmune protective effect of the phytoestrogenic isoflavonoid equol is partially due to its antioxidant activities. Photochem. Photobio. Sci. 2012, 11, 1186–1192. [Google Scholar] [CrossRef]
- Salley, T.N.; Mishra, M.; Tiwari, S.; Jadhav, A.; Ndisang, J.F. The heme oxygenase system rescues hepatic deterioration in the condition of obesity co-morbid with type-2 diabetes. PLoS ONE 2013, 8, e79270. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Green, C.J.; Foresti, R. Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid. Redox Signal. 2002, 4, 615–624. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorman, S.; Black, L.J.; Feelisch, M.; Hart, P.H.; Weller, R. Can Skin Exposure to Sunlight Prevent Liver Inflammation? Nutrients 2015, 7, 3219-3239. https://doi.org/10.3390/nu7053219
Gorman S, Black LJ, Feelisch M, Hart PH, Weller R. Can Skin Exposure to Sunlight Prevent Liver Inflammation? Nutrients. 2015; 7(5):3219-3239. https://doi.org/10.3390/nu7053219
Chicago/Turabian StyleGorman, Shelley, Lucinda J. Black, Martin Feelisch, Prue H. Hart, and Richard Weller. 2015. "Can Skin Exposure to Sunlight Prevent Liver Inflammation?" Nutrients 7, no. 5: 3219-3239. https://doi.org/10.3390/nu7053219
APA StyleGorman, S., Black, L. J., Feelisch, M., Hart, P. H., & Weller, R. (2015). Can Skin Exposure to Sunlight Prevent Liver Inflammation? Nutrients, 7(5), 3219-3239. https://doi.org/10.3390/nu7053219