
Ginsenoside Rh2 Mitigates Endoplasmic Reticulum Stress-Induced Apoptosis and Inflammation and Through Inhibition of Hepatocyte–Macrophage Inflammatory Crosstalk


Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
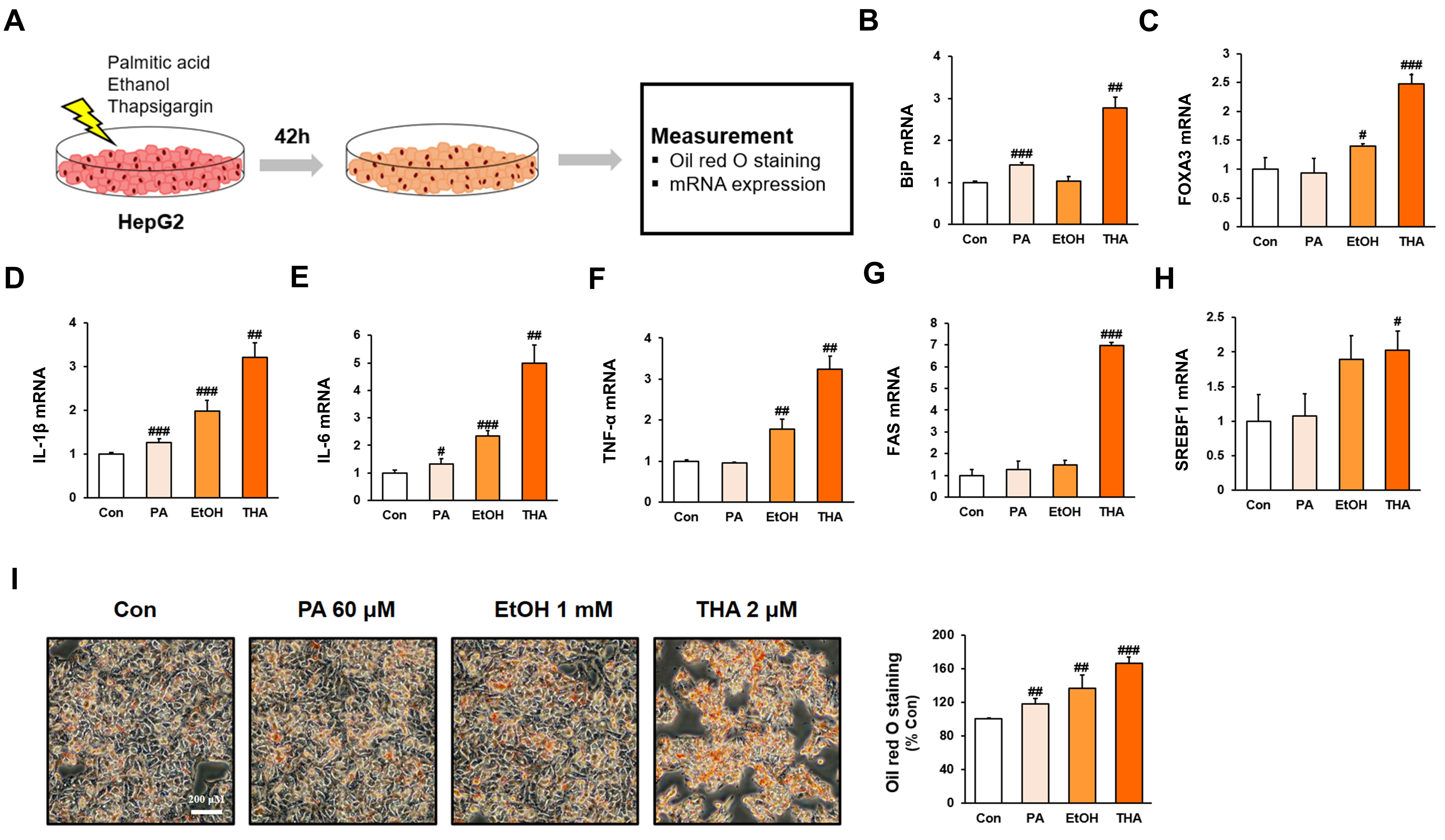
2.2. Cell Culture and Stimulation
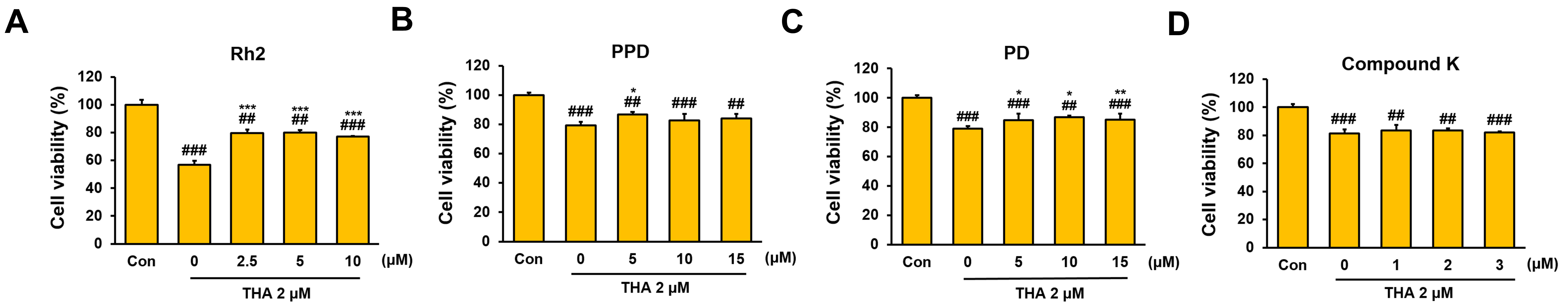
2.3. Cell Viability Assay
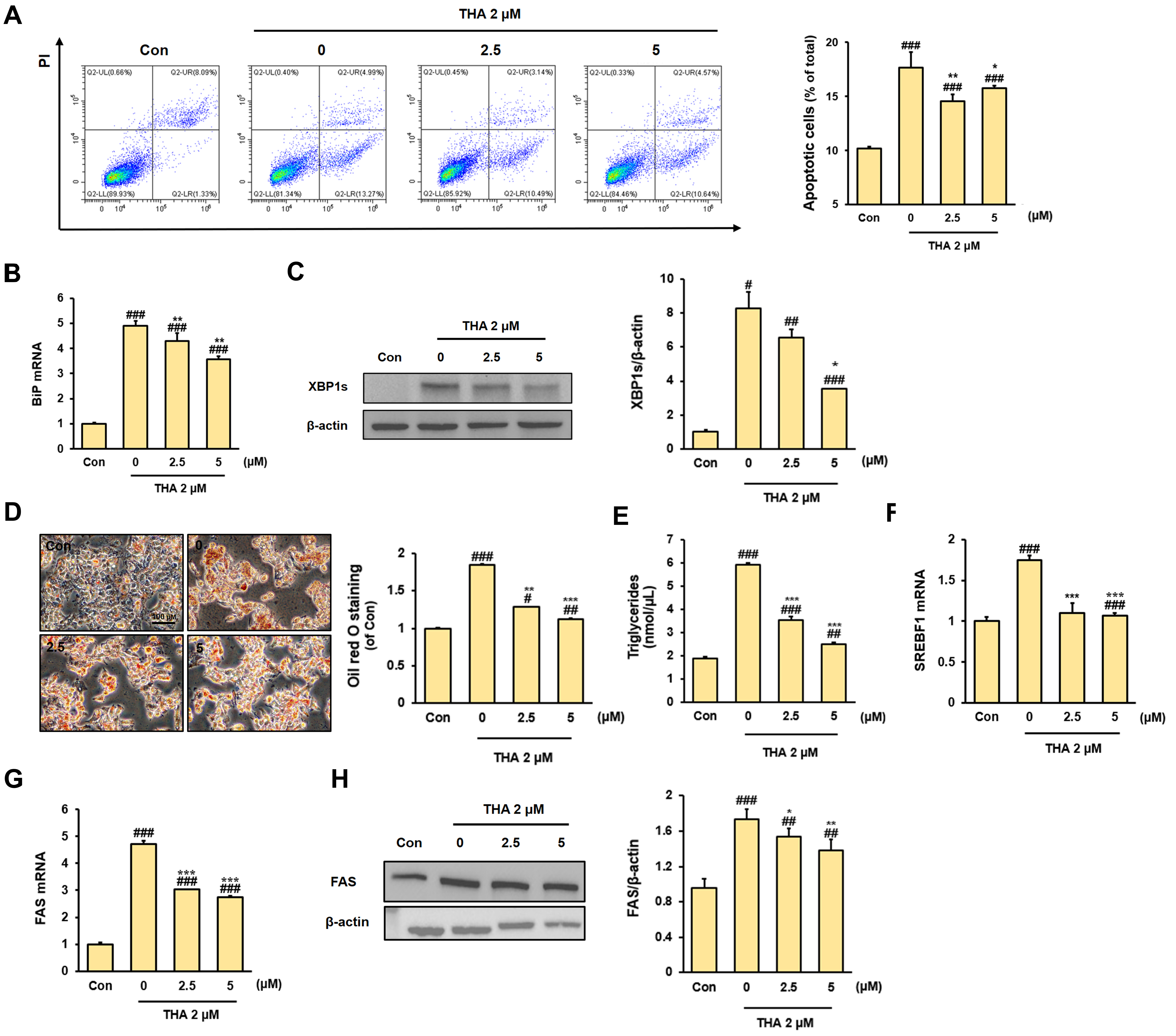
2.4. Apoptosis Assay
2.5. Oil Red O Staining
2.6. Triglycerides Assay
2.7. Protein Extraction and Immunoblotting
2.8. Harvest of Conditioned Medium (CM) from HepG2
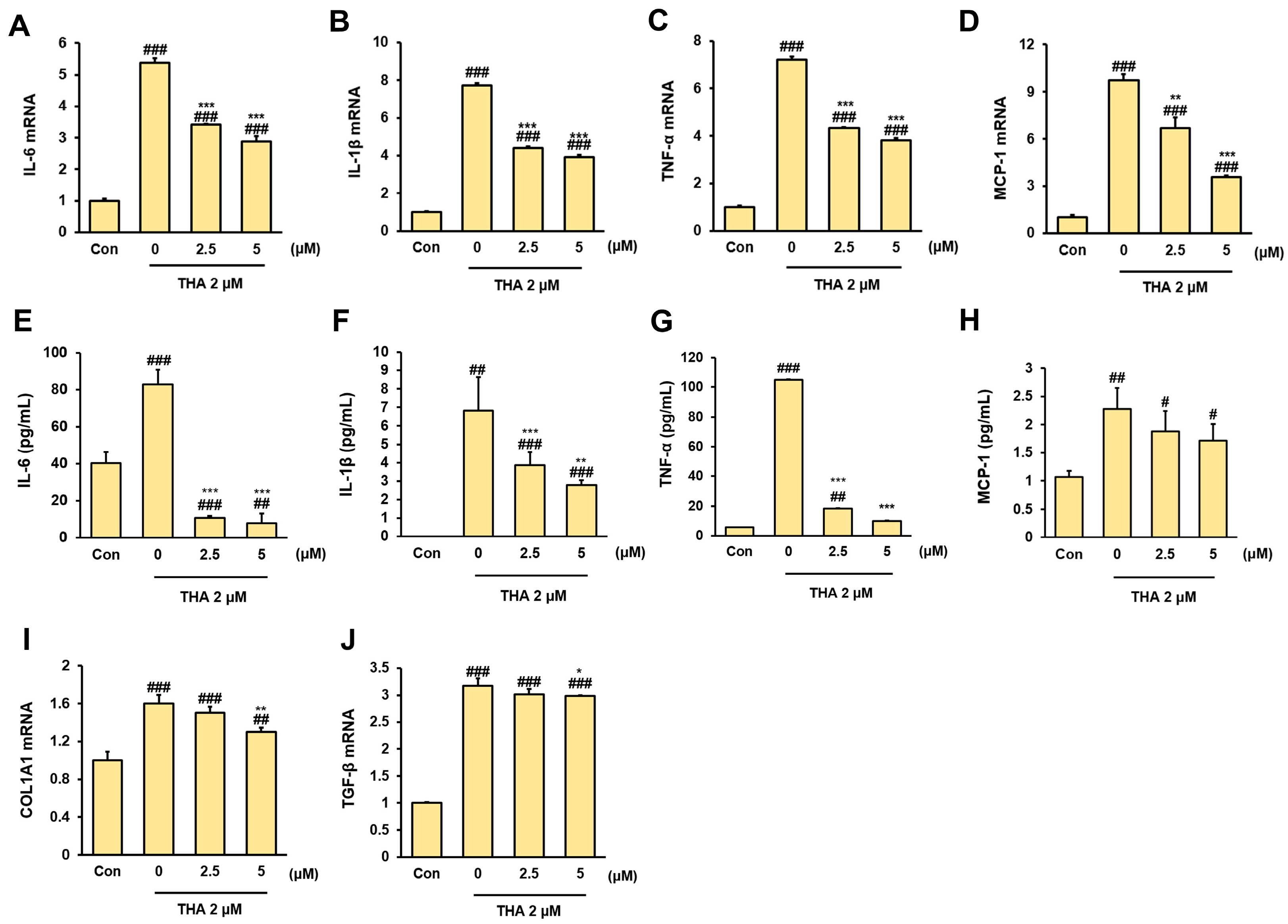
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
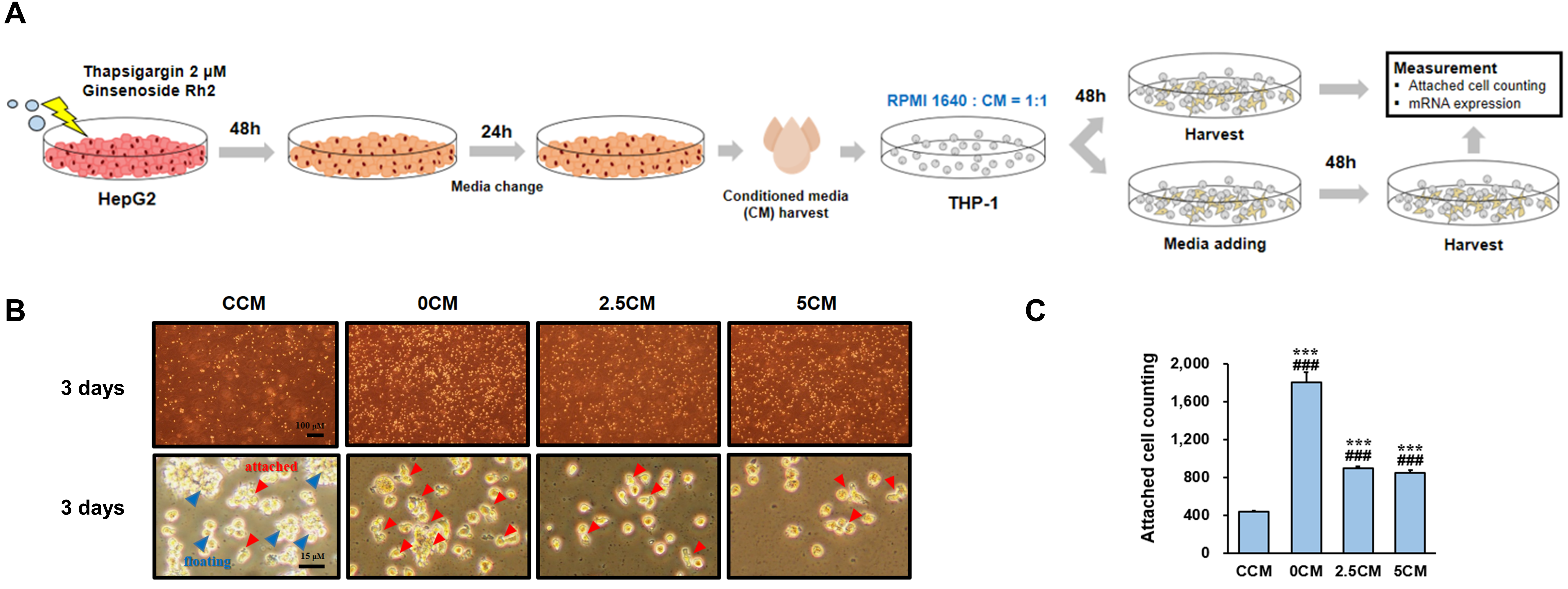
2.10. CM from HepG2 Cells Treatment in THP-1 Cells
2.11. RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.12. Statistical Analysis
3. Results
3.1. Inhibitory Effect of Rh2 Treatment on Apoptosis and Lipid Accumulation in ERS-Induced HepG2 Cells
3.2. Inhibitory Effect of Rh2 Treatment on Inflammation and Fibrosis in ERS-Induced HepG2 Cells
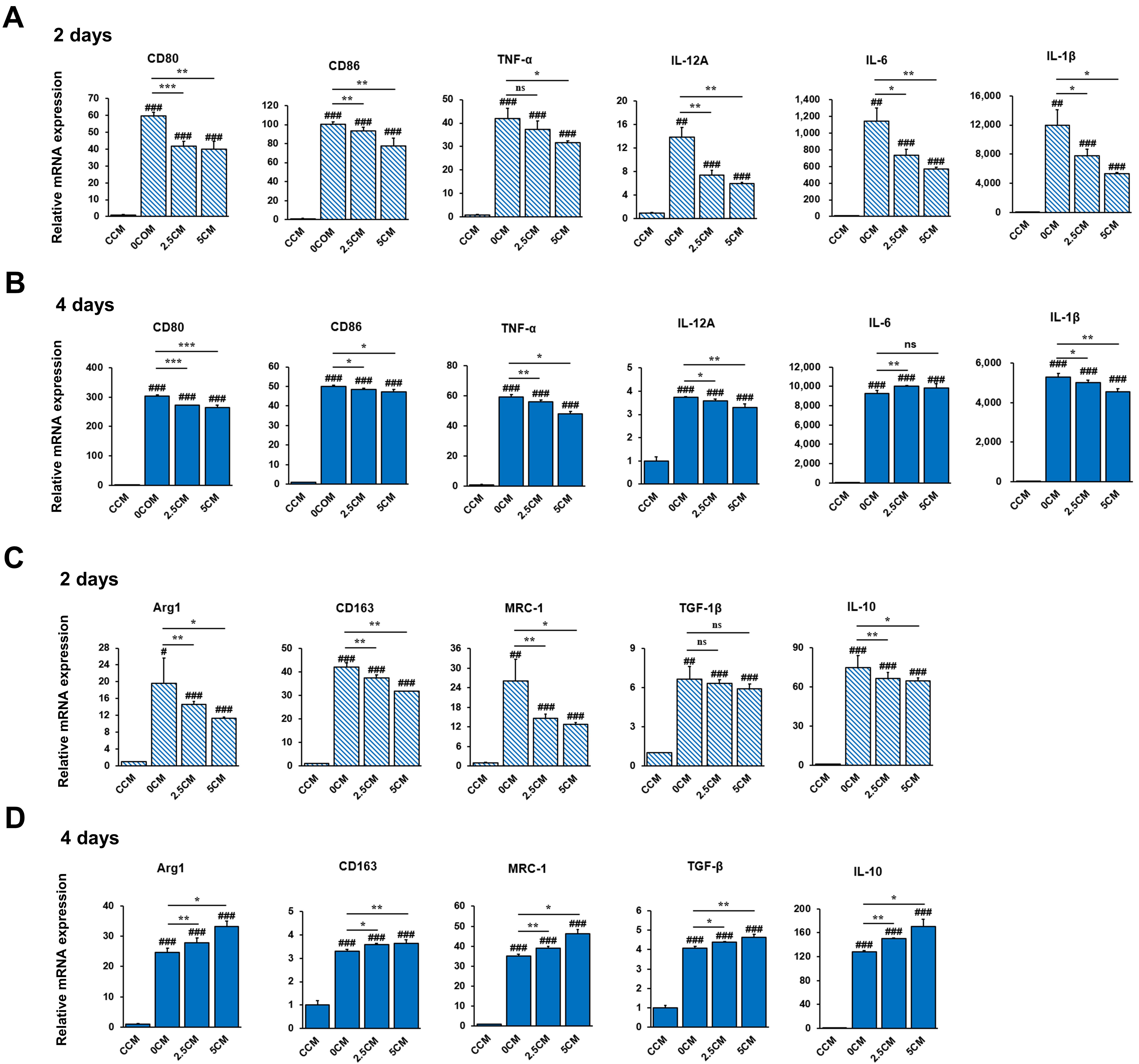
3.3. Inhibition of THP-1 Cell Activation by CM Derived from ERS-Induced HepG2 Cells Treated with Rh2
3.4. Regulation of THP-1 Cell Polarization by CM Derived from ERS-Induced HepG2 Cells Treated with Rh2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amini-Salehi, E.; Letafatkar, N.; Norouzi, N.; Joukar, F.; Habibi, A.; Javid, M.; Sattari, N.; Khorasani, M.; Farahmand, A.; Tavakoli, S.; et al. Global Prevalence of Nonalcoholic Fatty Liver Disease: An Updated Review Meta-Analysis comprising a Population of 78 million from 38 Countries. Arch. Med. Res. 2024, 55, 103043. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhou, H.; Bu, Q.; Wei, S.; Li, L.; Zhou, J.; Zhou, S.; Su, W.; Liu, M.; Liu, Z.; et al. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J. Hepatol. 2022, 77, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.L.; Ng, C.H.; Huang, D.Q.; Chan, K.E.; Tan, D.J.; Lim, W.H.; Yang, J.D.; Tan, E.; Muthiah, M.D. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S32–S42. [Google Scholar] [CrossRef]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145. [Google Scholar] [CrossRef]
- Fang, D.L.; Wan, Y.; Shen, W.; Cao, J.; Sun, Z.X.; Yu, H.H.; Zhang, Q.; Cheng, W.H.; Chen, J.; Ning, B. Endoplasmic reticulum stress leads to lipid accumulation through upregulation of SREBP-1c in normal hepatic and hepatoma cells. Mol. Cell. Biochem. 2013, 381, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Xu, C.F.; Yu, C.H.; Chen, W.X.; Li, Y.M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef]
- Peng, J.; Qin, C.; Ramatchandirin, B.; Pearah, A.; Guo, S.; Hussain, M.; Yu, L.; Wondisford, F.E.; He, L. Activation of the canonical ER stress IRE1-XBP1 pathway by insulin regulates glucose and lipid metabolism. J. Biol. Chem. 2022, 298, 102283. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Chen, Q.; Kovilakath, A.; Allegood, J.; Thompson, J.; Hu, Y.; Cowart, L.A.; Lesnefsky, E. Endoplasmic reticulum stress and mitochondrial dysfunction during aging: Role of sphingolipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2023, 1868, 159366. [Google Scholar] [CrossRef]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and Dysregulation of the Unfolded Protein Response in Nonalcoholic Fatty Liver Disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef]
- Colgan, S.M.; Hashimi, A.A.; Austin, R.C. Endoplasmic reticulum stress and lipid dysregulation. Expert. Rev. Mol. Med. 2011, 13, e4. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, A.; Hamidi-Zad, Z. Role of SREBPs in Liver Diseases: A Mini-review. J. Clin. Transl. Hepatol. 2018, 6, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Geier, A.; Tiniakos, D.; Denk, H.; Trauner, M. From the origin of NASH to the future of metabolic fatty liver disease. Gut 2021, 70, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Gong, J.; Tu, W.; Lin, J.; Tian, D. Hepatocytes: A key role in liver inflammation. Front. Immunol. 2023, 13, 1083780. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; He, K.; Li, J.; Gong, J. The role of Kupffer cells in hepatic diseases. Mol. Immunol. 2017, 85, 222–229. [Google Scholar] [CrossRef]
- Liu, X.L.; Pan, Q.; Cao, H.X.; Xin, F.Z.; Zhao, Z.H.; Yang, R.X.; Zeng, J.; Zhou, H.; Fan, J.G. Lipotoxic hepatocyte-derived exosomal microRNA 192-5p activates macrophages through rictor/Akt/forkhead box transcription factor O1 signaling in nonalcoholic fatty liver disease. Hepatology 2020, 72, 454–469. [Google Scholar] [CrossRef]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Hu, Q.R.; Pan, Y.; Wu, H.C.; Dai, Z.Z.; Huang, Q.X.; Luo, T.; Li, J.; Deng, Z.Y.; Chen, F. The ways for ginsenoside Rh2 to fight against cancer: The molecular evidences in vitro and in vivo. J. Ginseng. Res. 2023, 47, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Bai, L.P.; Wong, V.K.; Zhou, H.; Wang, J.R.; Liu, Y.; Jiang, Z.H.; Liu, L. The in Vitro Structure-Related Anti-Cancer Activity of Ginsenosides and Their Derivatives. Molecules 2011, 16, 10619–10630. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, T.; Liu, H.; Zhang, L. Ginsenoside Rh2 inhibits hepatocellular carcinoma through β-catenin and autophagy. Sci. Rep. 2016, 6, 19383. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Liu, Y.; Li, C.; Qi, P.; Bao, J. Antihyperglycemic effect of ginsenoside Rh2 by inducing islet β-cell regeneration in mice. Horm. Metab. Res. 2012, 44, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Vinoth Kumar, R.; Oh, T.W.; Park, Y.K. Anti-Inflammatory Effects of Ginsenoside-Rh2 Inhibits LPS-Induced Activation of Microglia and Overproduction of Inflammatory Mediators Via Modulation of TGF-β1/Smad Pathway. Neurochem. Res. 2016, 41, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Wu, Q.; Zhu, Y.; Guo, C.; Zheng, X. Ginsenoside Rh2 alleviates dextran sulfate sodium-induced colitis via augmenting TGFβ signaling. Mol. Biol. Rep. 2014, 41, 5485–5490. [Google Scholar] [CrossRef]
- Zhang, L.; Virgous, C.; Si, H. How Does Ginsenoside Rh2 Mitigate Adipogenesis in Cultured Cells and Obese Mice? Molecules 2020, 25, 2412. [Google Scholar] [CrossRef]
- Wang, L.; Hao, X.; Li, X.; Li, Q.; Fang, X. Effects of ginsenoside Rh2 on cisplatin-induced nephrotoxicity in renal tubular epithelial cells by inhibiting endoplasmic reticulum stress. J. Biochem. Mol. Toxicol. 2024, 38, e23768. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, Y. Role of ginsenoside Rh2 in tumor therapy and tumor microenvironment immunomodulation. Biomed. Pharmacother. 2022, 156, 113912. [Google Scholar]
- Zhang, H.; Park, S.; Huang, H.; Kim, E.; Yi, J.; Choi, S.K.; Ryoo, Z.; Kim, M. Anticancer effects and potential mechanisms of ginsenoside Rh2 in various cancer types (Review). Oncol. Rep. 2021, 45, 33. [Google Scholar] [CrossRef]
- Yu, S.; Wang, J.; Zheng, H.; Wang, R.; Johnson, N.; Li, T.; Li, P.; Lin, J.; Li, Y.; Yan, J.; et al. Pathogenesis from Inflammation to Cancer in NASH-Derived HCC. J. Hepatocell. Carcinoma 2022, 9, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.R.; Liu, J.; Plumeri, D.; Cao, Y.B.; He, T.; Lin, L.; Li, Y.; Jiang, Y.Y.; Li, J.; Shang, J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am. J. Transl. Res. 2011, 3, 284–291. [Google Scholar]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Ge, G.; Yan, Y.; Cai, H. Ginsenoside Rh2 Inhibited Proliferation by Inducing ROS Mediated ER Stress Dependent Apoptosis in Lung Cancer Cells. Biol. Pharm. Bull. 2017, 40, 2117–2124. [Google Scholar] [CrossRef]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1312. [Google Scholar] [CrossRef]
- Fukushima, H.; Yamashina, S.; Arakawa, A.; Taniguchi, G.; Aoyama, T.; Uchiyama, A.; Kon, K.; Ikejima, K.; Watanabe, S. Formation of p62-positive inclusion body is associated with macrophage polarization in non-alcoholic fatty liver disease. Hepatol. Res. 2018, 48, 757–767. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 16, 577. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, F.; Cheng, H.; Su, M.; Wang, Y. Macrophage polarization: An important role in inflammatory diseases. Front. Immunol. 2024, 15, 1352946. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.K.; Das, S.; Pourhoseini, S.; Dattaroy, D.; Igwe, S.; Ray, J.B.; Fan, D.; Michelotti, G.A.; Diehl, A.M.; Chatterijee, S. M1 polarization bias and subsequent nonalcoholic steatohepatitis progression is attenuated by nitric oxide donor DETA NONOate via inhibition of CYP2E1-induced oxidative stress in obese mice. J. Pharmacol. Exp. Ther. 2015, 352, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wu, S.; Li, Y.; Shao, P.; Jiang, J. Inhibition of macrophage polarization and pyroptosis in collagen-induced arthritis through MSC-exo and ginsenoside Rh2. Arthritis Res. Ther. 2025, 27, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, J.; Du, F.; Gao, X.; Ma, X.; Huang, Y.; Xu, F.; Niu, W.; Wang, F.; Mao, Y.; et al. Absorption and disposition of ginsenosides after oral administration of Panax notoginseng extract to rats. Drug Metab. Dispos. 2009, 37, 2290–2298. [Google Scholar] [CrossRef]
- Yang, Z.; Gao, S.; Wang, J.; Yin, T.; Teng, Y.; Wu, B.; You, M.; Jiang, Z.; Hu, M. Enhancement of oral bioavailability of 20(S)-ginsenoside Rh2 through improved understanding of its absorption and efflux mechanisms. Drug Metab. Dispos. 2011, 39, 1866–1872. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Qiao, J.; Wang, J.; Jiang, L.; Wang, C.; Yu, S.; Zhang, P.; Zhao, D.; Fan, M.; et al. Ginsenoside Rh2 Induces HeLa Apoptosis through Upregulating Endoplasmic Reticulum Stress-Related and Downstream Apoptotic Gene Expression. Molecules 2022, 27, 7865. [Google Scholar] [CrossRef]
- Yun, T.K. Experimental and epidemiological evidence on non-organ specific cancer preventive effect of Korean ginseng and identification of active compounds. Mutat. Res. 2003, 523–524, 63–74. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequences | Accession Number | |
|---|---|---|---|
| β-actin | Forward (5′-3′) | GATTCCTATGTGGGCGACGA | NM_001101.5 |
| Reverse (5′-3′) | TCTCCATGTCGTCCCAGTTG | ||
| IL-1β | Forward (5′-3′) | CTCTGTCATTCGCTCCCACA | XM_054341810.1 |
| Reverse (5′-3′) | ACACTGCTACTTCTTGCCCC | ||
| IL-6 | Forward (5′-3′) | AGTGAGGAACAAGCCAGAGC | NM_000600.5 |
| Reverse (5′-3′) | ATTTGTGGTTGGGTCAGGGG | ||
| TNF-α | Forward (5′-3′) | GTCCTCTTCAAGGGCCAAGG | NM_000594.4 |
| Reverse (5′-3′) | GGCTCTTGATGGCAGAGAGG | ||
| FAS | Forward (5′-3′) | GGCCCACAAGAGCTACATCA | XM_054315477.1 |
| Reverse (5′-3′) | GGAGCGAGAAGTCAACACGA | ||
| SREBF1 | Forward (5′-3′) | TGACCGACATCGAAGGTGAA | NM_001005291.3 |
| Reverse (5′-3′) | AAAGTGCAATCCATGGCTCC | ||
| FOXA3 | Forward (5′-3′) | TCTTGGGGCCTGATCCTTCT | NM_004497.3 |
| Reverse (5′-3′) | GGATCAACACCATGCCCACT | ||
| BiP | Forward (5′-3′) | TGAAAGAAACCGCTGAGGCT | NM_005347.5 |
| Reverse (5′-3′) | TCTTTGGTTGCTTGGCGTTG | ||
| MCP-1 | Forward (5′-3′) | GCAGTAAGTGTCCCAAAGAAGC | NM_002982.4 |
| Reverse (5′-3′) | TGGGTTTGCTTGTCCAGGTG | ||
| COL1A1 | Forward (5′-3′) | GCTGGTGCTCGTGGAAAT | NM_000088.4 |
| Reverse (5′-3′) | ACCCTTAGCACCAACAGC | ||
| CD86 | Forward (5′-3′) | CTTCCTGCTCTCTGGTGCTG | NM_176892.2 |
| Reverse (5′-3′) | GCTCACTCAGGCTTTGGTTC | ||
| CD80 | Forward (5′-3′) | CCACAACCTTTGGAGACCCA | NM_005191.4 |
| Reverse (5′-3′) | AGGCAGGGCTGATGACAATC | ||
| IL-12A | Forward (5′-3′) | GCTCCAGAAGGCCAGACAAA | NM_000882.4 |
| Reverse (5′-3′) | TAAACAGGCCTCCACTGTGC | ||
| CD163 | Forward (5′-3′) | GGACCCACTTCCTGTTCTGG | XM_054373862.1 |
| Reverse (5′-3′) | TGACACACCACCTGAGCATC | ||
| ARG1 | Forward (5′-3′) | GGGTTGACTGACTGGAGAGC | NM_000045.4 |
| Reverse (5′-3′) | CGTGGCTGTCCCTTTGAGAA | ||
| MRC1 | Forward (5′-3′) | GGGACGTGGCTGTGGATAAA | NM_002438.4 |
| Reverse (5′-3′) | TCCAAAACCCAGAAGACGCA | ||
| IL-10 | Forward (5′-3′) | CGAGATGCCTTCAGCAGAGT | NM_000572.3 |
| Reverse (5′-3′) | GGCAACCCAGGTAACCCTTA | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Jeong, I.; Kim, O.-K. Ginsenoside Rh2 Mitigates Endoplasmic Reticulum Stress-Induced Apoptosis and Inflammation and Through Inhibition of Hepatocyte–Macrophage Inflammatory Crosstalk. Nutrients 2025, 17, 1682. https://doi.org/10.3390/nu17101682
Park S, Jeong I, Kim O-K. Ginsenoside Rh2 Mitigates Endoplasmic Reticulum Stress-Induced Apoptosis and Inflammation and Through Inhibition of Hepatocyte–Macrophage Inflammatory Crosstalk. Nutrients. 2025; 17(10):1682. https://doi.org/10.3390/nu17101682
Chicago/Turabian StylePark, Shinjung, Inae Jeong, and Ok-Kyung Kim. 2025. "Ginsenoside Rh2 Mitigates Endoplasmic Reticulum Stress-Induced Apoptosis and Inflammation and Through Inhibition of Hepatocyte–Macrophage Inflammatory Crosstalk" Nutrients 17, no. 10: 1682. https://doi.org/10.3390/nu17101682
APA StylePark, S., Jeong, I., & Kim, O.-K. (2025). Ginsenoside Rh2 Mitigates Endoplasmic Reticulum Stress-Induced Apoptosis and Inflammation and Through Inhibition of Hepatocyte–Macrophage Inflammatory Crosstalk. Nutrients, 17(10), 1682. https://doi.org/10.3390/nu17101682