

Efficacy and Safety of Ketogenic Diet Treatment in Pediatric Patients with Mitochondrial Disease
 , , , , , , and
, , , , , , and
Abstract
1. Introduction
1.1. Diagnosis of Mitochondrial Diseases
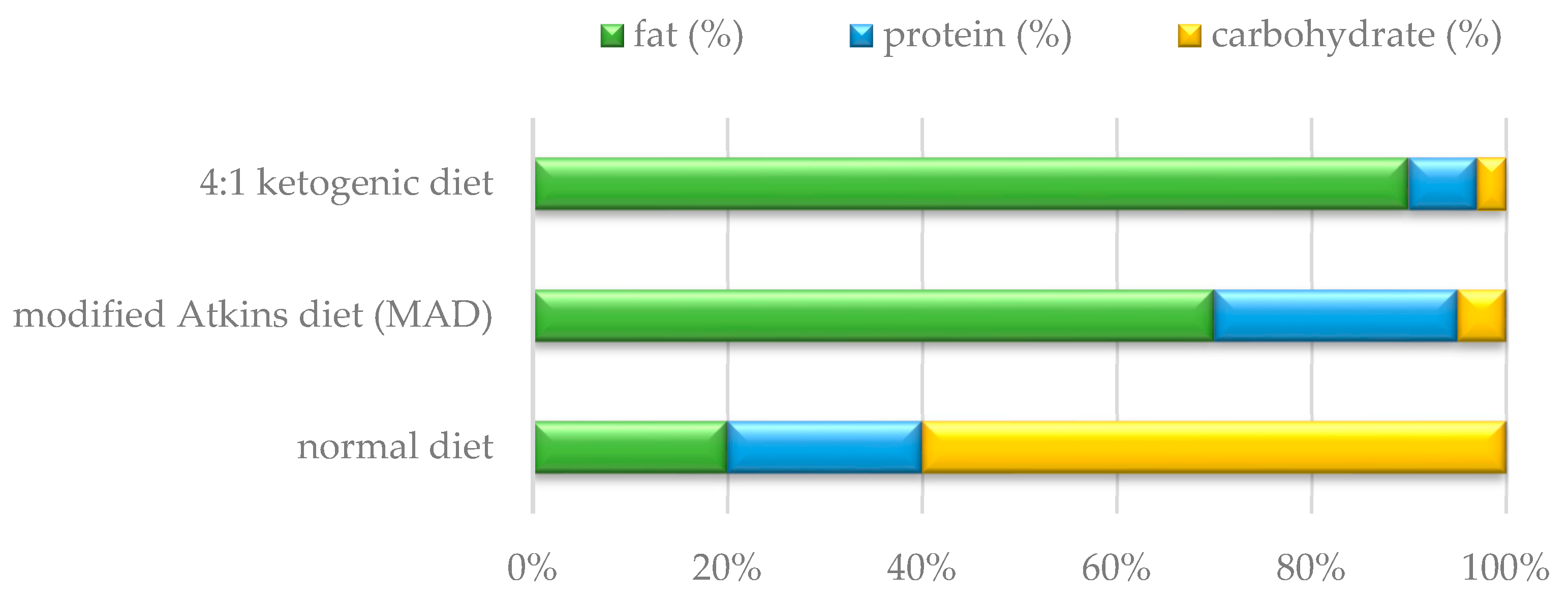
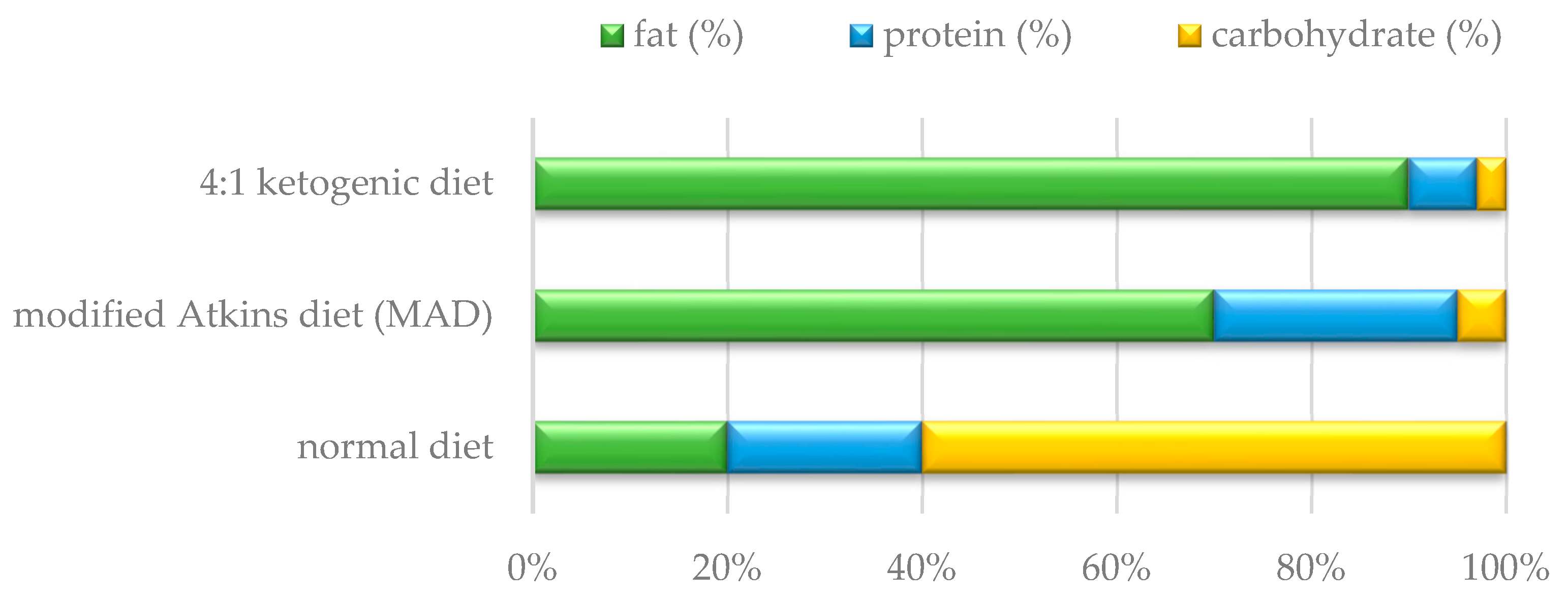
1.2. Use of Ketogenic Diet in Patients with Mitochondrial Disease
2. Materials and Methods
2.1. Course of Study
2.2. Statistical Methods
3. Results
3.1. Efficacy of the Ketogenic Diet in Particular Groups
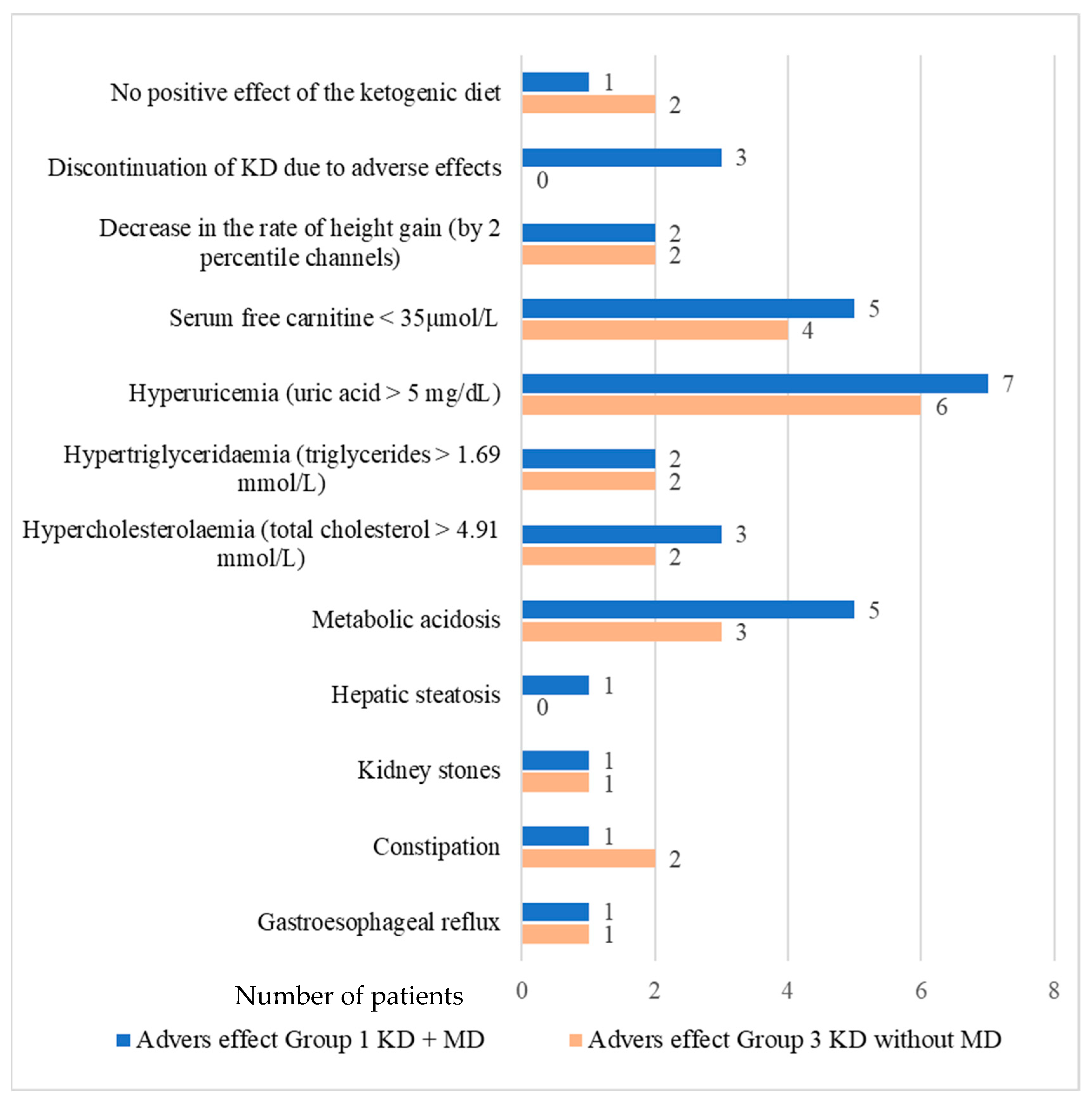
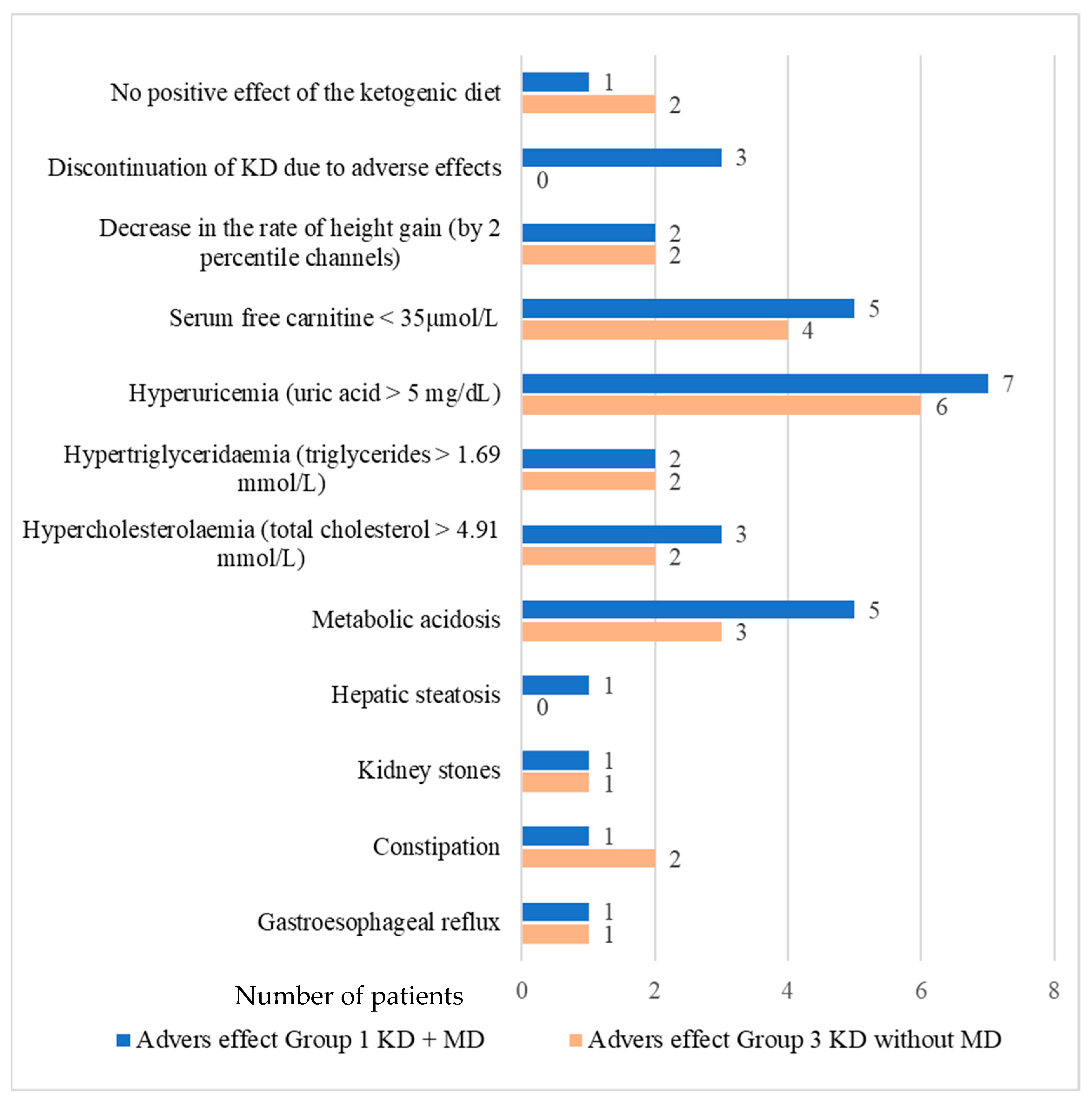
3.2. Adverse Effects of the Ketogenic Diet
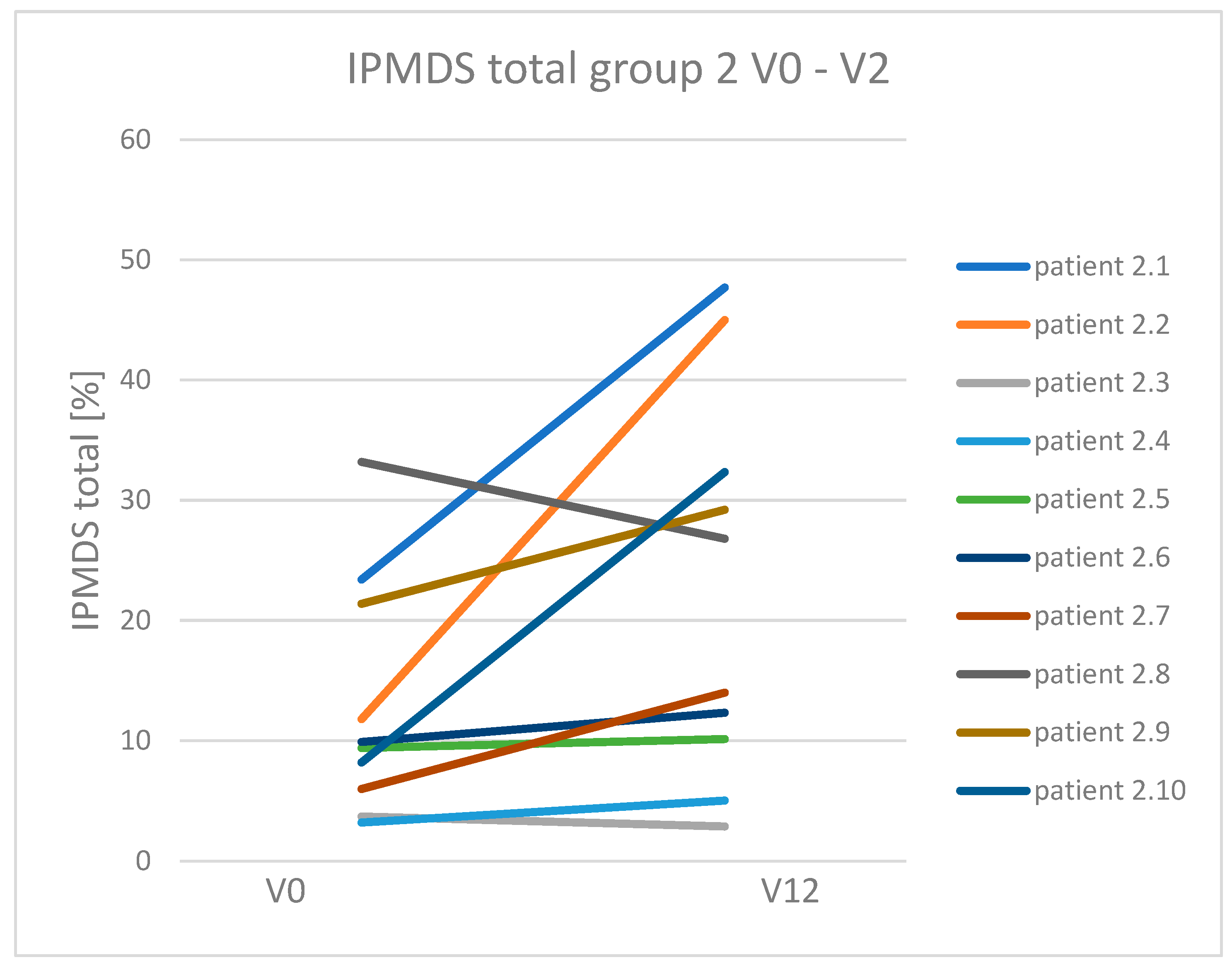
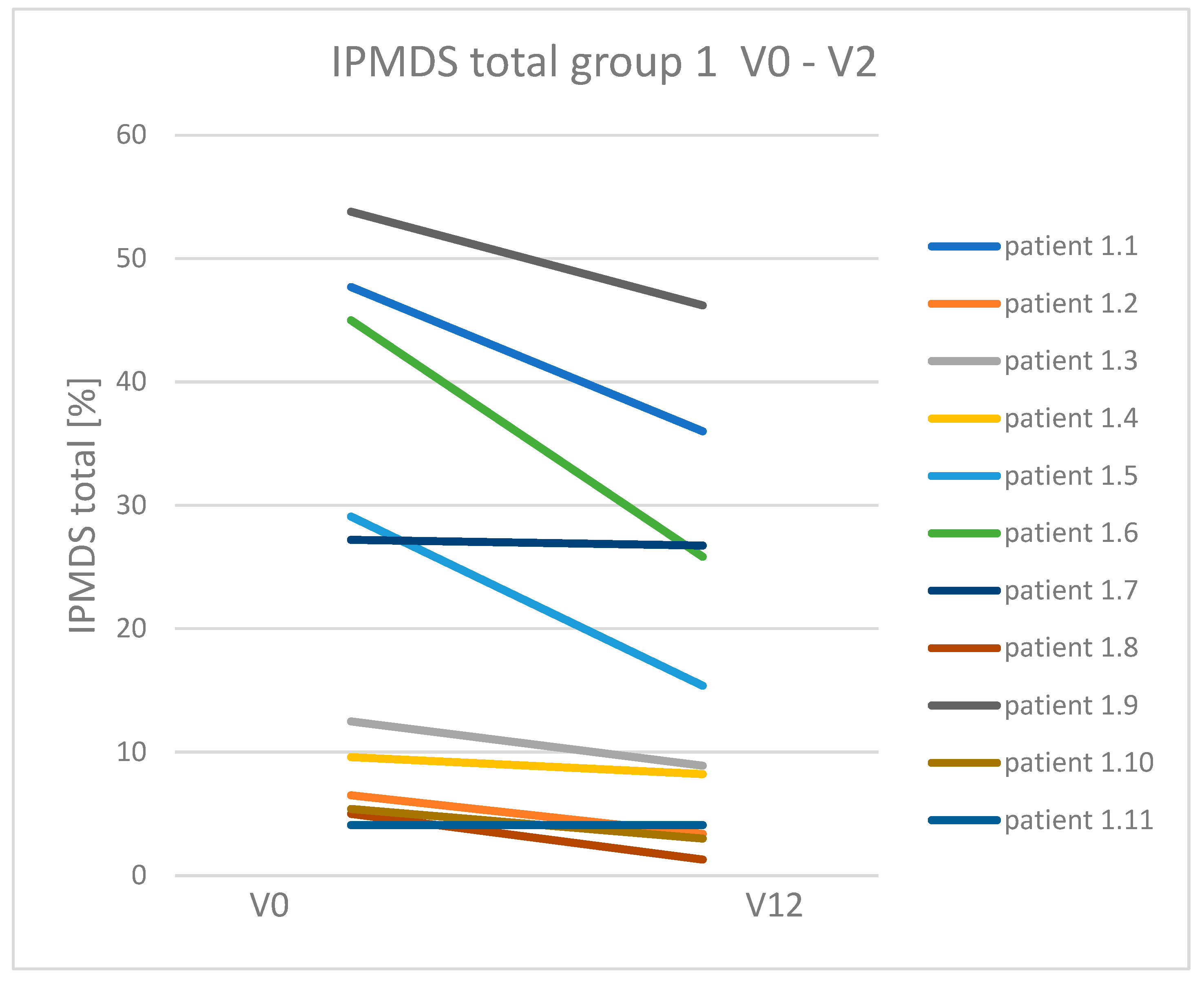
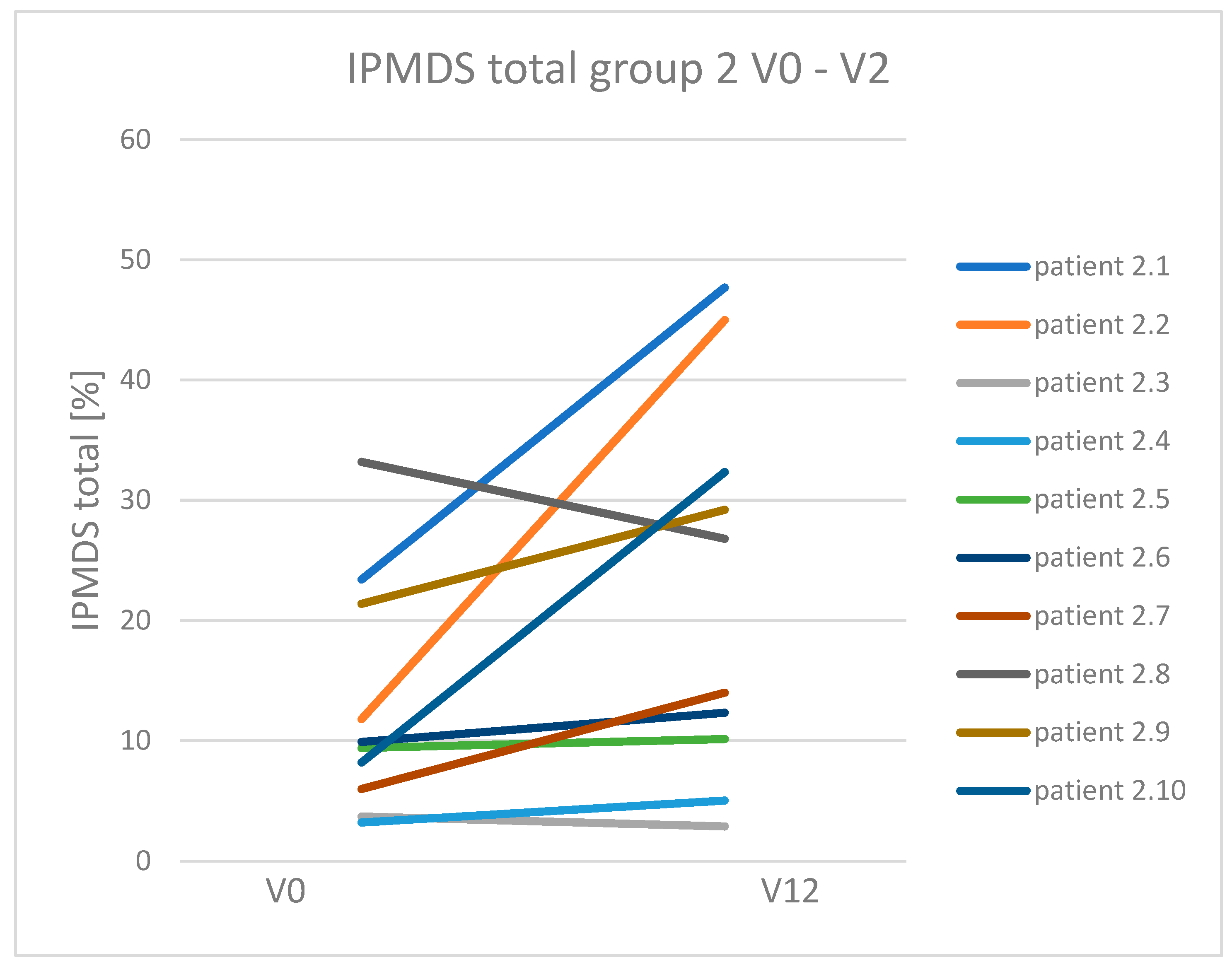
3.3. Dynamics of Clinical Symptoms Occurring in Patients with Mitochondrial Disease Treated and Non-Treated with a Ketogenic Diet
3.4. Analysis of Mitochondrial Disease Biomarker Concentration in Particular Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ala | alanine |
| BHB | β-hydroxybutyric acid |
| CMHI- | the Children’s Memorial Health Institute |
| CNS | central nervous system |
| FGF21 | fibroblast growth factor 21 |
| IPMDS | International Pediatric Mitochondrial Disease Scale |
| MAD | modified Atkins diet |
| OXPHOS | oxidative phosphorylation process |
| PDHD | pyruvic acid dehydrogenase deficiency |
References
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef]
- Paiva Coelho, M.; Martins, E.; Vilarinho, L. Diagnosis, management, and follow-up of mitochondrial disorders in childhood: A personalized medicine in the new era of genome sequence. Eur. J. Pediatr. 2019, 178, 21–32. [Google Scholar] [CrossRef]
- Davison, J.E.; Rahman, S. Recognition, investigation and management of mitochondrial disease. Arch. Dis. Child. 2017, 102, 1082–1090. [Google Scholar] [CrossRef]
- Honzik, T.; Tesarova, M.; Magner, M.; Mayr, J.; Jesina, P.; Vesela, K.; Wenchich, L.; Szentivanyi, K.; Hansikova, H.; Sperl, W.; et al. Neonatal onset of mitochondrial disorders in 129 patients: Clinical and laboratory characteristics and a new approach to diagnosis. J. Inherit. Metab. Dis. 2012, 35, 749–759. [Google Scholar] [CrossRef]
- Barca, E.; Long, Y.; Cooley, V.; Schoenaker, R.; Emmanuele, V.; DiMauro, S.; Cohen, B.H.; Karaa, A.; Vladutiu, G.D.; Haas, R.; et al. Mitochondrial diseases in North America: An analysis of the NAMDC Registry. Neurol. Genet. 2020, 2, e402. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; Rodenburg, R.J.; van der Knaap, M.S.; Willemsen, M.A.; Sperl, W.; Laugel, V.; Ostergaard, E.; Tarnopolsky, M.; Martin, M.A.; Nesbitt, V.; et al. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: What we learned from 130 cases. J. Inherit. Metab. Dis. 2012, 35, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; van den Heuvel, L.; Hol, F.; de Vries, M.C.; Hogeveen, M.; Rodenburg, R.J.; Smeitink, J.A.M. Mitochondrial disease criteria: Diagnostic applications in children. Neurology 2006, 67, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Pajdowska, M.; Gradowska, W.; Piekutowska-Abramczuk, D.; Baczyńska, A.; Iwanicka-Pronicka, K.; Sykut-Cegielska, J.; Pronicka, E.l. Rola badania profilu kwasów organicznych w moczu metodą chromatografi gazowej sprzężonej ze spektrometrią mas (GC-MS) w detekcji chorób mitochondrialnych. Standardy Medyczne. Pediatria 2012, 9, 552–561. [Google Scholar]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ji, K.; Ma, X.; Liu, S.; Li, W.; Zhao, Y.; Yan, C. Accuracy of FGF-21 and GDF-15 for the diagnosis of mitochondrial disorders: A meta-analysis. Ann. Clin. Transl. Neurol. 2020, 7, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Alban, C.; Fatale, E.; Joulani, A.; Ilin, P.; Saada, A. The Relationship between Mitochondrial Respiratory Chain Activities in Muscle and Metabolites in Plasma and Urine: A Retrospective Study. J. Clin. Med. 2017, 10, 31. [Google Scholar] [CrossRef]
- Suomalainen, A.; Elo, J.M.; Pietiläinen, K.H.; Hakonen, A.H.; Sevastianova, K.; Korpela, M.; Isohanni, P.; Marjavaara, S.K.; Tyni, T.; Kiuru-Enari, S.; et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: A diagnostic study. Lancet Neurol. 2011, 10, 806–818. [Google Scholar] [CrossRef]
- Formichi, P.; Cardone, N.; Taglia, I.; Cardaioli, E.; Salvatore, S.; Gerfo, A.L.; Simoncini, C.; Montano, V.; Siciliano, G.; Mancuso, M.; et al. Fibroblast growth factor 21 and grow differentiation factor 15 are sensitive biomarkers of mitochondrial diseases due to mitochondrial transfer-RNA mutations and mitochondrial DNA deletions. Neurol. Sci. 2020, 41, 3653–3662. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkilä, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjøsund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 15, 3948–3958. [Google Scholar] [CrossRef]
- Koga, Y.; Povalko, N.; Inoue, E.; Nashiki, K.; Tanaka, M. Biomarkers and clinical rating scales for sodium pyruvate therapy in patients with mitochondrial disease. Mitochondrion 2019, 48, 11–15. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Qiu, Y.; Zhou, M.; Chen, M.; Hu, Y.; Hong, S.; Jiang, L.; Guo, Y. Circulating FGF21 and GDF15 as Biomarkers for Screening, Diagnosis, and Severity Assessment of Primary Mitochondrial Disorders in Children. Front. Pediatr. 2022, 14, 851534. [Google Scholar] [CrossRef]
- van der Louw, E.; van den Hurk, D.; Neal, E.; Leiendecker, B.; Fitzsimmon, G.; Dority, L.; Thompson, L.; Marchió, M.; Dudzińska, M.; Dressler, A.; et al. Ketogenic diet guidelines for infants with refractory epilepsy. Eur. J. Paediatr. Neurol. 2016, 20, 798–809. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 21, 175–192. [Google Scholar] [CrossRef]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef]
- Garone, C.; Viscomi, C. Towards a therapy for mitochondrial disease: An update. Biochem. Soc. Trans. 2018, 19, 1247–1261. [Google Scholar] [CrossRef]
- Geffroy, G.; Benyahia, R.; Frey, S.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Inisan, A.; Renaud, A.; Chevrollier, A.; et al. The accumulation of assembly intermediates of the mitochondrial complex I matrix arm is reduced by limiting glucose uptake in a neuronal-like model of MELAS syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1596–1608. [Google Scholar] [CrossRef]
- Martin-McGill, K.J.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2020, 24, CD001903. [Google Scholar] [CrossRef]
- Paleologou, E.; Ismayilova, N.; Kinali, M. Use of the Ketogenic Diet to Treat Intractable Epilepsy in Mitochondrial Disorders. J. Clin. Med. 2017, 26, 56. [Google Scholar] [CrossRef]
- Newmaster, K.; Zhu, Z.; Bolt, E.; Chang, R.J.; Day, C.; Mhanna, A.; Paudel, S.; Farooq, O.; Swaminathan, A.; Acharya, P.; et al. A Review of the Multi-Systemic Complications of a Ketogenic Diet in Children and Infants with Epilepsy. Children 2022, 10, 1372. [Google Scholar] [CrossRef]
- Kang, H.C.; Chung, D.E.; Kim, D.W.; Kim, H.D. Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia 2004, 45, 1116–1123. [Google Scholar] [CrossRef]
- Koene, S.; Hendriks, J.C.M.; Dirks, I.; de Boer, L.; de Vries, M.C.; Janssen, M.C.H.; Smuts, I.; Fung, C.; Wong, V.C.N.; de Coo, I.R.F.M.; et al. International Paediatric Mitochondrial Disease Scale. J. Inherit. Metab. Dis. 2016, 39, 705–712. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucińska-Więckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosińska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 12, 174. [Google Scholar] [CrossRef]
- Frey, S.; Geffroy, G.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Amati-Bonneau, P.; Chevrollier, A.; Barth, M.; Henrion, D.; et al. The addition of ketone bodies alleviates mitochondrial dysfunction by restoring complex I assembly in a MELAS cellular model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 284–291. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef]
- Wall, C.E.; Whyte, J.; Suh, J.M.; Fan, W.; Collins, B.; Liddle, C.; Yu, R.T.; Atkins, A.R.; Naviaux, J.C.; Li, K.; et al. High-fat diet and FGF21 cooperatively promote aerobic thermogenesis in mtDNA mutator mice. Proc. Natl. Acad. Sci. USA 2015, 14, 8714–8719. [Google Scholar] [CrossRef]
- Santra, S.; Gilkerson, R.W.; Davidson, M.; Schon, E.A. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann. Neurol. 2004, 56, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Ahola-Erkkilä, S.; Carroll, C.J.; Peltola-Mjösund, K.; Tulkki, V.; Mattila, I.; Seppänen-Laakso, T.; Orešič, M.; Tyynismaa, H.; Suomalainen, A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum. Mol. Genet. 2010, 15, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Ahola, S.; Auranen, M.; Isohanni, P.; Niemisalo, S.; Urho, N.; Buzkova, J.; Velagapudi, V.; Lundbom, N.; Hakkarainen, A.; Muurinen, T.; et al. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol. Med. 2016, 2, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; Sabouny, R.; Villa, B.R.; Yee, N.C.; Mychasiuk, R.; Uddin, G.M.; Rho, J.M.; Shutt, T.E. Aberrant Mitochondrial Morphology and Function in the BTBR Mouse Model of Autism Is Improved by Two Weeks of Ketogenic Diet. Int. J. Mol. Sci. 2020, 5, 3266. [Google Scholar] [CrossRef]
- Zweers, H.; van Wegberg, A.M.J.; Janssen, M.C.H.; Wortmann, S.B. Ketogenic diet for mitochondrial disease: A systematic review on efficacy and safety. Orphanet J. Rare Dis. 2021, 3, 295. [Google Scholar] [CrossRef]
- Falk, R.E.; Cederbaum, S.D.; Blass, J.P.; Gibson, G.E.; Kark, R.A. Ketonic diet in the management of pyruvate dehydrogenase deficiency. Pediatrics 1976, 58, 713–721. [Google Scholar] [CrossRef]
- Sofou, K.; Dahlin, M.; Hallböök, T.; Lindefeldt, M.; Viggedal, G. Ketogenic diet in pyruvate dehydrogenase complex deficiency: Short- and long-term outcomes. J. Inherit. Metab. Dis. 2017, 40, 237–245. [Google Scholar] [CrossRef]
- Inui, T.; Wada, Y.; Shibuya, M.; Arai-Ichinoi, N.; Okubo, Y.; Endo, W.; Uchida, T.; Togashi, N.; Naito, E.; Haginoya, K. Intravenous ketogenic diet therapy for neonatal-onset pyruvate dehydrogenase complex deficiency. Brain Dev. 2022, 44, 244–248. [Google Scholar] [CrossRef]
- Shelkowitz, E.; Ficicioglu, C.; Stence, N.; Van Hove, J.; Larson, A. Serial Magnetic Resonance Imaging (MRI) in Pyruvate Dehydrogenase Complex Deficiency. J. Child. Neurol. 2020, 35, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Kang, H.C.; Lee, J.S.; Kim, S.H.; Kim, E.Y.; Lee, S.K.; Slama, A.; Kim, H.D. Mitochondrial respiratory chain defects: Underlying etiology in various epileptic conditions. Epilepsia 2008, 49, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Na, J.H.; Kim, H.D.; Lee, Y.M. Effective and safe diet therapies for Lennox-Gastaut syndrome with mitochondrial dysfunction. Ther. Adv. Neurol. Disord. 2020, 6, 1756286419897813. [Google Scholar] [CrossRef] [PubMed]
- Joshi, C.N.; Greenberg, C.R.; Mhanni, A.A.; Salman, M.S. Ketogenic diet in Alpers-Huttenlocher syndrome. Pediatr. Neurol. 2009, 40, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Koessler, M.; Haberlandt, E.; Karall, D.; Baumann, M.; Höller, A.; Scholl-Bürgi, S. Ketogenic diet in a patient with refractory status epilepticus due to POLG mutation. JIMD Rep. 2020, 1, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, H.; Zhong, J.; Yang, L.; Chen, G.; Wang, D.; Zheng, G.; Han, H.; Han, X.; Long, Y.; et al. Efficacy and Safety of the Ketogenic Diet for Mitochondrial Disease with Epilepsy: A Prospective, Open-labeled, Controlled Study. Front. Neurol. 2022, 1, 880944. [Google Scholar] [CrossRef]
- Bölsterli, B.K.; Boltshauser, E.; Palmieri, L.; Spenger, J.; Brunner-Krainz, M.; Distelmaier, F.; Freisinger, P.; Geis, T.; Gropman, A.L.; Häberle, J.; et al. Ketogenic Diet Treatment of Defects in the Mitochondrial Malate Aspartate Shuttle and Pyruvate Carrier. Nutrients 2022, 31, 3605. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group 1 (MD + KD) | Group 2 (MD without KD) | Group 3 (KD without MD) | Group 4 (without MD, without KD) | All Groups | |
|---|---|---|---|---|---|
| Number of people recruited | 14 | 21 | 11 | 17 | 63 |
| Number of people who did not complete the study | 3 | 11 | 1 | 6 | 21 |
| Number of people who completed the study | 11 | 10 | 10 | 11 | 42 |
| Median age at study entry (months); (range) | 60 (10–181) | 35 (2–175) | 69 (18–202) | 31 (8–78) | 51 (2–202) |
| Median follow-up (months); (range) | 12 (5–15) | 12 (9–16) | 12 (3–13) | 12 (7–16) | 12 (3–16) |
| Number of boys (%) | 7 (64%) | 6 (60%) | 7 (70%) | 6 (55%) | 26 (62%) |
| Positive Effects of the KD | No. of Patients in Whom Improvement Was Observed/No. of Patients with a Problem (%) | All n = 21 | |
|---|---|---|---|
| Group 1 MD + KD n = 11 | Group 3 KD without MD n = 10 | ||
| Seizure reduction (total) | 2/2 (100%) | 6/8 (75%) | 8/10 (80%) |
| 100% reduction in seizures | 1/2 (50%) | 1/8 (12.5%) | 2/10 (20%) |
| Reduction in seizures by >90% | 0 | 0 | 0 |
| Seizure reduction by 50–90% | 1/2 (50%) | 4/8 (50%) | 5/10 (50%) |
| <50% reduction in seizures | 0 | 1/8 (12.5%) | 1/10 (10%) |
| Reduction in the number of antiepileptic drugs administered | 2/5 (40%) | 0/8 | 2/13 (15.4%) |
| Improvement of muscle tone | 8/9 (88.8%) * | 1/7 (14.3%) | 9/16 (56.2%) |
| Reduction in extra movements (tremor, ataxia, dystonia) | 5/6 (83.3%) | 3/6 (50%) | 8/12 (66.7%) |
| Clinical Symptoms | No. of Patients in Whom Symptoms Was Observed/No. of All Patients (%) | All n = 21 | |
|---|---|---|---|
| Group 1 MD + KD n = 11 | Group 2 MD without KD n = 10 | ||
| Total IPMDS score reduction (clinical improvement) | 9/11 (81.8%) * | 2/10 (20%) | 11/21 (52.4%) |
| Reducing the number of seizures | 2/2 (100%) | 0/3 | 2/5 (40%) |
| Increased number of seizures | 0/5 | 2/5 | 2/10 (20%) |
| Reduction in the number of antiepileptic drugs | 2/5 (40%) | 0/5 | 2/10 (20%) |
| Improvement of muscle tone | 8/9 (88.8%) ** | 2/6 (33.3%) | 10/15 (66.6%) |
| Reduction in movement disorders (tremor, ataxia, dystonia) | 5/6 (83.3%) | 2/7 (28.6%) | 7/13 (53.8%) |
| Improved exercise tolerance | 8/11 (72.7%) ** | 1/7 (14.3%) | 9/18 (50%) |
| Decrease in rate of weight gain (by 2 percentiles or <3c) | 0/11 | 4/10 (40%) | 4/21 (19%) |
| Decrease in height gain rate (by 2 percentile channels or <3c) | 2/11 (18.2%) | 0/10 | 2/21 (9.5%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wesół-Kucharska, D.; Greczan, M.; Kaczor, M.; Ehmke vel Emczyńska-Seliga, E.; Hajdacka, M.; Czekuć-Kryśkiewicz, E.; Piekutowska-Abramczuk, D.; Halat-Wolska, P.; Ciara, E.; Jaworski, M.; et al. Efficacy and Safety of Ketogenic Diet Treatment in Pediatric Patients with Mitochondrial Disease. Nutrients 2024, 16, 812. https://doi.org/10.3390/nu16060812
Wesół-Kucharska D, Greczan M, Kaczor M, Ehmke vel Emczyńska-Seliga E, Hajdacka M, Czekuć-Kryśkiewicz E, Piekutowska-Abramczuk D, Halat-Wolska P, Ciara E, Jaworski M, et al. Efficacy and Safety of Ketogenic Diet Treatment in Pediatric Patients with Mitochondrial Disease. Nutrients. 2024; 16(6):812. https://doi.org/10.3390/nu16060812
Chicago/Turabian StyleWesół-Kucharska, Dorota, Milena Greczan, Magdalena Kaczor, Ewa Ehmke vel Emczyńska-Seliga, Małgorzata Hajdacka, Edyta Czekuć-Kryśkiewicz, Dorota Piekutowska-Abramczuk, Paulina Halat-Wolska, Elżbieta Ciara, Maciej Jaworski, and et al. 2024. "Efficacy and Safety of Ketogenic Diet Treatment in Pediatric Patients with Mitochondrial Disease" Nutrients 16, no. 6: 812. https://doi.org/10.3390/nu16060812
APA StyleWesół-Kucharska, D., Greczan, M., Kaczor, M., Ehmke vel Emczyńska-Seliga, E., Hajdacka, M., Czekuć-Kryśkiewicz, E., Piekutowska-Abramczuk, D., Halat-Wolska, P., Ciara, E., Jaworski, M., Jezela-Stanek, A., & Rokicki, D. (2024). Efficacy and Safety of Ketogenic Diet Treatment in Pediatric Patients with Mitochondrial Disease. Nutrients, 16(6), 812. https://doi.org/10.3390/nu16060812