
Sulforaphane Inhibits Adhesion and Migration of Cisplatin- and Gemcitabine-Resistant Bladder Cancer Cells In Vitro

 ,
,  ,
,  and
and
Abstract
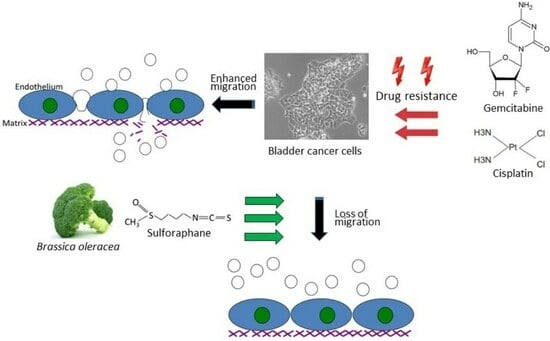
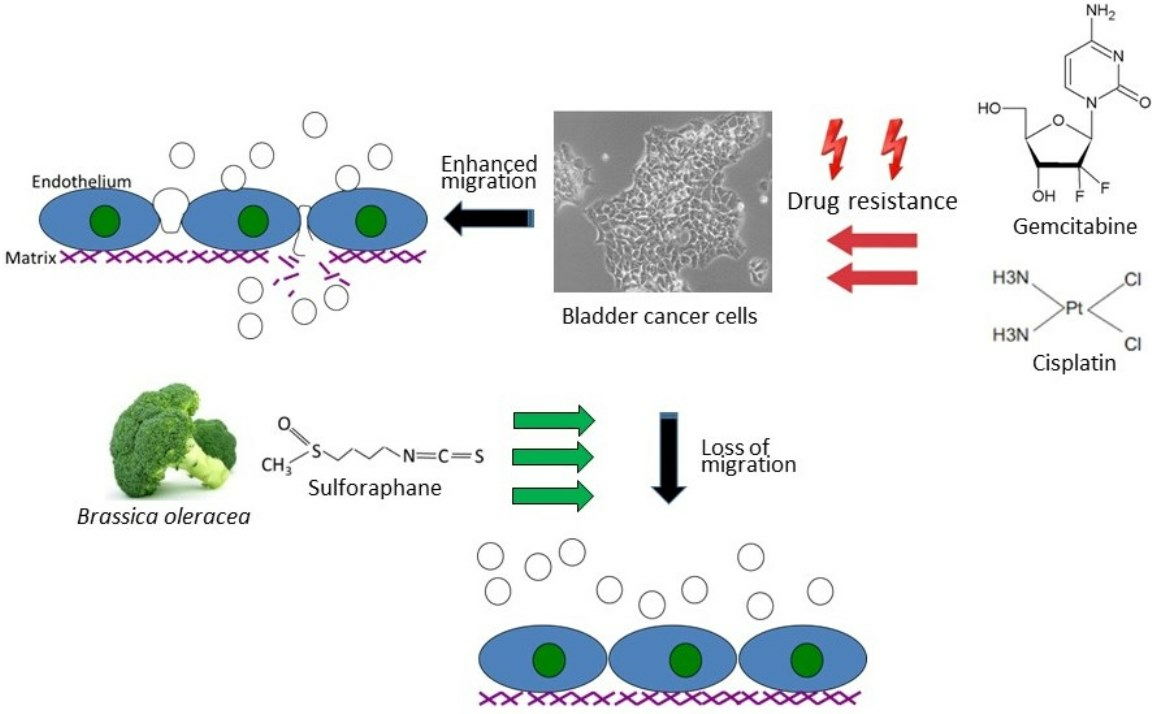{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Resistance Induction
2.2. Sulforaphane (SFN)
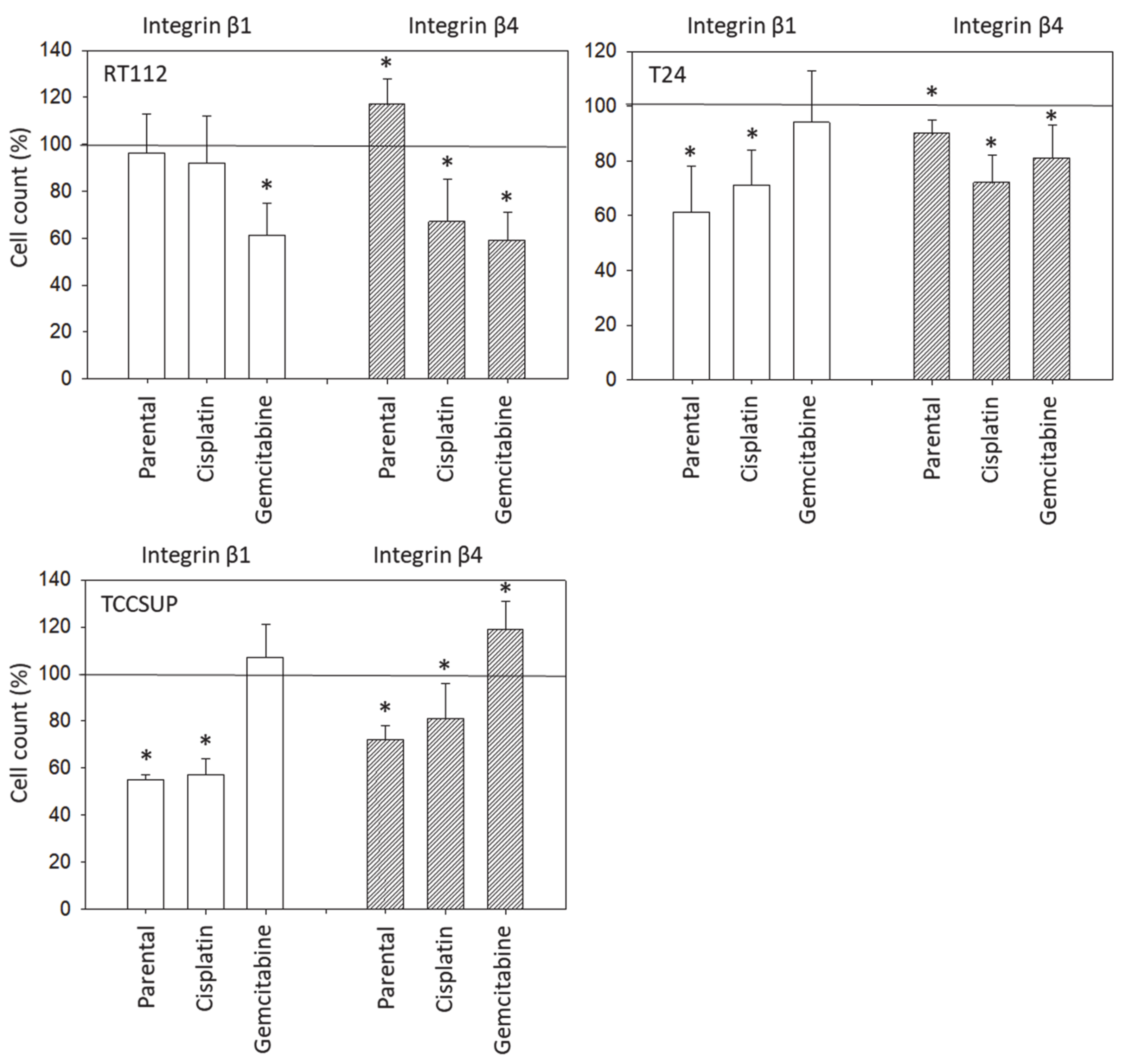
2.3. Integrin Expression
2.4. Cell Attachment to Collagen and Fibronectin
2.5. Chemotaxis
2.6. Western Blot
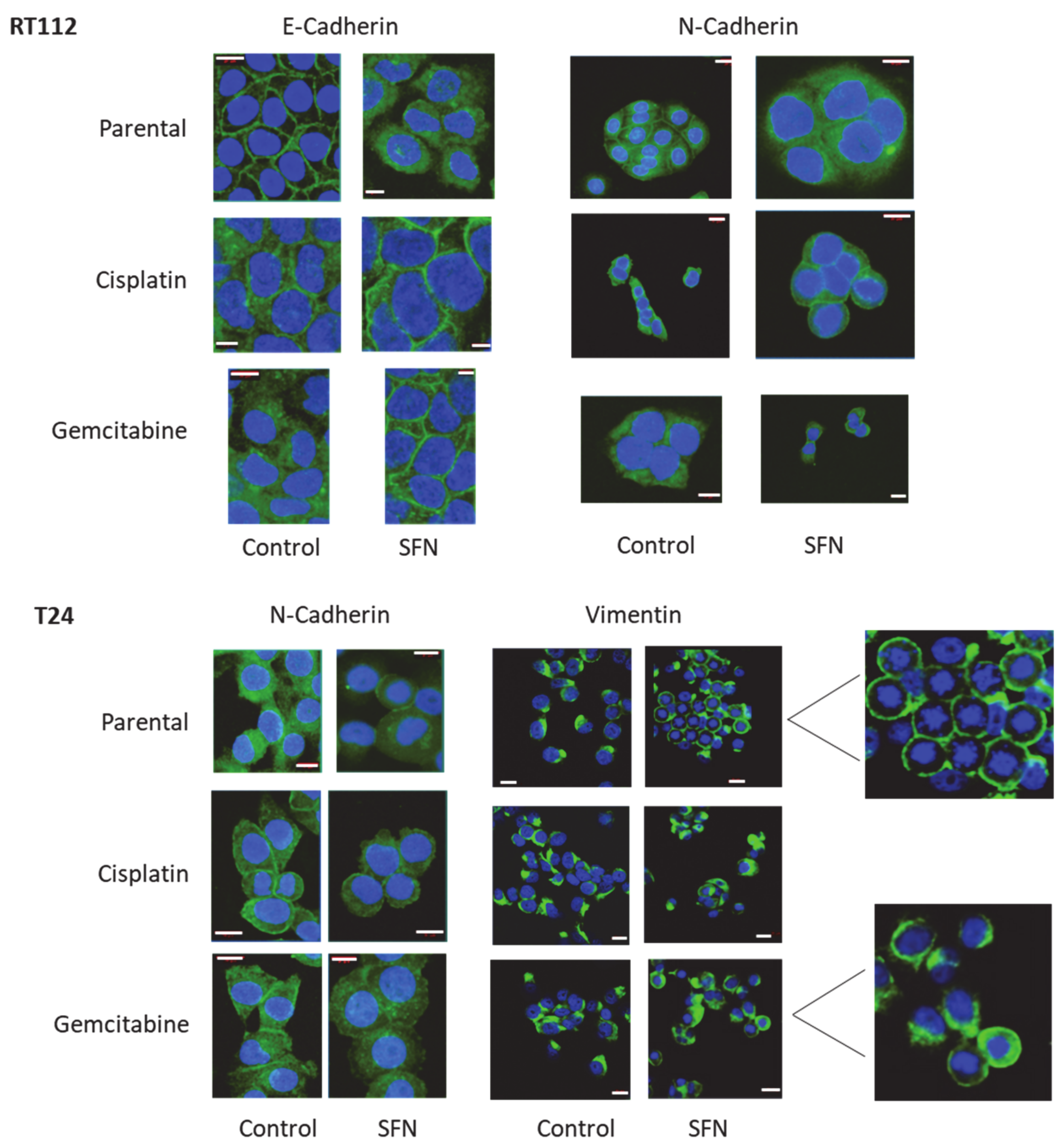
2.7. Cadherin and Vimentin Localization
2.8. Blocking
2.9. Statistics
3. Results
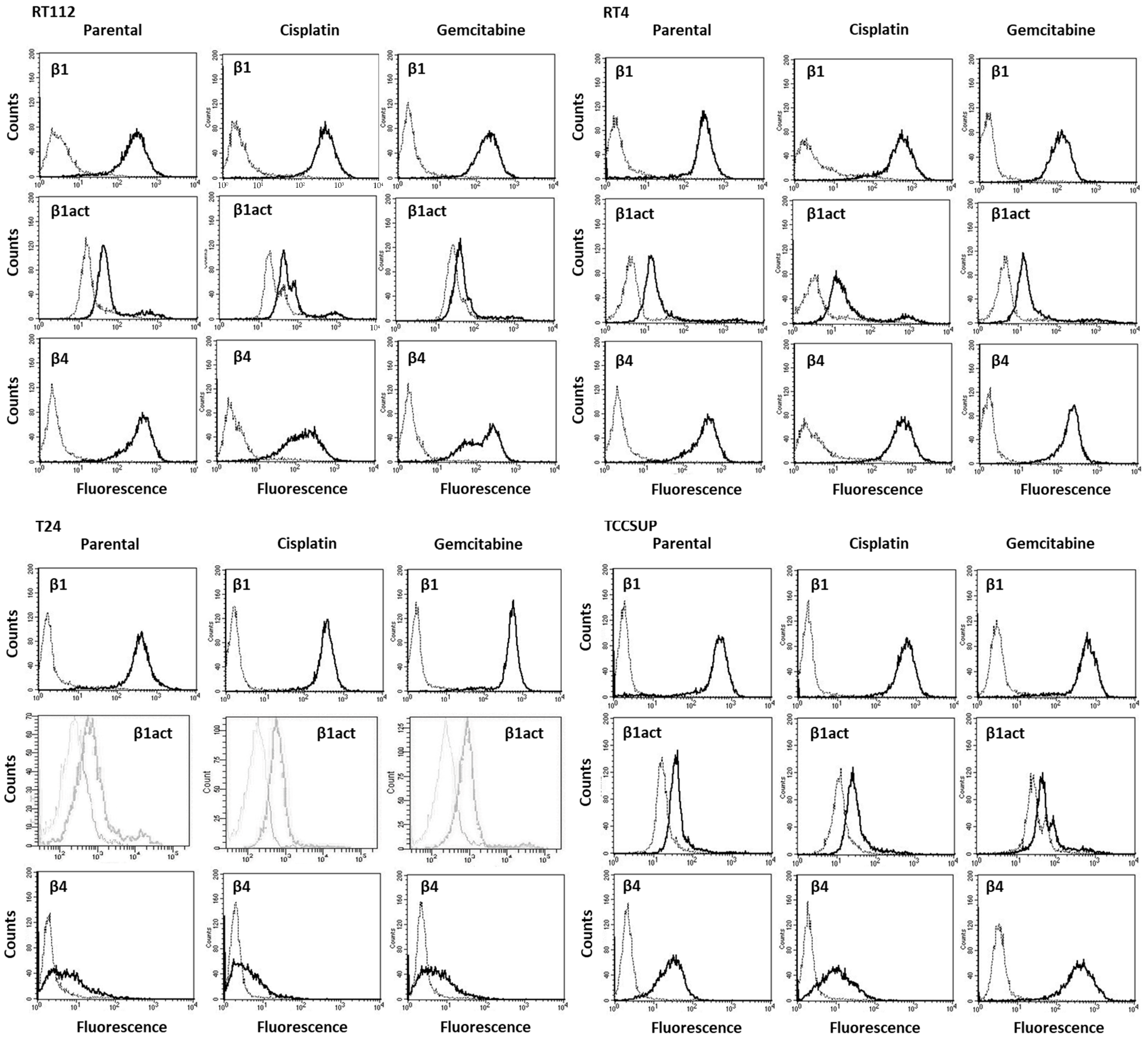
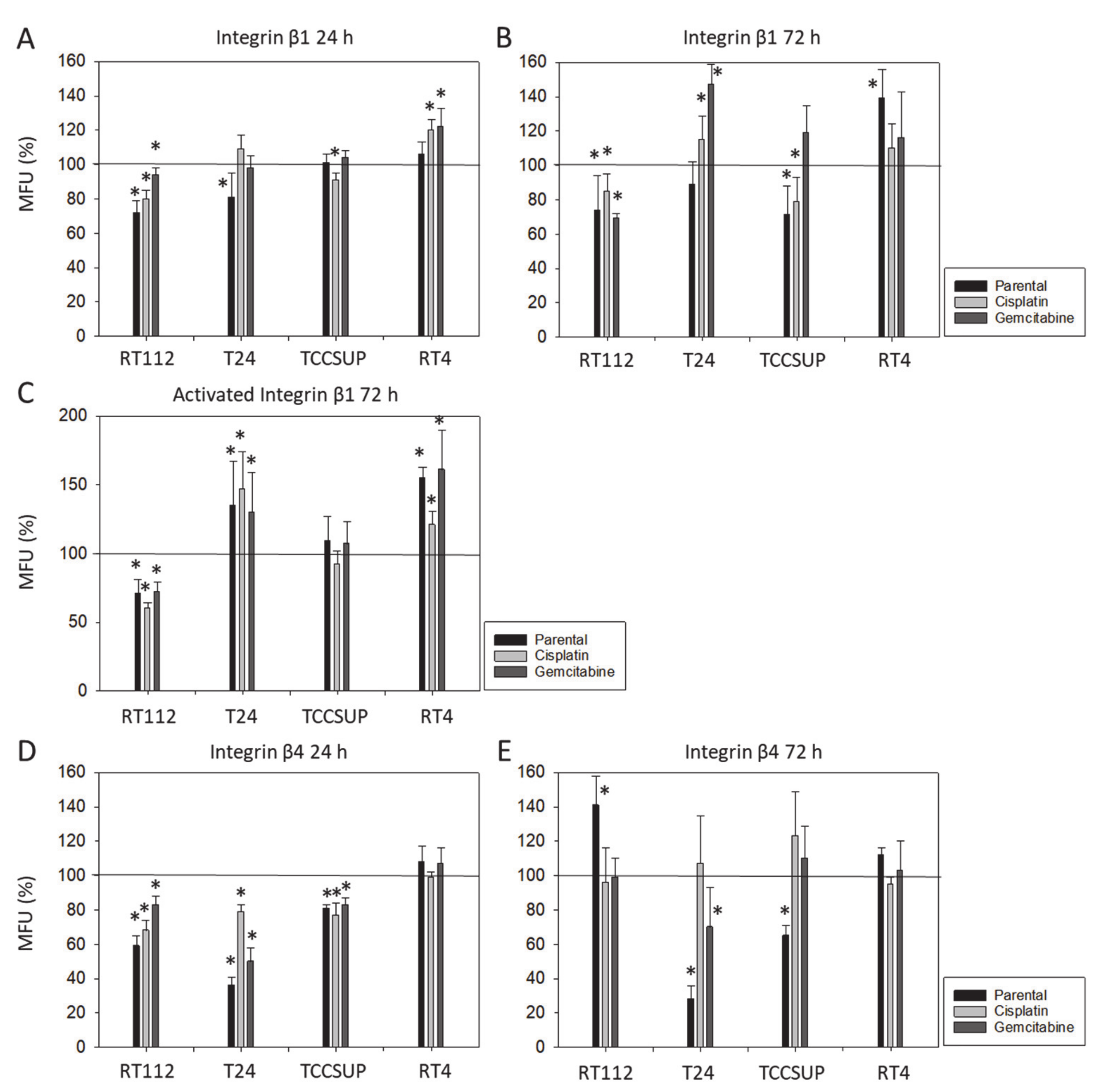
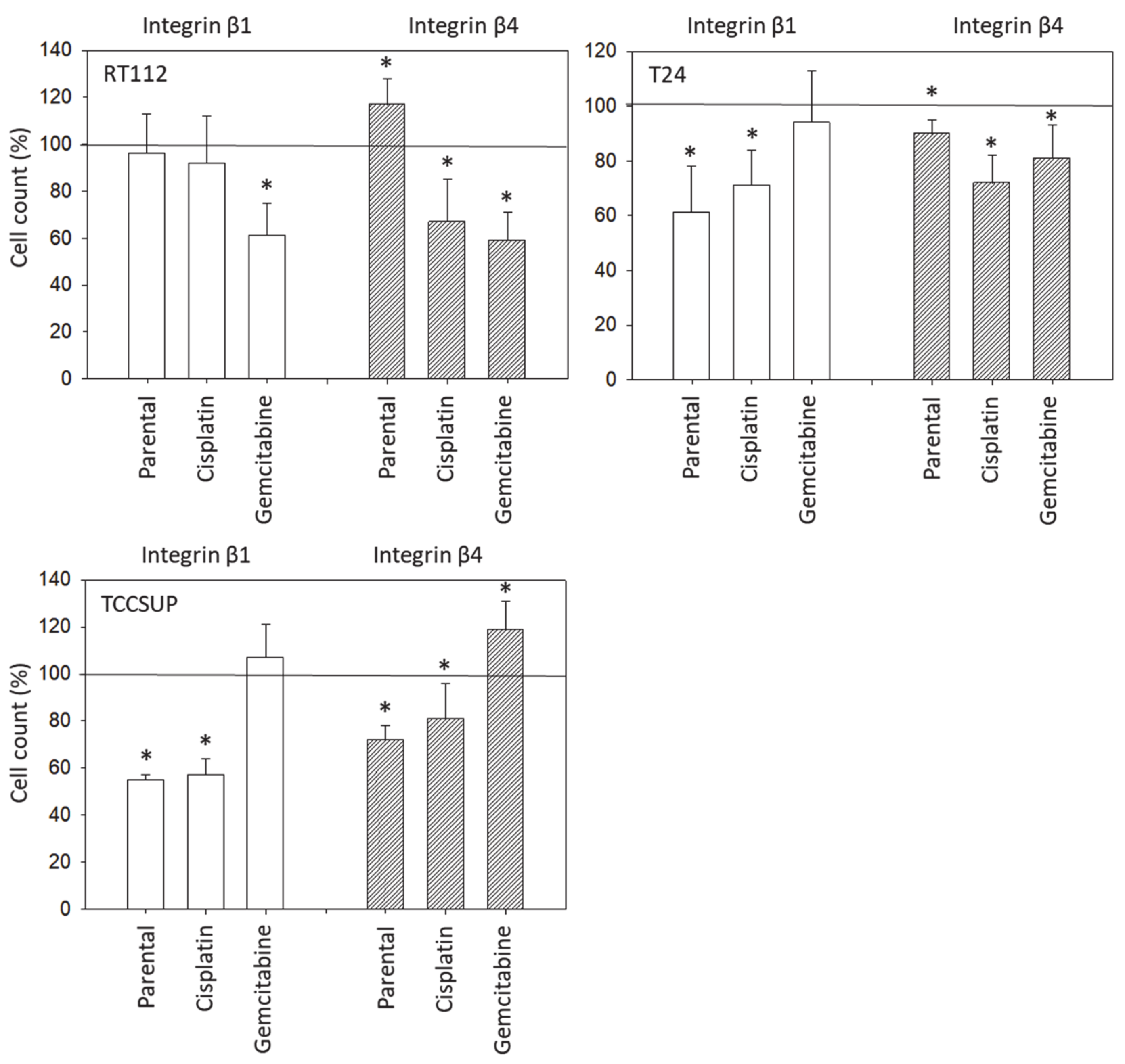
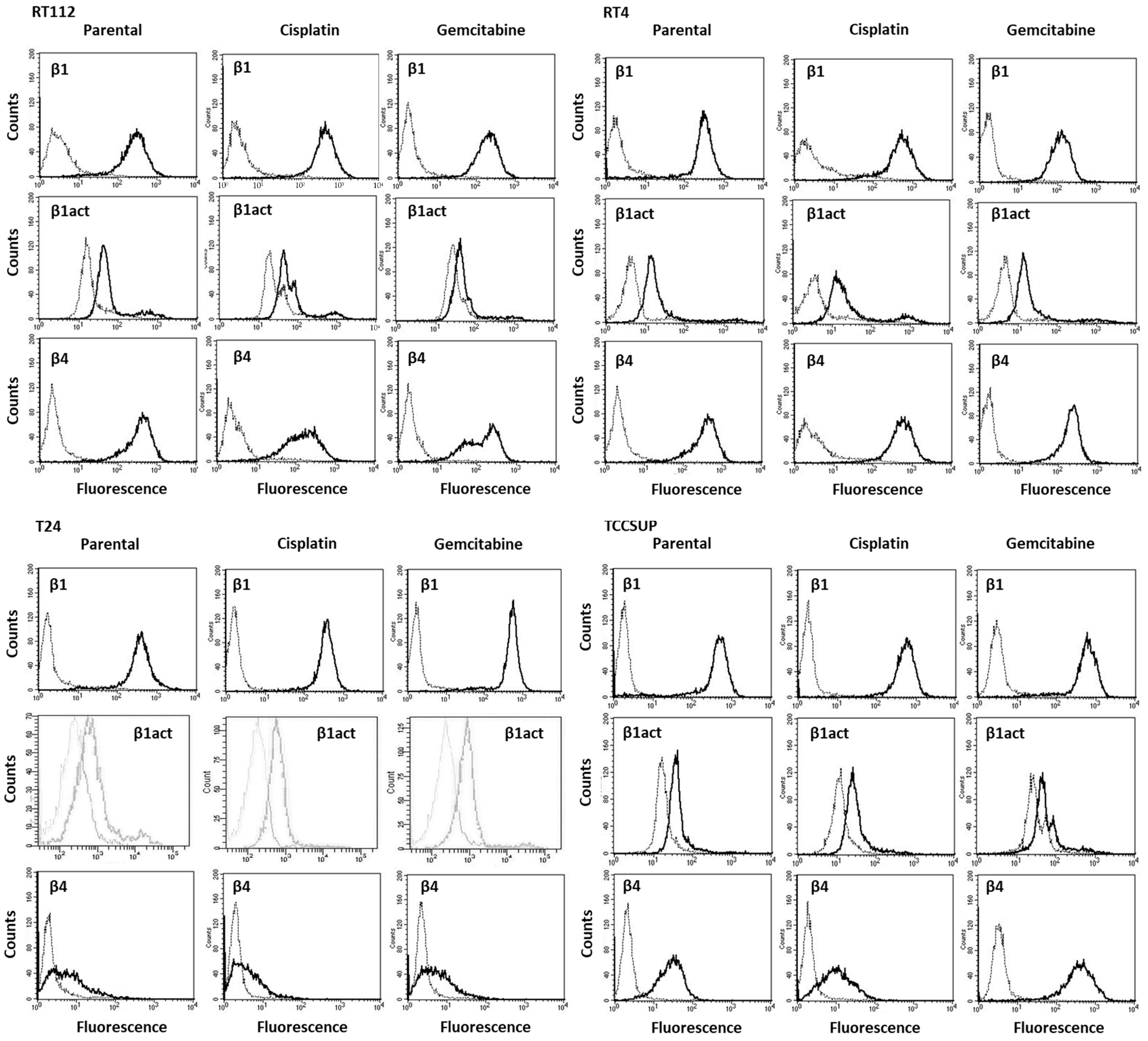
3.1. Integrin Expression
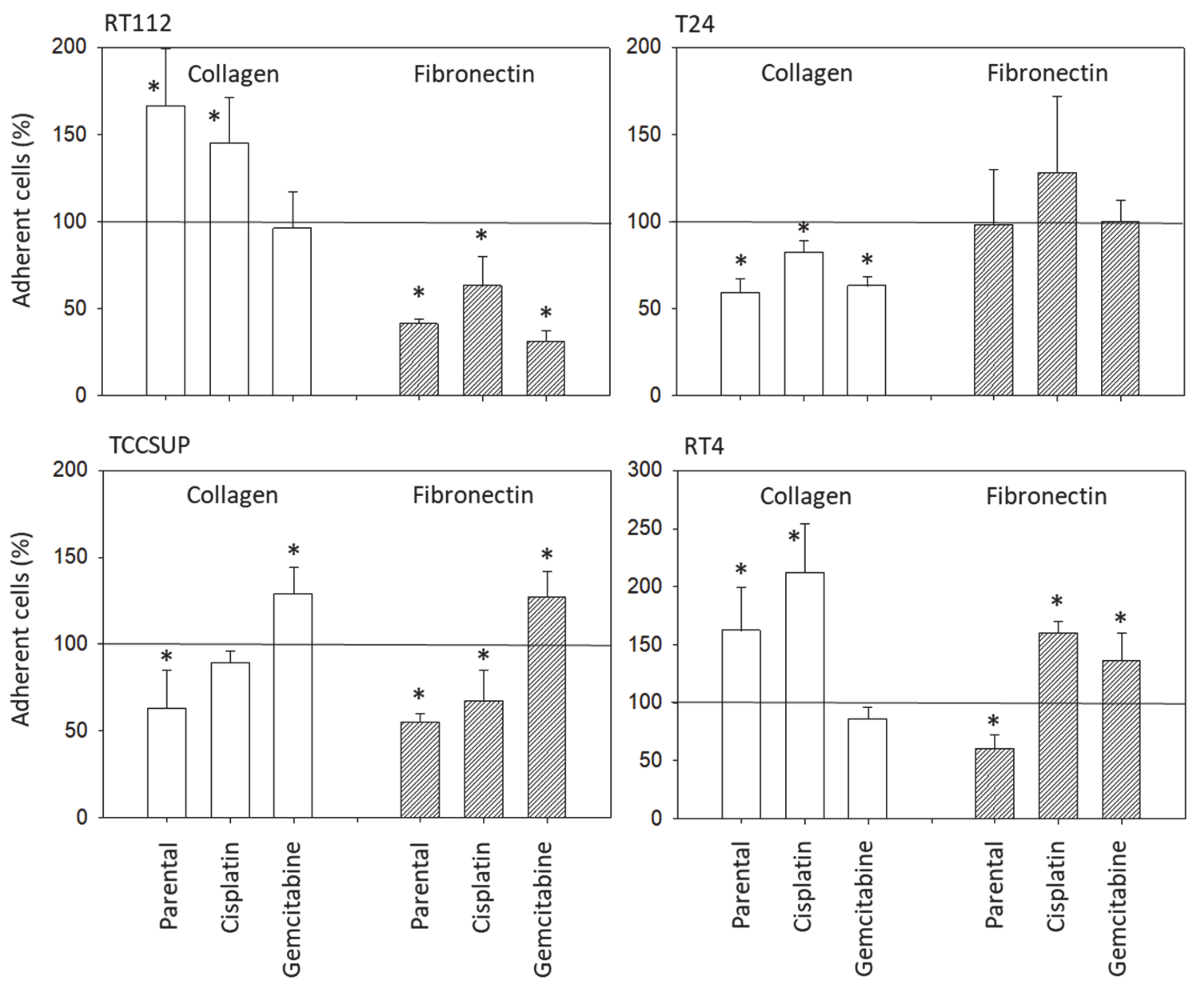
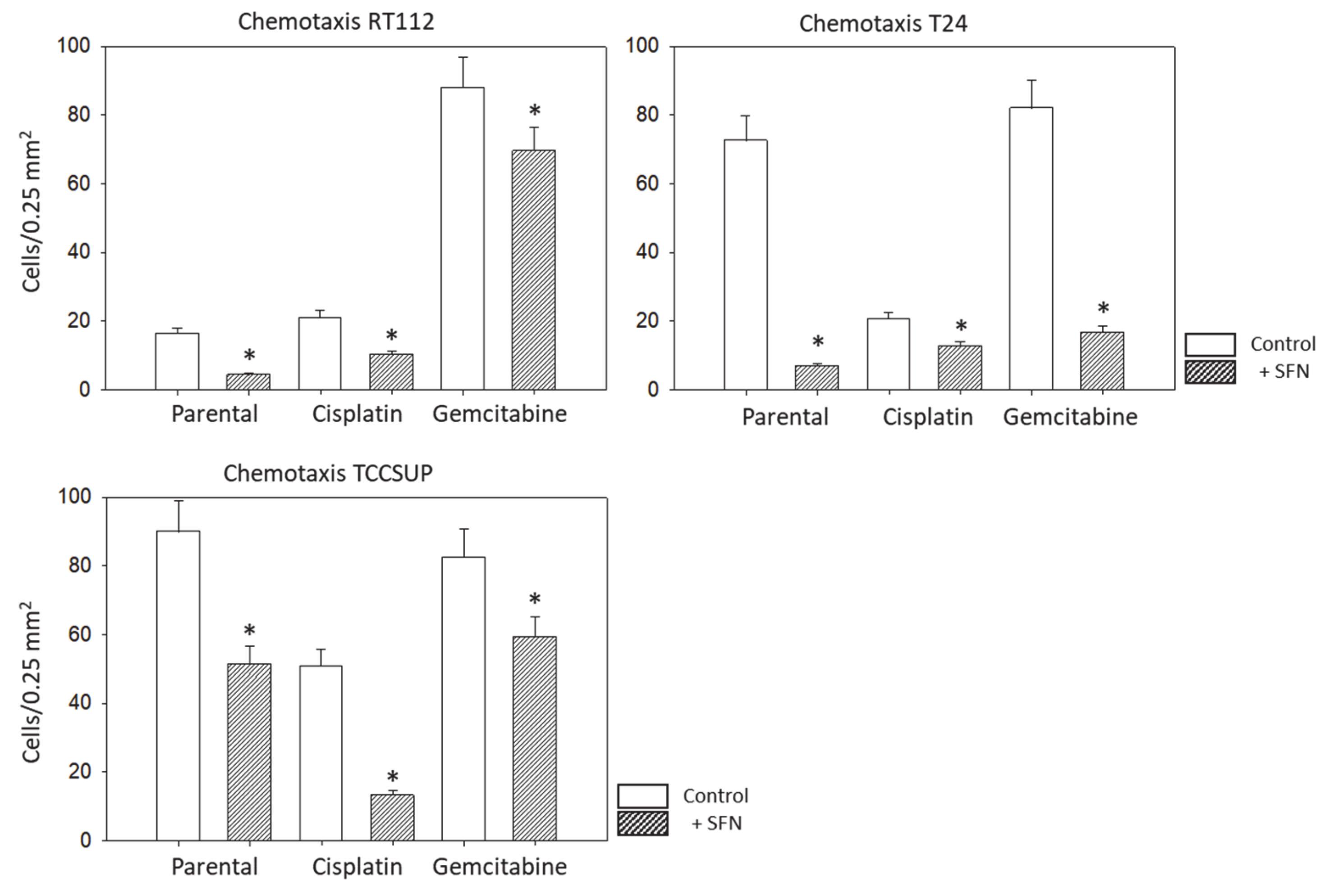
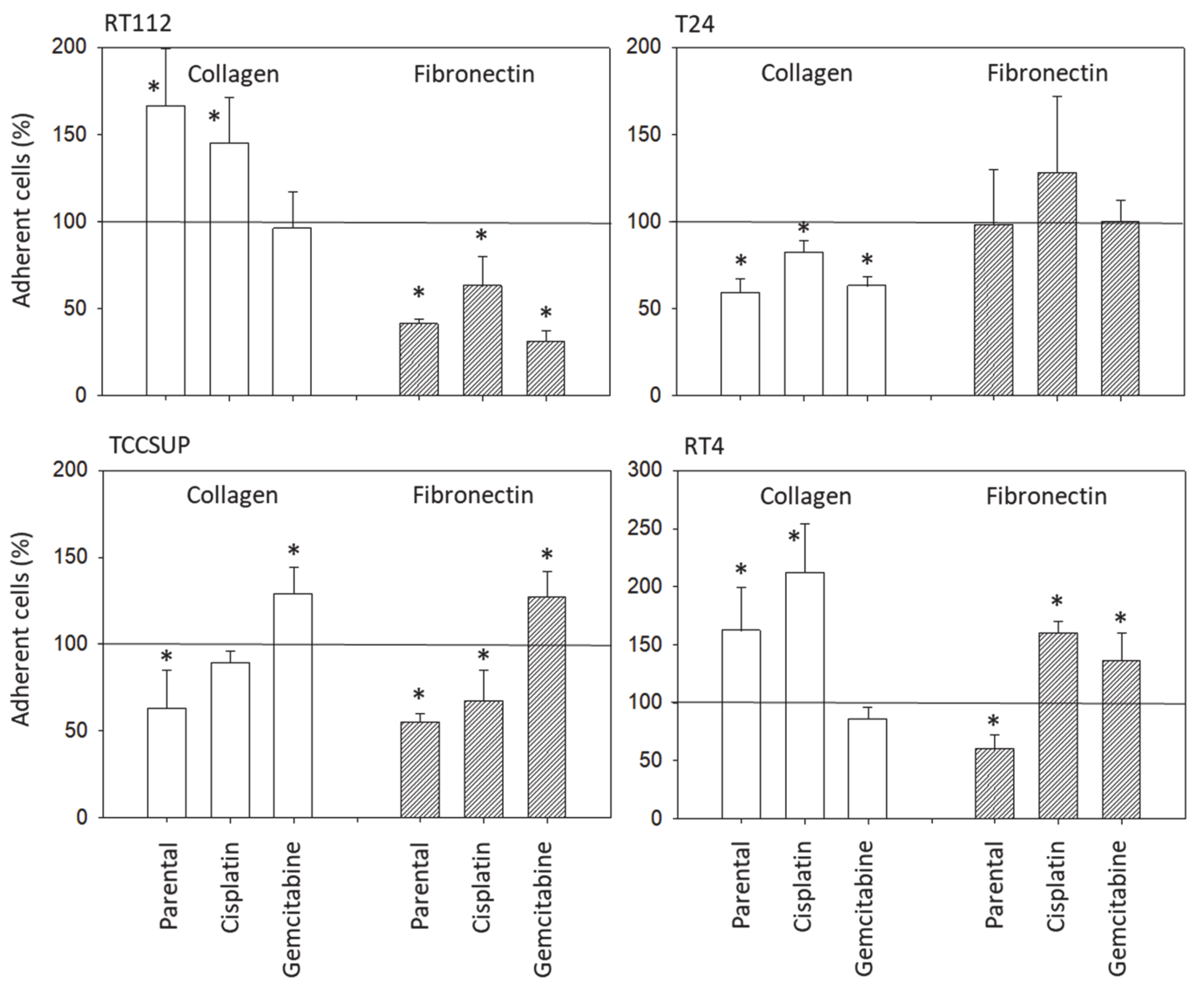
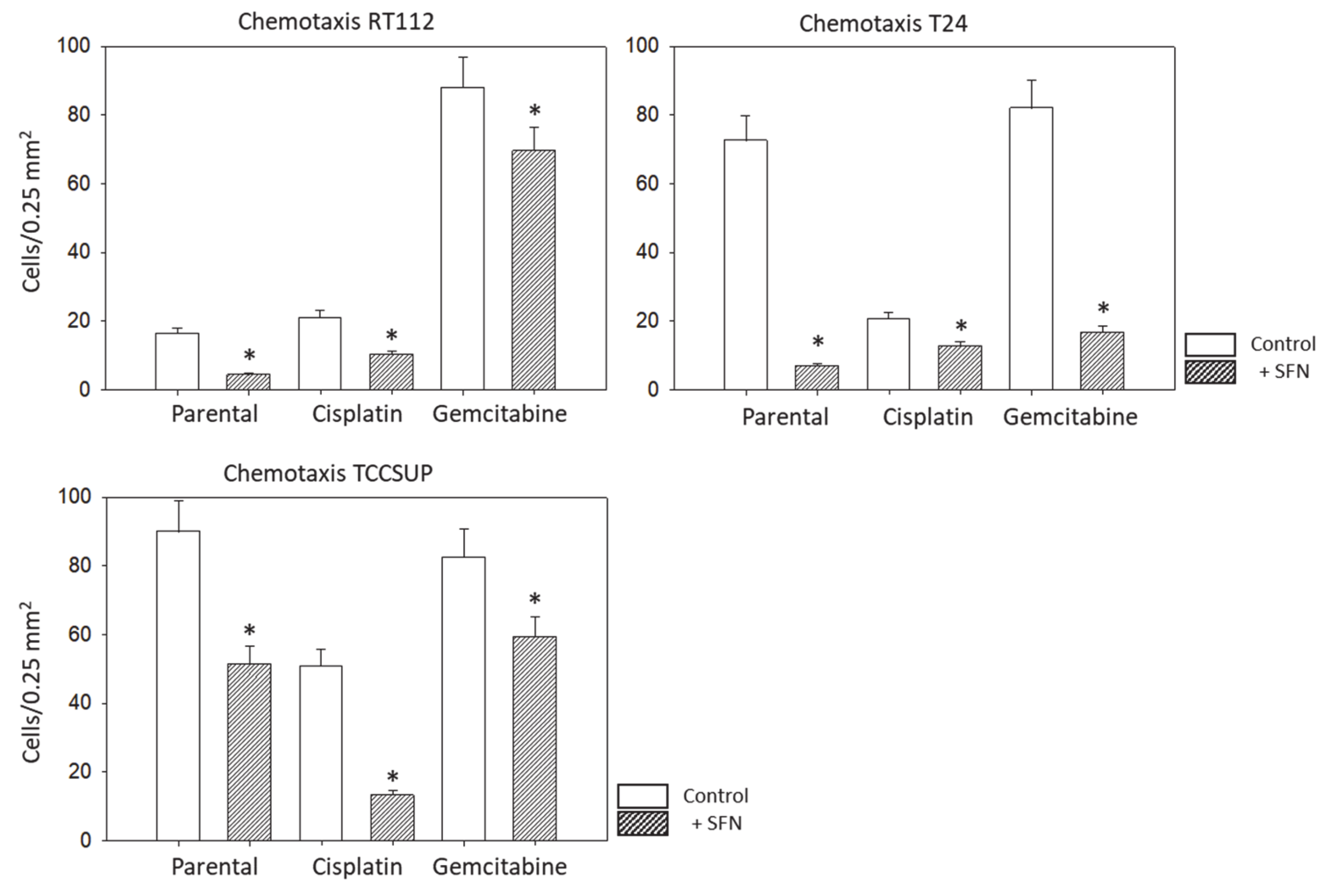
3.2. Tumor Cell Adhesion and Chemotaxis
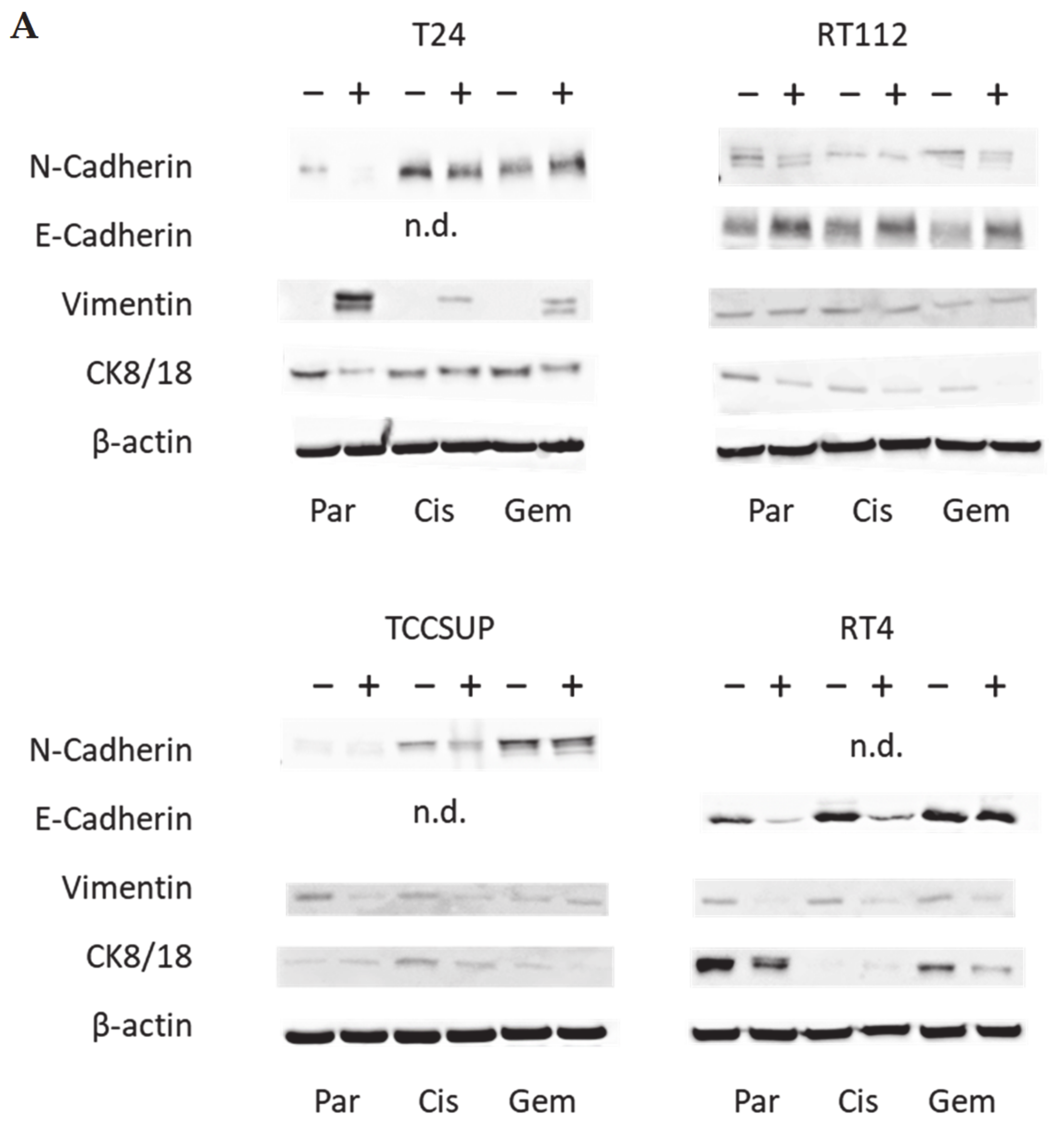
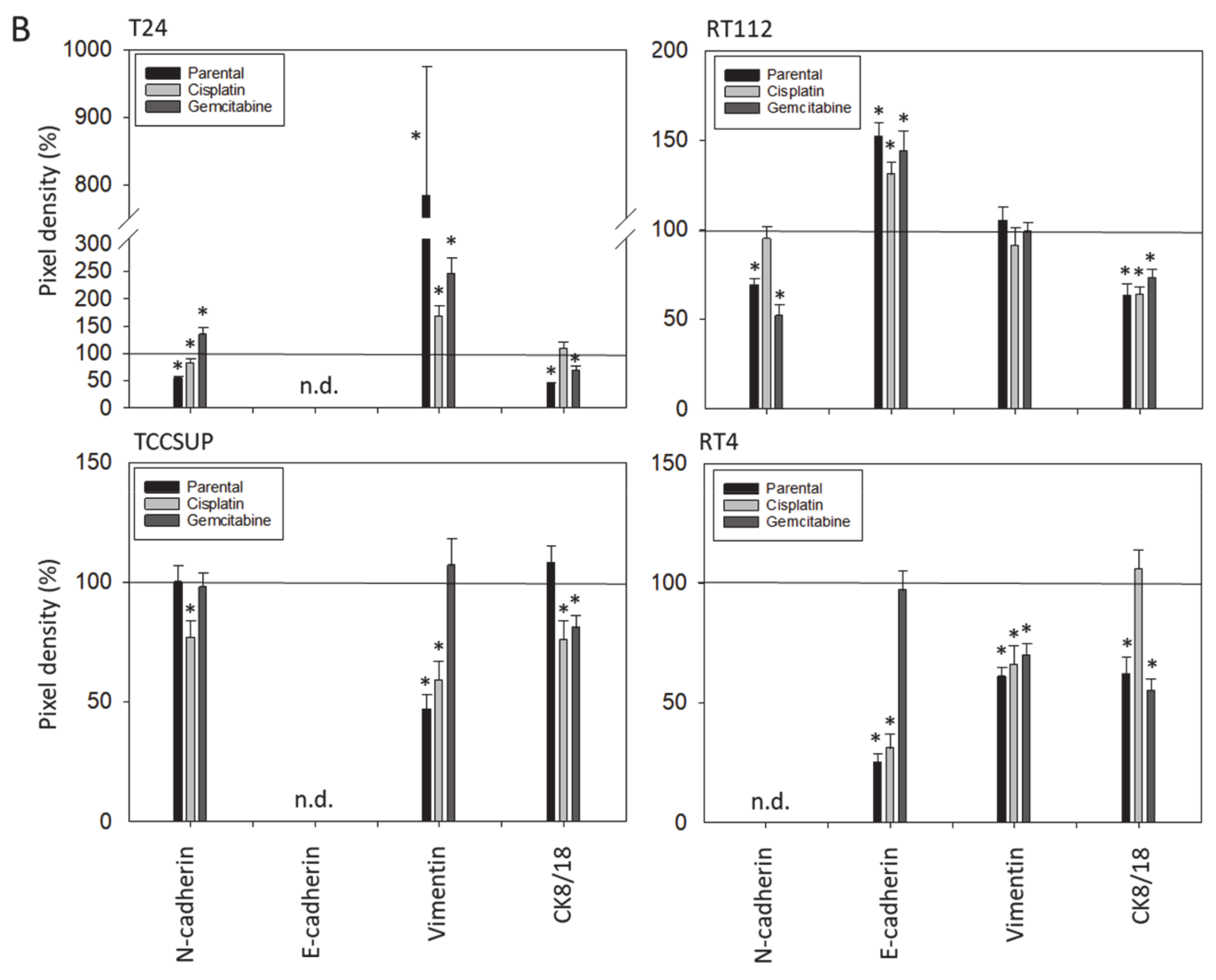
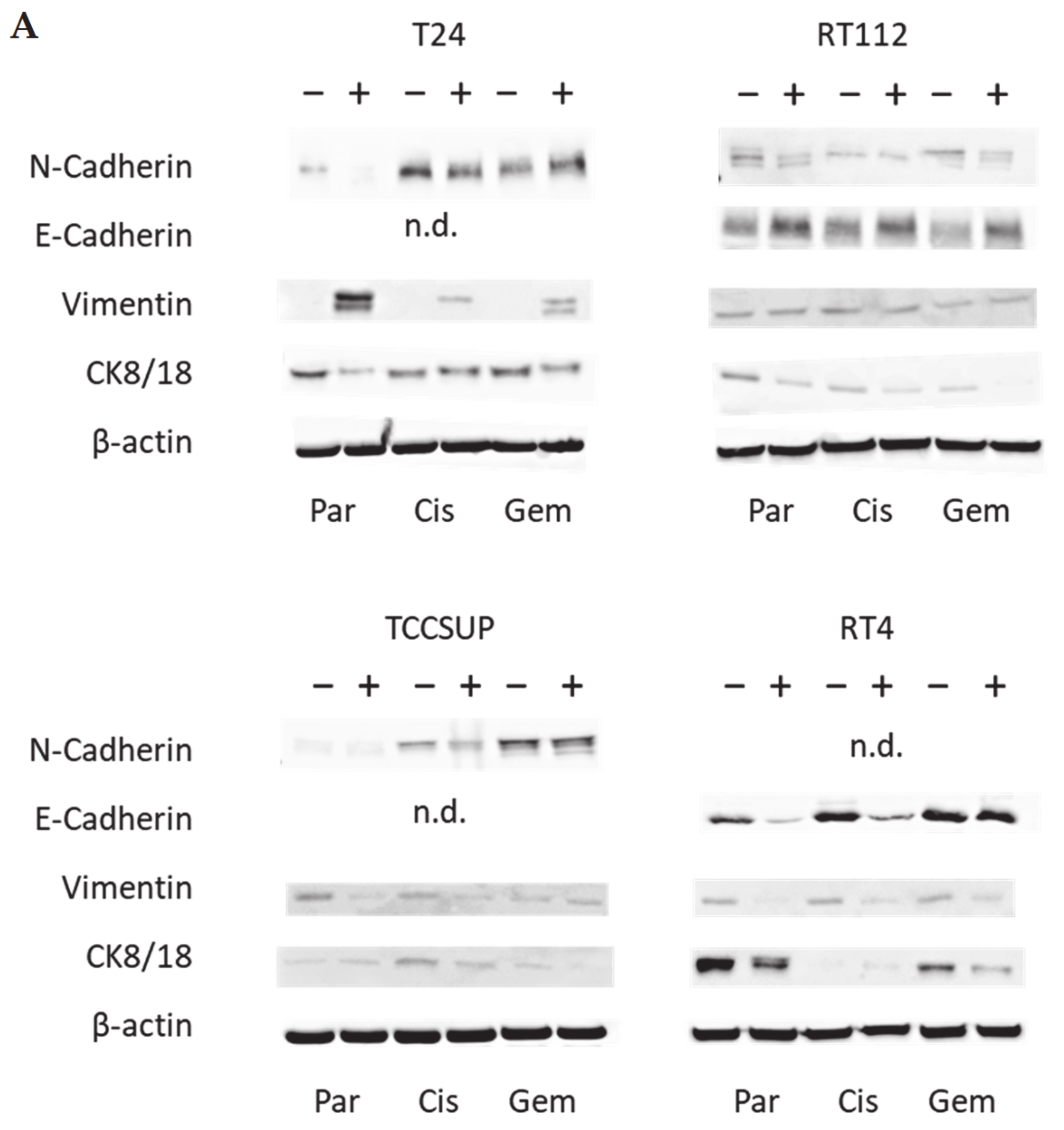
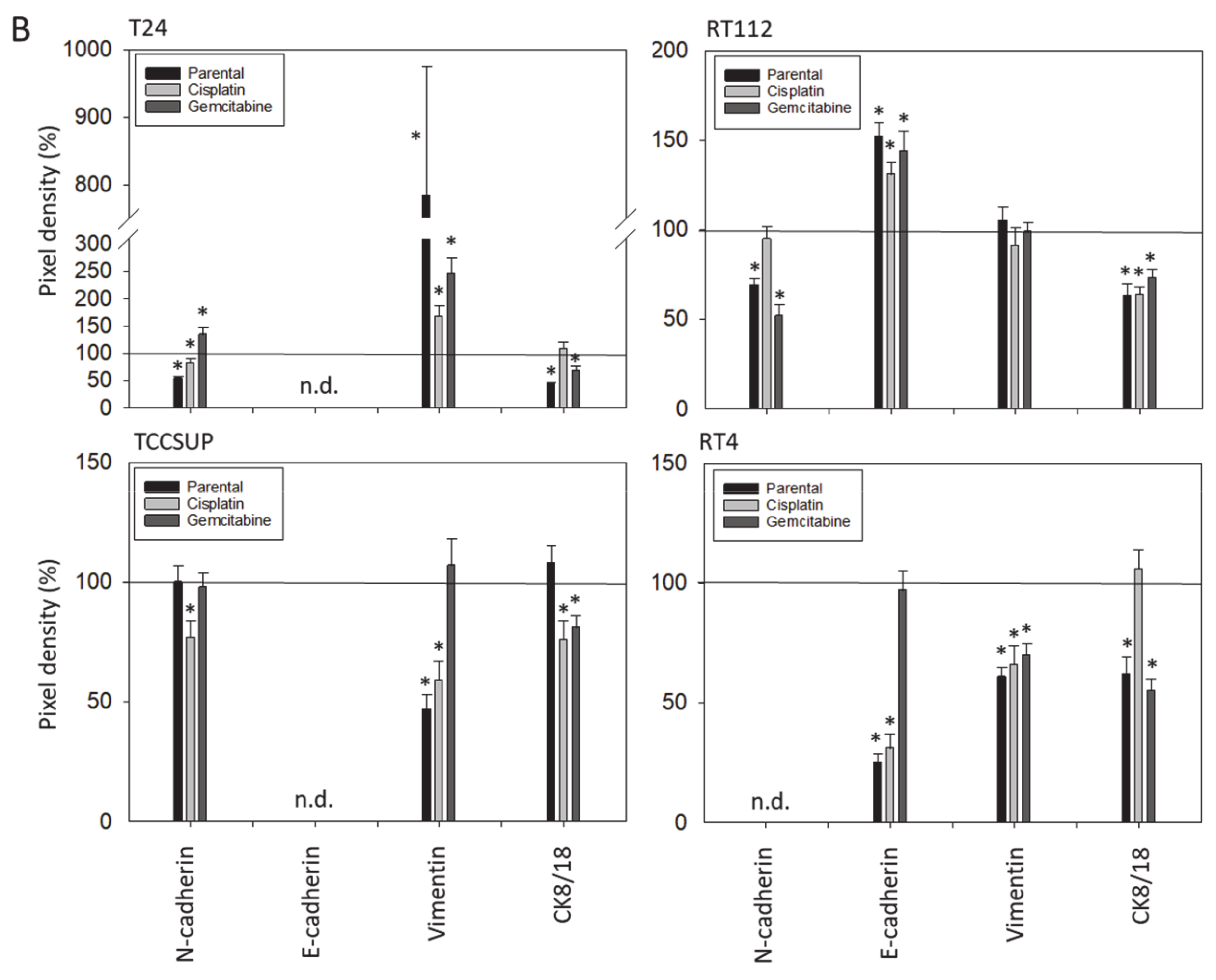
3.3. Tumor Cell Differentiation
3.4. Cadherin and Vimentin Translocation
3.5. Integrin Blocking Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Z.; Li, Z.; Zhu, Y.; Xu, C. E3 ubiquitin ligases and deubiquitinases in bladder cancer tumorigenesis and implications for immunotherapies. Front. Immunol. 2023, 14, 1226057. [Google Scholar] [CrossRef]
- Bonucci, M.; Geraci, A.; Pero, D.; Villivà, C.; Cordella, D.; Condello, M.; Meschini, S.; Del Campo, L.; Tomassi, F.; Porcu, A.; et al. Complementary and Integrative Approaches to Cancer: A Pilot Survey of Attitudes and Habits among Cancer Patients in Italy. Evid. Based Complement. Altern. Med. 2022, 2022, 2923967. [Google Scholar] [CrossRef]
- Kanimozhi, T.; Hindu, K.; Maheshvari, Y.; Khushnidha, Y.G.; Kumaravel, M.; Srinivas, K.S.; Manickavasagam, M.; Mangathayaru, K. Herbal supplement usage among cancer patients: A questionnaire-based survey. J. Cancer Res. Ther. 2021, 17, 136–141. [Google Scholar]
- Kennelley, G.E.; Amaye-Obu, T.; Foster, B.A.; Tang, L.; Paragh, G.; Huss, W.J. Mechanistic review of sulforaphane as a chemoprotective agent in bladder cancer. Am. J. Clin. Exp. Urol. 2023, 11, 103–120. [Google Scholar]
- Schepici, G.; Bramanti, P.; Mazzon, E. Efficacy of Sulforaphane in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8637. [Google Scholar] [CrossRef]
- Xie, H.; Rutz, J.; Maxeiner, S.; Grein, T.; Thomas, A.; Juengel, E.; Chun, F.K.; Cinatl, J.; Haferkamp, A.; Tsaur, I.; et al. Plant-Derived Sulforaphane Suppresses Growth and Proliferation of Drug-Sensitive and Drug-Resistant Bladder Cancer Cell Lines In Vitro. Cancers 2022, 14, 4682. [Google Scholar] [CrossRef]
- Wang, F.; Liu, P.; An, H.; Zhang, Y. Sulforaphane suppresses the viability and metastasis, and promotes the apoptosis of bladder cancer cells by inhibiting the expression of FAT-1. Int. J. Mol. Med. 2020, 46, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Justin, S.; Rutz, J.; Maxeiner, S.; Chun, F.K.; Juengel, E.; Blaheta, R.A. Bladder Cancer Metastasis Induced by Chronic Everolimus Application Can Be Counteracted by Sulforaphane In Vitro. Int. J. Mol. Sci. 2020, 21, 5582. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Buongiorno, L.P.; Wang, W.; Tang, J.C.Y.; Miceli, N.; Taviano, M.F.; Shan, Y.; Bao, Y. The Inhibitory Effect of Sulforaphane on Bladder Cancer Cell Depends on GSH Depletion-Induced by Nrf2 Translocation. Molecules 2021, 26, 4919. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Qorri, B.; Baluch, N.; Sparaneo, A.; Fabrizio, F.P.; Muscarella, L.A.; Tyker, A.; Kumar, S.; Cheng, H.M.; Szewczuk, M.R.; et al. Next-generation multimodality of nutrigenomic cancer therapy: Sulforaphane in combination with acetazolamide actively target bronchial carcinoid cancer in disabling the PI3K/Akt/mTOR survival pathway and inducing apoptosis. Oncotarget 2021, 12, 1470–1489. [Google Scholar] [CrossRef]
- Liu, F.; Lv, R.B.; Liu, Y.; Hao, Q.; Liu, S.J.; Zheng, Y.Y.; Li, C.; Zhu, C.; Wang, M. Salinomycin and Sulforaphane Exerted Synergistic Antiproliferative and Proapoptotic Effects on Colorectal Cancer Cells by Inhibiting the PI3K/Akt Signaling Pathway in vitro and in vivo. OncoTargets Ther. 2020, 13, 4957–4969. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Fazli, M.; Saeednia, S.; Gholami Kharanagh, M.; Ahmadiankia, N. Sulforaphane Modulates Cell Migration and Expression of β-Catenin and Epithelial Mesenchymal Transition Markers in Breast Cancer Cells. Iran. J. Public Health 2020, 49, 77–85. [Google Scholar]
- Gong, T.T.; Liu, X.D.; Zhan, Z.P.; Wu, Q.J. Sulforaphane enhances the cisplatin sensitivity through regulating DNA repair and accumulation of intracellular cisplatin in ovarian cancer cells. Exp. Cell Res. 2020, 393, 112061. [Google Scholar] [CrossRef]
- Račkauskas, R.; Zhou, D.; Ūselis, S.; Strupas, K.; Herr, I.; Schemmer, P. Sulforaphane sensitizes human cholangiocarcinoma to cisplatin via the downregulation of anti-apoptotic proteins. Oncol. Rep. 2017, 37, 3660–3666. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Xie, Y.K.; Wu, Y.; Li, L.L.; Liu, Y.; Miao, X.B.; Liu, Q.Z.; Yao, K.T.; Xiao, G.H. Sulforaphane inhibits cancer stem-like cell properties and cisplatin resistance through miR-214-mediated downregulation of c-MYC in non-small cell lung cancer. Oncotarget 2017, 8, 12067–12080. [Google Scholar] [CrossRef]
- Tomooka, F.; Kaji, K.; Nishimura, N.; Kubo, T.; Iwai, S.; Shibamoto, A.; Suzuki, J.; Kitagawa, K.; Namisaki, T.; Akahane, T.; et al. Sulforaphane Potentiates Gemcitabine-Mediated Anti-Cancer Effects against Intrahepatic Cholangiocarcinoma by Inhibiting HDAC Activity. Cells 2023, 12, 687. [Google Scholar] [CrossRef]
- Chatterjee, S.; Rhee, Y.H.; Ahn, J.C. Sulforaphene-Carboplatin Combination Synergistically Enhances Apoptosis by Disruption of Mitochondrial Membrane Potential and Cell Cycle Arrest in Human Non-Small Cell Lung Carcinoma. J. Med. Food 2016, 19, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Siech, C.; Rutz, J.; Maxeiner, S.; Grein, T.; Sonnenburg, M.; Tsaur, I.; Chun, F.K.; Blaheta, R.A. Insulin-like Growth Factor-1 Influences Prostate Cancer Cell Growth and Invasion through an Integrin α3, α5, αV, and β1 Dependent Mechanism. Cancers 2022, 14, 363. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Su, L.J.; Flaig, T.W. VEGFR and EGFR inhibition increases epithelial cellular characteristics and chemotherapy sensitivity in mesenchymal bladder cancer cells. Oncol. Rep. 2010, 24, 1019–1028. [Google Scholar]
- Mehus, A.A.; Bergum, N.; Knutson, P.; Shrestha, S.; Kalonick, M.; Zhou, X.; Garrett, S.H.; Sens, D.A.; Sens, M.A.; Somji, S. Chronic Arsenic Exposure Upregulates the Expression of Basal Transcriptional Factors and Increases Invasiveness of the Non-Muscle Invasive Papillary Bladder Cancer Line RT4. Int. J. Mol. Sci. 2022, 23, 12313. [Google Scholar] [CrossRef] [PubMed]
- Rubtsova, S.N.; Zhitnyak, I.Y.; Gloushankova, N.A. Dual role of E-cadherin in cancer cells. Tissue Barriers 2022, 10, 2005420. [Google Scholar] [CrossRef] [PubMed]
- Bendardaf, R.; Sharif-Askari, F.S.; Sharif-Askari, N.S.; Syrjänen, K.; Pyrhönen, S. Cytoplasmic E-Cadherin Expression Is Associated With Higher Tumour Level of VEGFA, Lower Response Rate to Irinotecan-based Treatment and Poorer Prognosis in Patients With Metastatic Colorectal Cancer. Anticancer Res. 2019, 39, 1953–1957. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Nie, J.H.; Lin, X.M.; Wu, J.B.; Chen, Z.; Xu, B.; Liu, J. Biological implications of PTEN upregulation and altered sodium/iodide symporter intracellular distribution in resveratrol-suppressed anaplastic thyroid cancer cells. J. Cancer 2020, 11, 6883–6891. [Google Scholar] [CrossRef] [PubMed]
- Zang, W.; Liu, J.; Geng, F.; Liu, D.; Zhang, S.; Li, Y.; Pan, Y. Butyrate promotes oral squamous cell carcinoma cells migration, invasion and epithelial-mesenchymal transition. PeerJ 2022, 10, e12991. [Google Scholar] [CrossRef] [PubMed]
- Li, L.N.; Zhang, H.D.; Yuan, S.J.; Yang, D.X.; Wang, L.; Sun, Z.X. Differential sensitivity of colorectal cancer cell lines to artesunate is associated with expression of beta-catenin and E-cadherin. Eur. J. Pharmacol. 2008, 588, 1–8. [Google Scholar] [CrossRef]
- Wint, H.; Li, J.; Abe, T.; Yamada, H.; Higaki, T.; Nasu, Y.; Watanabe, M.; Takei, K.; Takeda, T. Pacsin 2-dependent N-cadherin internalization regulates the migration behaviour of malignant cancer cells. J. Cell Sci. 2023, 136, jcs260827. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Reis, S.; Miranda-Gonçalves, V.; Guimarães-Teixeira, C.; Martins-Lima, C.; Lobo, J.; Montezuma, D.; Dias, P.C.; Neyret-Kahn, H.; Bernard-Pierrot, I.; Henrique, R.; et al. Vimentin epigenetic deregulation in Bladder Cancer associates with acquisition of invasive and metastatic phenotype through epithelial-to-mesenchymal transition. Int. J. Biol. Sci. 2023, 19, 1–12. [Google Scholar] [CrossRef]
- Wang, Y.; Gong, J.; Yao, Y. Extracellular nanofiber-orchestrated cytoskeletal reorganization and mediated directional migration of cancer cells. Nanoscale 2020, 12, 3183–3193. [Google Scholar] [CrossRef]
- Li, S.; Xiong, N.; Peng, Y.; Tang, K.; Bai, H.; Lv, X.; Jiang, Y.; Qin, X.; Yang, H.; Wu, C.; et al. Acidic pHe regulates cytoskeletal dynamics through conformational integrin β1 activation and promotes membrane protrusion. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2395–2408. [Google Scholar] [CrossRef]
- Frescas, D.; Roux, C.M.; Aygun-Sunar, S.; Gleiberman, A.S.; Krasnov, P.; Kurnasov, O.V.; Strom, E.; Virtuoso, L.P.; Wrobel, M.; Osterman, A.L.; et al. Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody. Proc. Natl. Acad. Sci. USA 2017, 114, E1668–E1677. [Google Scholar] [CrossRef] [PubMed]
- Kuburich, N.A.; den Hollander, P.; Pietz, J.T.; Mani, S.A. Vimentin and cytokeratin: Good alone, bad together. Semin. Cancer Biol. 2022, 86, 816–826. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.; Rutz, J.; Maxeiner, S.; Grein, T.; Thomas, A.; Juengel, E.; Chun, F.K.-H.; Cinatl, J.; Haferkamp, A.; Tsaur, I.; et al. Sulforaphane Inhibits Adhesion and Migration of Cisplatin- and Gemcitabine-Resistant Bladder Cancer Cells In Vitro. Nutrients 2024, 16, 623. https://doi.org/10.3390/nu16050623
Xie H, Rutz J, Maxeiner S, Grein T, Thomas A, Juengel E, Chun FK-H, Cinatl J, Haferkamp A, Tsaur I, et al. Sulforaphane Inhibits Adhesion and Migration of Cisplatin- and Gemcitabine-Resistant Bladder Cancer Cells In Vitro. Nutrients. 2024; 16(5):623. https://doi.org/10.3390/nu16050623
Chicago/Turabian StyleXie, Hui, Jochen Rutz, Sebastian Maxeiner, Timothy Grein, Anita Thomas, Eva Juengel, Felix K.-H. Chun, Jindrich Cinatl, Axel Haferkamp, Igor Tsaur, and et al. 2024. "Sulforaphane Inhibits Adhesion and Migration of Cisplatin- and Gemcitabine-Resistant Bladder Cancer Cells In Vitro" Nutrients 16, no. 5: 623. https://doi.org/10.3390/nu16050623
APA StyleXie, H., Rutz, J., Maxeiner, S., Grein, T., Thomas, A., Juengel, E., Chun, F. K.-H., Cinatl, J., Haferkamp, A., Tsaur, I., & Blaheta, R. A. (2024). Sulforaphane Inhibits Adhesion and Migration of Cisplatin- and Gemcitabine-Resistant Bladder Cancer Cells In Vitro. Nutrients, 16(5), 623. https://doi.org/10.3390/nu16050623