
Efficacy of Butyrate to Inhibit Colonic Cancer Cell Growth Is Cell Type-Specific and Apoptosis-Dependent


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials, Cell Culture, and Cell Proliferation Assay
2.2. Cell Apoptosis Analysis
2.3. Western Blotting Analysis
2.4. Immunofluorescent Staining
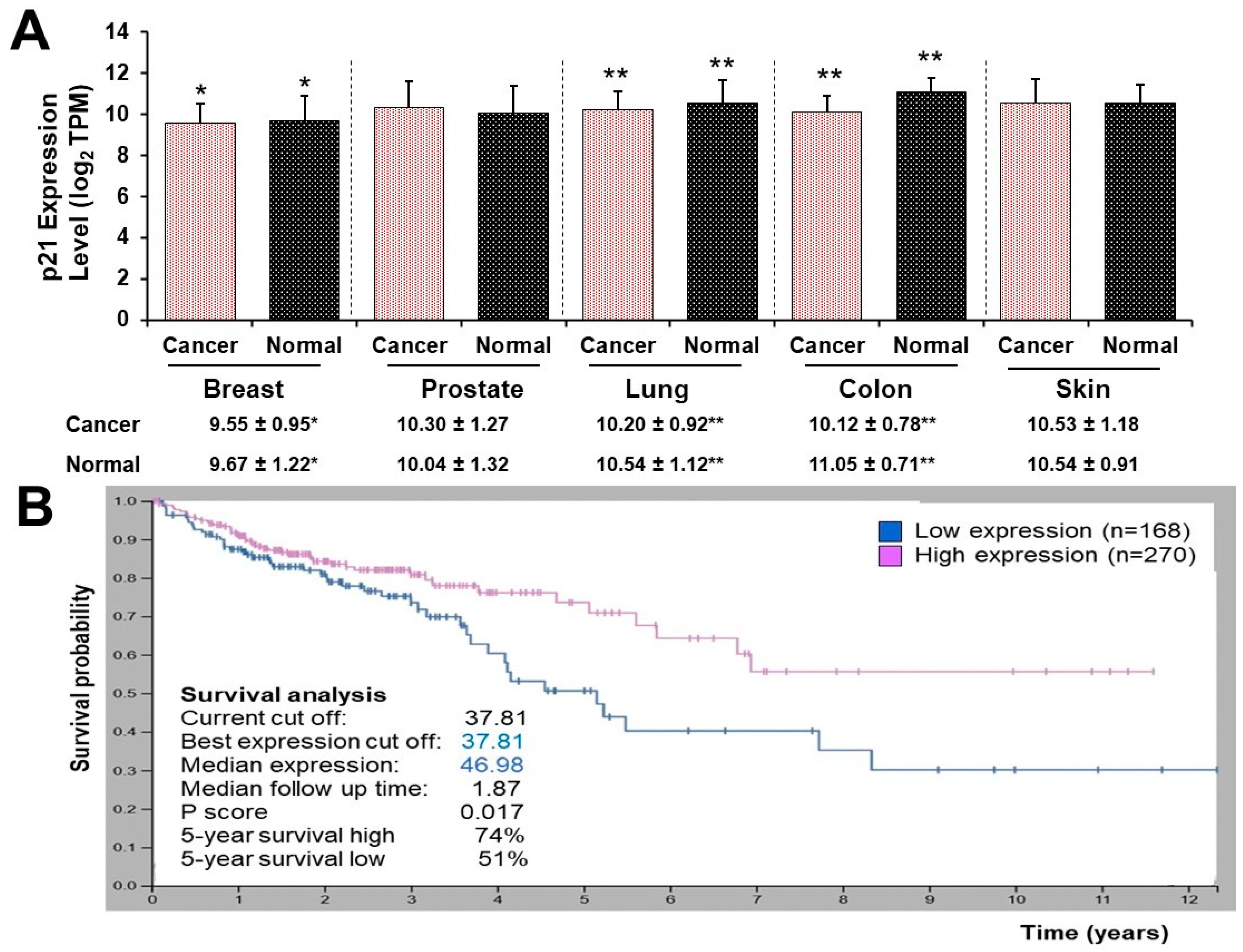
2.5. Analysis p21 Gene Expression in Human Tumor Tissues and Survival Analysis
2.6. Statistical Analysis
3. Results
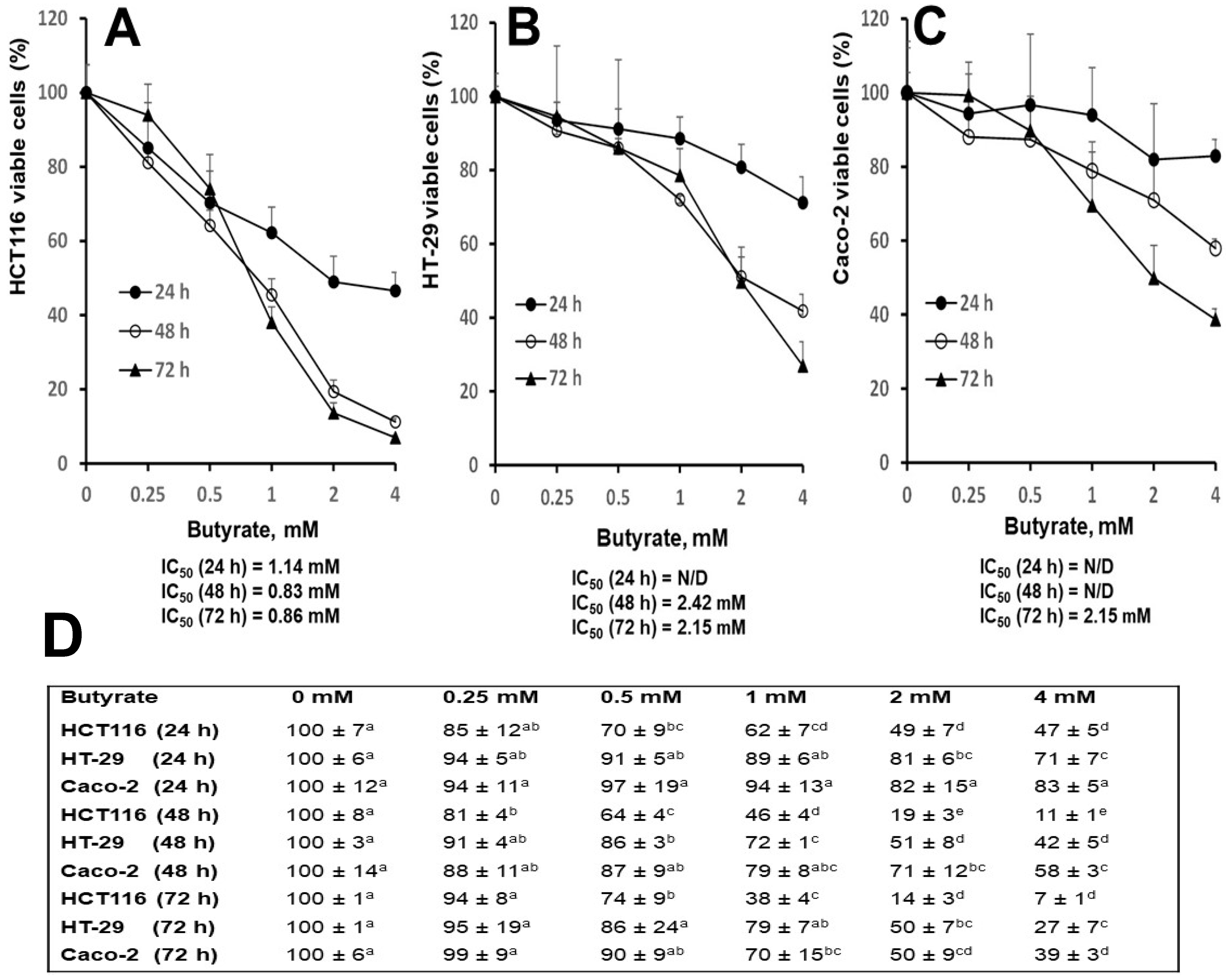
3.1. Distinct Effects of Butyrate on Cell Proliferation among Three Cell Types
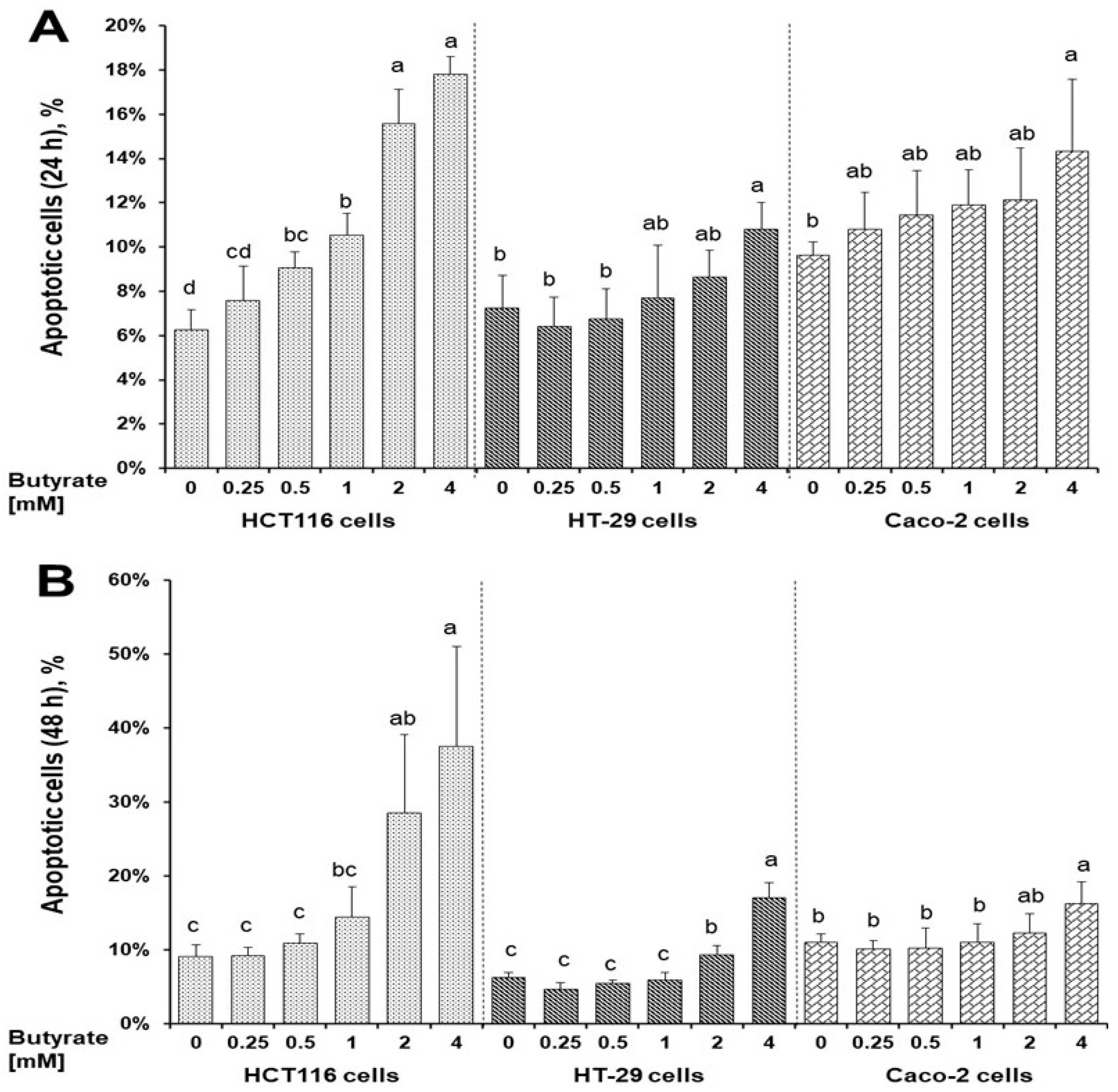
3.2. Distinct Effects of Butyrate on Apoptosis among Three Cell Types
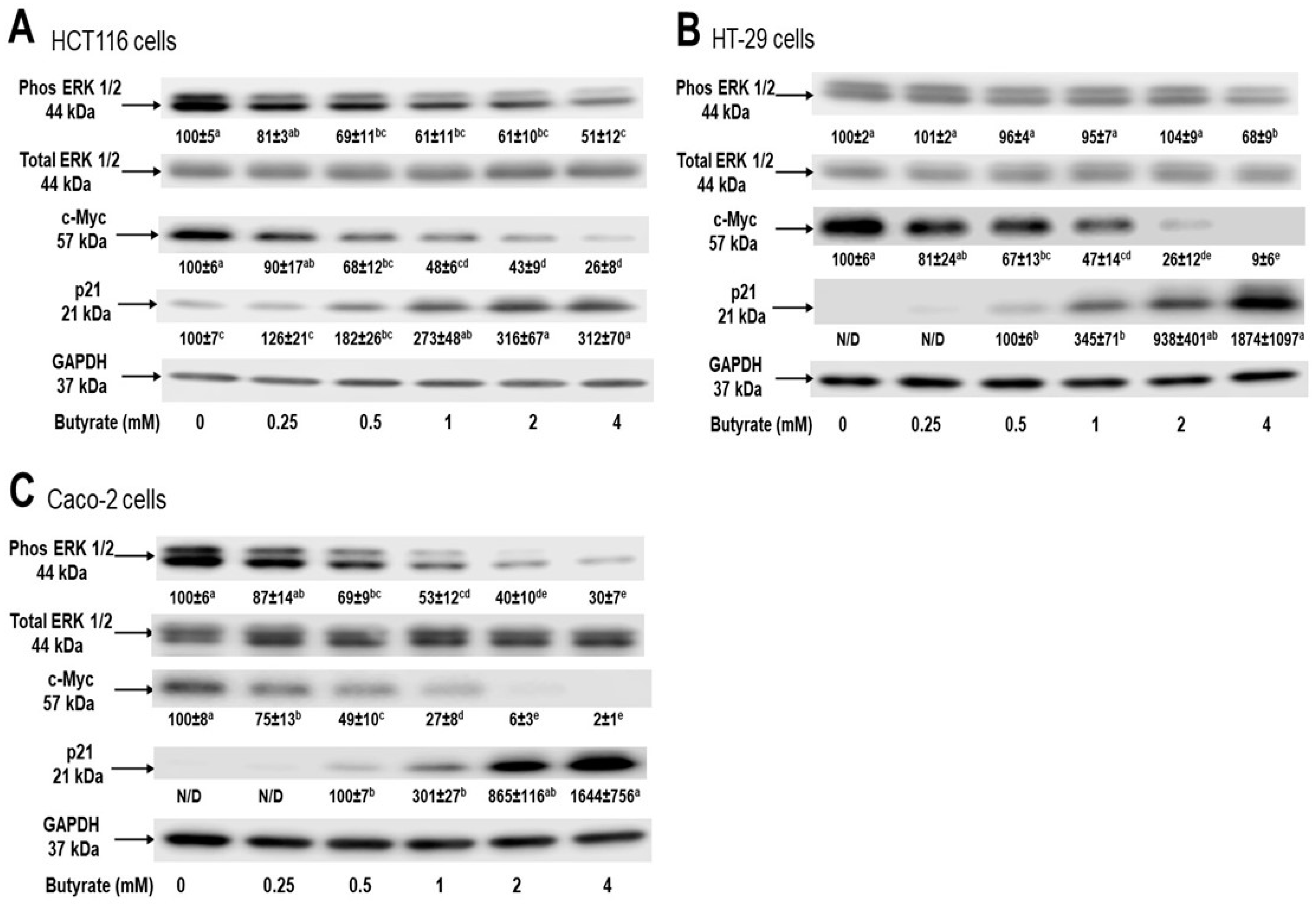
3.3. Distinct Effects of Butyrate on Signaling Proteins among Three Cell Types
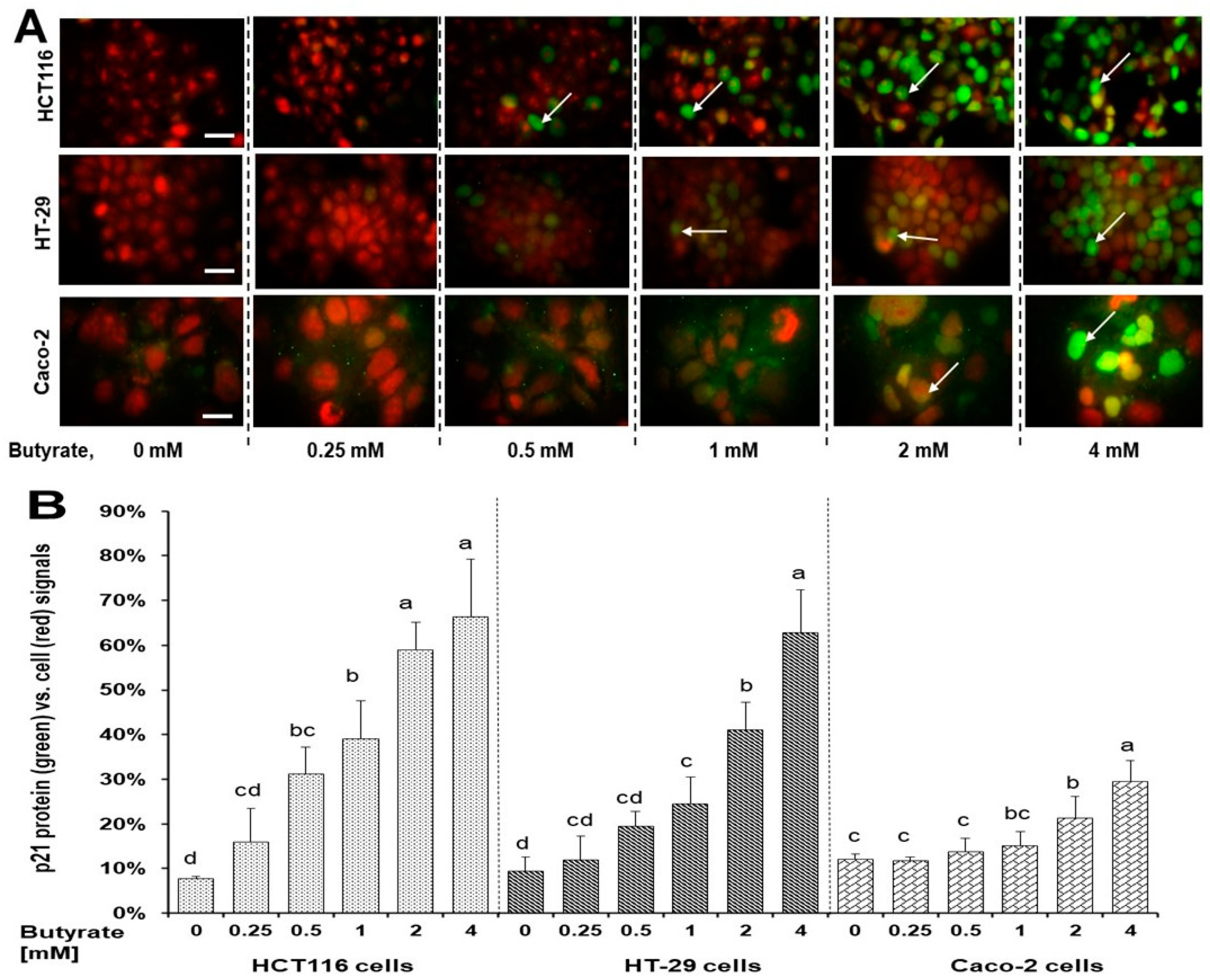
3.4. Distinct Effects of Butyrate on p21 Protein’s Subcellular Localization among Three Cell Types
3.5. High and Low p21 Expression Cohorts Differ in Survival Time after Diagnosis with Colon Cancer
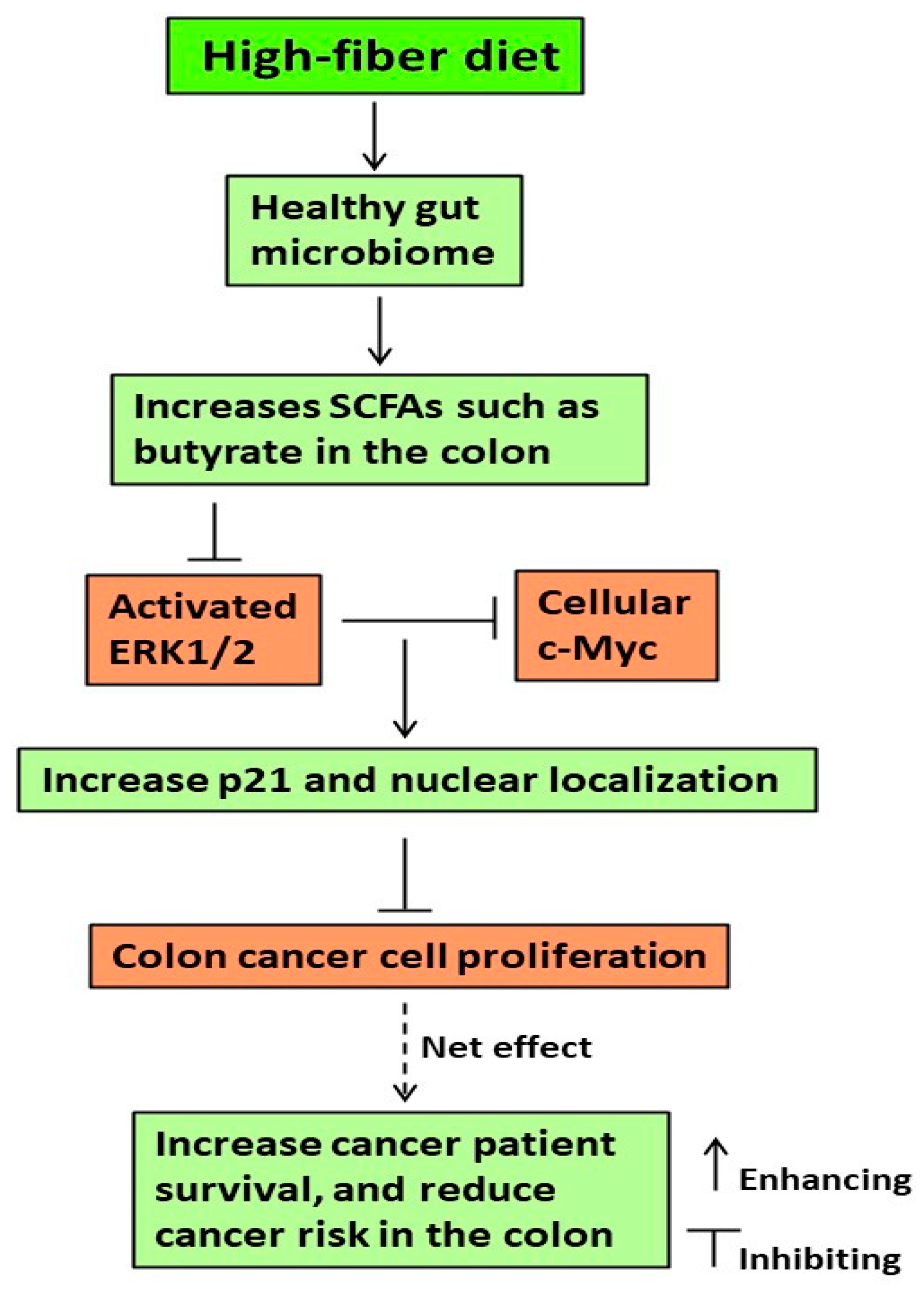
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef]
- Satia, J.A.; Keku, T.; Galanko, J.A.; Martin, C.; Doctolero, R.T.; Tajima, A.; Sandler, R.S.; Carethers, J.M. Diet, lifestyle, and genomic instability in the North Carolina Colon Cancer Study. Cancer Epidemiol. Biomark. Prev. 2005, 14, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Perrin, P.; Pierre, F.; Patry, Y.; Champ, M.; Berreur, M.; Pradal, G.; Bornet, F.; Meflah, K.; Menanteau, J. Only fibres promoting a stable butyrate producing colonic ecosystem decrease the rate of aberrant crypt foci in rats. Gut 2001, 48, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Hirose, Y.; Cohen, L.A.; Simi, B.; Cooma, I.; Rao, C.V. Preventive potential of wheat bran fractions against experimental colon carcinogenesis: Implications for human colon cancer prevention. Cancer Res. 2000, 60, 4792–4797. [Google Scholar] [PubMed]
- Slavin, J.L. Position of the American Dietetic Association: Health implications of dietary fiber. J. Am. Diet. Assoc. 2008, 108, 1716–1731. [Google Scholar] [PubMed]
- Papathanasopoulos, A.; Camilleri, M. Dietary fiber supplements: Effects in obesity and metabolic syndrome and relationship to gastrointestinal functions. Gastroenterology 2010, 138, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Raninen, K.; Lappi, J.; Mykkanen, H.; Poutanen, K. Dietary fiber type reflects physiological functionality: Comparison of grain fiber, inulin, and polydextrose. Nutr. Rev. 2011, 69, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Lazarova, D.L.; Bordonaro, M. Mechanisms linking dietary fiber, gut microbiota and colon cancer prevention. World J. Gastrointest. Oncol. 2014, 6, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.E.; Marthinsen, D.; Kuhnlein, H. Colonic function and fermentation in men consuming high fiber diets. J. Nutr. 1983, 113, 2535–2544. [Google Scholar] [CrossRef]
- Fleming, S.E.; O’Donnell, A.U.; Perman, J.A. Influence of frequent and long-term bean consumption on colonic function and fermentation. Am. J. Clin. Nutr. 1985, 41, 909–918. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Degirolamo, C.; Modica, S.; Palasciano, G.; Moschetta, A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol. Med. 2011, 17, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.H.; Wright, N.A. The gastrointestinal tract stem cell niche. Stem Cell Rev. 2006, 2, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Hamlin, S.K.; Safratowich, B.D.; Cheng, W.H.; Johnson, L.K. Superior inhibitory efficacy of butyrate over propionate and acetate against human colon cancer cell proliferation via cell cycle arrest and apoptosis: Linking dietary fiber to cancer prevention. Nutr. Res. 2020, 83, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Lannagan, T.R.; Jackstadt, R.; Leedham, S.J.; Sansom, O.J. Advances in colon cancer research: In vitro and animal models. Curr. Opin. Genet. Dev. 2021, 66, 50–56. [Google Scholar] [CrossRef]
- National Cancer Institue. The Cancer Genome Atlas Program. 2024. Available online: https://www.cancer.gov/ccg/research/genome-sequencing/tcga (accessed on 30 January 2024).
- Yeung, T.M.; Gandhi, S.C.; Wilding, J.L.; Muschel, R.; Bodmer, W.F. Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. USA 2010, 107, 3722–3727. [Google Scholar] [CrossRef]
- Lea, T. Caco-2 Cell Line. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 103–111. [Google Scholar]
- Yang, B.; Cao, L.; Liu, B.; McCaig, C.D.; Pu, J. The transition from proliferation to differentiation in colorectal cancer is regulated by the calcium activated chloride channel A1. PLoS ONE 2013, 8, e60861. [Google Scholar] [CrossRef]
- Monittola, F.; Bianchi, M.; Nasoni, M.G.; Luchetti, F.; Magnani, M.; Crinelli, R. Gastric cancer cell types display distinct proteasome/immunoproteasome patterns associated with migration and resistance to proteasome inhibitors. J. Cancer Res. Clin. Oncol. 2023, 149, 10085–10097. [Google Scholar] [CrossRef]
- Luo, Q.; Zhou, P.; Chang, S.; Huang, Z.; Zeng, X. Characterization of butyrate-metabolism in colorectal cancer to guide clinical treatment. Sci. Rep. 2023, 13, 5106. [Google Scholar] [CrossRef]
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; DePinho, R.A. Genetic and biological hallmarks of colorectal cancer. Genes. Dev. 2021, 35, 787–820. [Google Scholar] [CrossRef]
- Louis, K.S.; Siegel, A.C. Cell viability analysis using trypan blue: Manual and automated methods. Methods Mol. Biol. 2011, 740, 7–12. [Google Scholar]
- Chen, T.R. In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33,258 stain. Exp. Cell Res. 1977, 104, 255–262. [Google Scholar] [CrossRef]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Vedenov, D.; Pesti, G.M. A comparison of methods of fitting several models to nutritional response data. J. Anim. Sci. 2008, 86, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wray, J.; Meng, X.; Shen, Z. BCCIP is required for the nuclear localization of the p21 protein. Cell Cycle 2009, 8, 3019–3024. [Google Scholar] [CrossRef] [PubMed][Green Version]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, B.F.; Meng, S.; Wu, J.T.; Archer, S.Y.; Hodin, R.A. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J. Nutr. 2002, 132, 1012–1017. [Google Scholar] [CrossRef]
- Scharlau, D.; Borowicki, A.; Habermann, N.; Hofmann, T.; Klenow, S.; Miene, C.; Munjal, U.; Stein, K.; Glei, M. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat. Res. 2009, 682, 39–53. [Google Scholar] [CrossRef]
- Schlörmann, W.; Horlebein, C.; Hübner, S.M.; Wittwer, E.; Glei, M. Potential Role of ROS in Butyrate- and Dietary Fiber-Mediated Growth Inhibition and Modulation of Cell Cycle-, Apoptosis- and Antioxidant-Relevant Proteins in LT97 Colon Adenoma and HT29 Colon Carcinoma Cells. Cancers 2023, 15, 440. [Google Scholar] [CrossRef]
- Bultman, S.J. Molecular pathways: Gene-environment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clin. Cancer Res. 2014, 20, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak-Siedlecka, K.; Marano, L.; Merola, E.; Roviello, F.; Połom, K. Sodium butyrate in both prevention and supportive treatment of colorectal cancer. Front. Cell. Infect. Microbiol. 2022, 12, 1023806. [Google Scholar] [CrossRef]
- Zeng, H.; Briske-Anderson, M. Prolonged butyrate treatment inhibits the migration and invasion potential of HT1080 tumor cells. J. Nutr. 2005, 135, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2022, 85, 123–154. [Google Scholar] [CrossRef]
- Zuo, Z.; Liu, J.; Sun, Z.; Cheng, Y.W.; Ewing, M.; Bugge, T.H.; Finkel, T.; Leppla, S.H.; Liu, S. ERK and c-Myc signaling in host-derived tumor endothelial cells is essential for solid tumor growth. Proc. Natl. Acad. Sci. USA 2023, 120, e2211927120. [Google Scholar] [CrossRef]
- Tan, L.; Peng, D.; Cheng, Y. Significant position of C-myc in colorectal cancer: A promising therapeutic target. Clin. Transl. Oncol. 2022, 24, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Augenlicht, L.H.; Wadler, S.; Corner, G.; Richards, C.; Ryan, L.; Multani, A.S.; Pathak, S.; Benson, A.; Haller, D.; Heerdt, B.G. Low-level c-myc amplification in human colonic carcinoma cell lines and tumors: A frequent, p53-independent mutation associated with improved outcome in a randomized multi-institutional trial. Cancer Res. 1997, 57, 1769–1775. [Google Scholar]
- Wang, Z.; Liu, M.; Zhu, H.; Zhang, W.; He, S.; Hu, C.; Quan, L.; Bai, J.; Xu, N. Suppression of p21 by c-Myc through members of miR-17 family at the post-transcriptional level. Int. J. Oncol. 2010, 37, 1315–1321. [Google Scholar] [PubMed]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A.L. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [PubMed]
- Kreis, N.N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Acquaviva, R.; Tomasello, B.; Di Giacomo, C.; Santangelo, R.; La Mantia, A.; Naletova, I.; Sarpietro, M.G.; Castelli, F.; Malfa, G.A. Protocatechuic Acid, a Simple Plant Secondary Metabolite, Induced Apoptosis by Promoting Oxidative Stress through HO-1 Downregulation and p21 Upregulation in Colon Cancer Cells. Biomolecules 2021, 11, 1485. [Google Scholar] [CrossRef]
- Parveen, A.; Akash, M.S.; Rehman, K.; Kyunn, W.W. Dual Role of p21 in the Progression of Cancer and Its Treatment. Crit. Rev. Eukaryot. Gene Expr. 2016, 26, 49–62. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oncel, S.; Safratowich, B.D.; Lindlauf, J.E.; Liu, Z.; Palmer, D.G.; Briske-Anderson, M.; Zeng, H. Efficacy of Butyrate to Inhibit Colonic Cancer Cell Growth Is Cell Type-Specific and Apoptosis-Dependent. Nutrients 2024, 16, 529. https://doi.org/10.3390/nu16040529
Oncel S, Safratowich BD, Lindlauf JE, Liu Z, Palmer DG, Briske-Anderson M, Zeng H. Efficacy of Butyrate to Inhibit Colonic Cancer Cell Growth Is Cell Type-Specific and Apoptosis-Dependent. Nutrients. 2024; 16(4):529. https://doi.org/10.3390/nu16040529
Chicago/Turabian StyleOncel, Sema, Bryan D. Safratowich, James E. Lindlauf, Zhenhua Liu, Daniel G. Palmer, Mary Briske-Anderson, and Huawei Zeng. 2024. "Efficacy of Butyrate to Inhibit Colonic Cancer Cell Growth Is Cell Type-Specific and Apoptosis-Dependent" Nutrients 16, no. 4: 529. https://doi.org/10.3390/nu16040529
APA StyleOncel, S., Safratowich, B. D., Lindlauf, J. E., Liu, Z., Palmer, D. G., Briske-Anderson, M., & Zeng, H. (2024). Efficacy of Butyrate to Inhibit Colonic Cancer Cell Growth Is Cell Type-Specific and Apoptosis-Dependent. Nutrients, 16(4), 529. https://doi.org/10.3390/nu16040529