
Dynamics of the Gut Microbiota and Faecal and Serum Metabolomes during Pregnancy—A Longitudinal Study
Abstract
1. Introduction
2. Materials and Methods
2.1. Pregnancy Cohort
2.2. Sample Collection
2.3. Clinical Measurements and Blood Index Evaluation
2.4. Faecal DNA Extraction, 16S rRNA Sequencing, and GroEL Gene Sequencing
2.5. Sequence Data Processing and Bioinformatic Analysis
2.6. Untargeted Metabolomics for Faecal and Serum Samples
2.7. Targeted Metabolomics of Short-Chain Fatty Acids (SCFAs) by Gas Chromatography–Mass Spectrometry (GC-MS)
2.8. Statistical Analysis
3. Results
3.1. Participants’ Clinical Characteristics
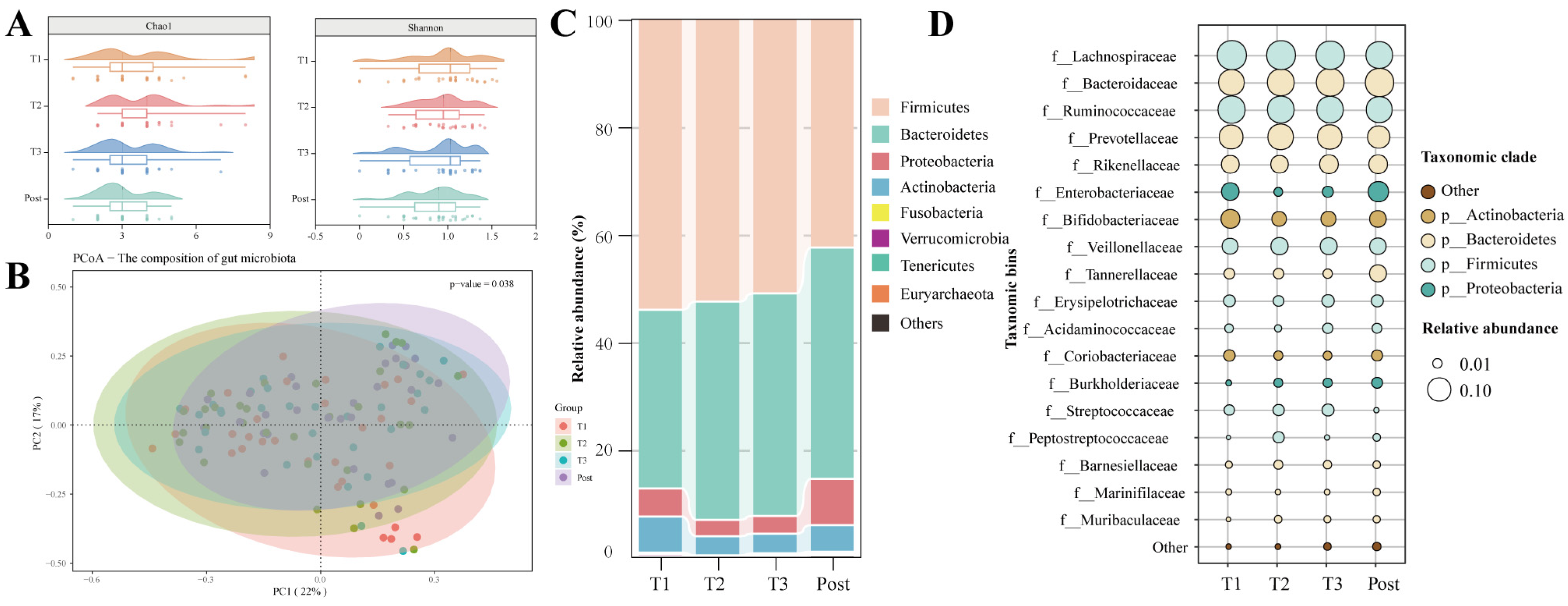
3.2. Dramatic Changes in the Gut Microbiota with Advancing Gestation
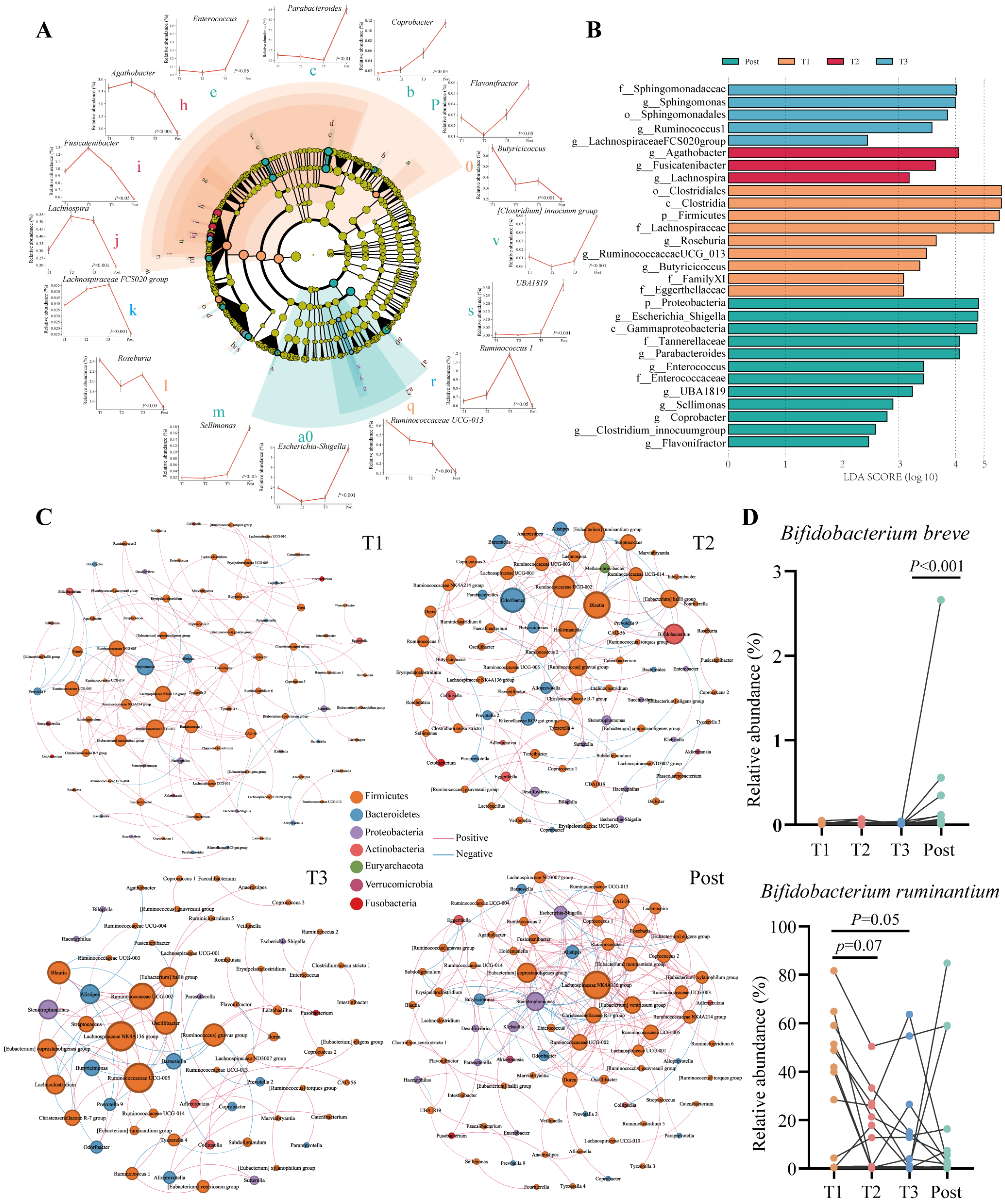
3.3. Symbiotic Network Analysis of the Gut Microbiota in Pregnant Women
3.4. Minimal Changes in Bifidobacterium Communities at the Species Level with Advancing Gestation
3.5. Changes in Functional Pathways of Gut Microbiota during Pregnancy
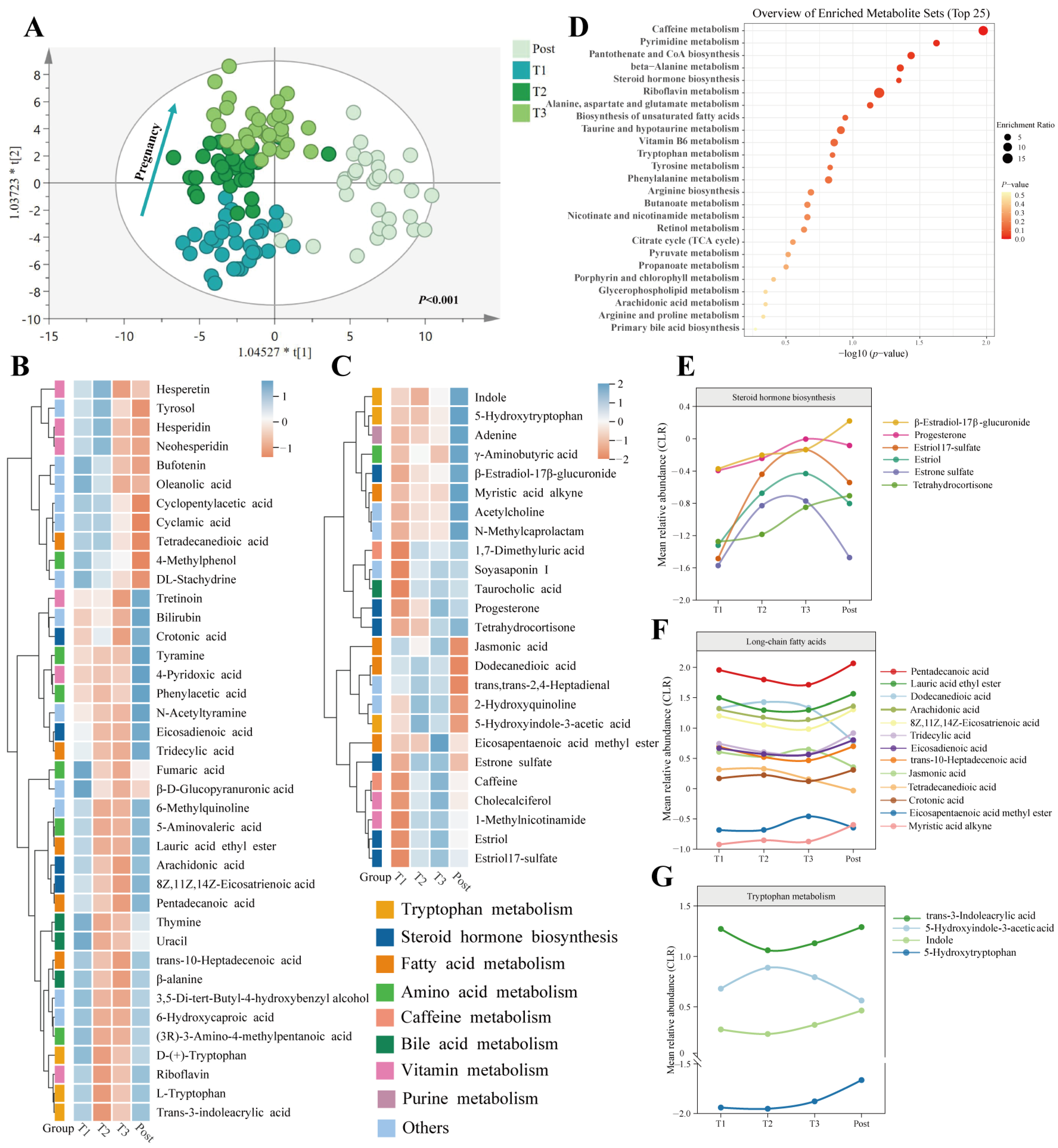
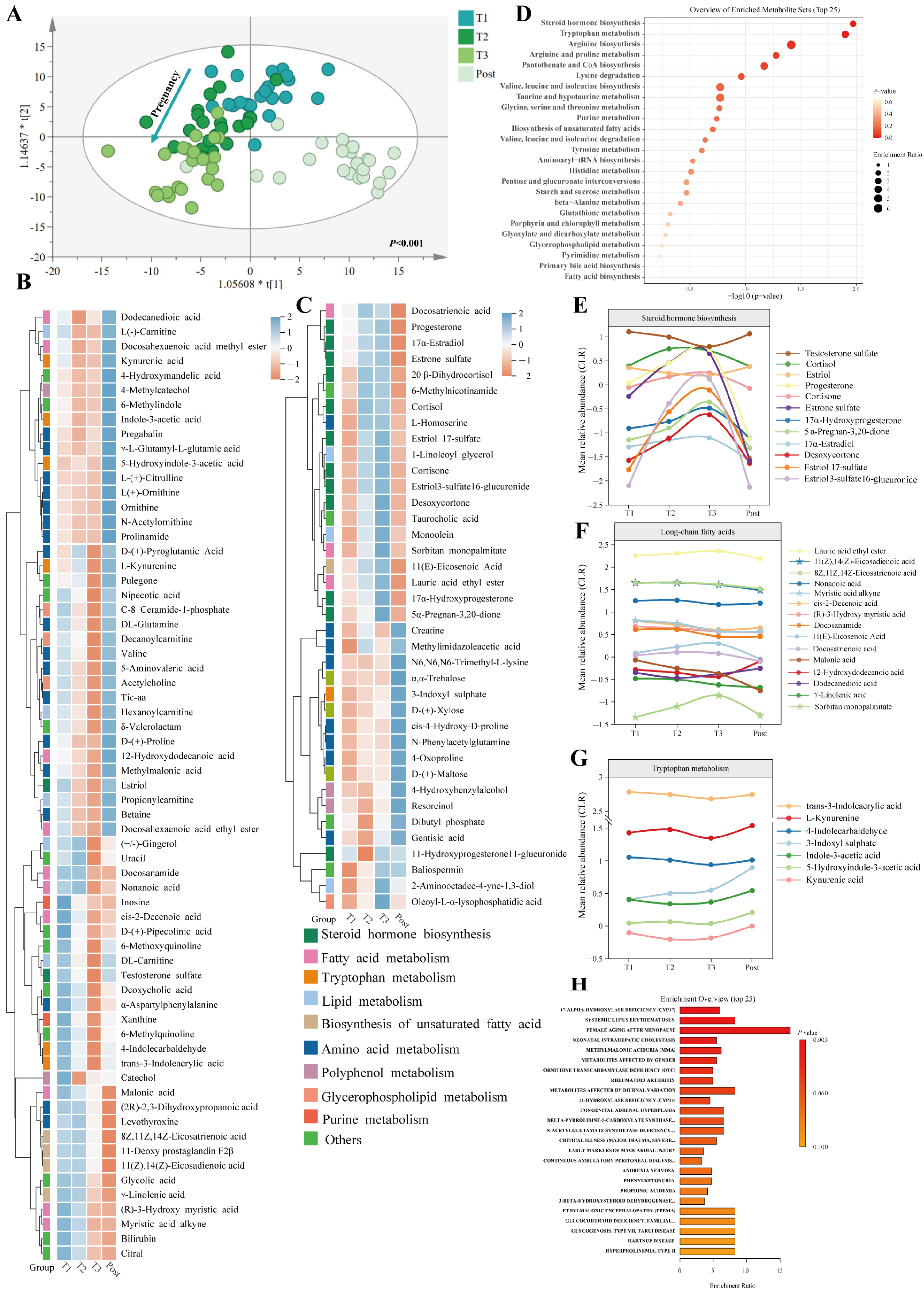
3.6. Temporal Changes in Faecal and Serum Metabolomic Profiles during Pregnancy and the Postpartum Period
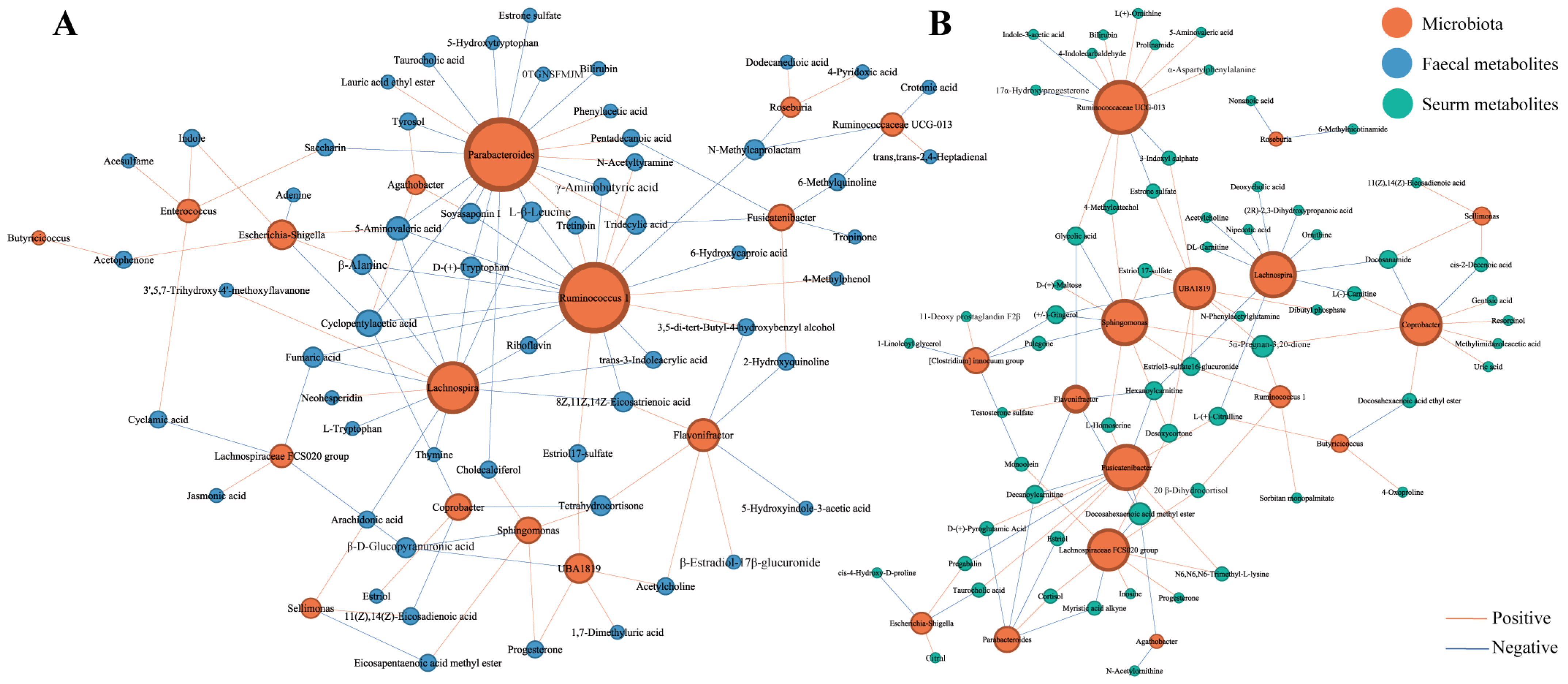
3.7. Correlations between Pregnancy-Related Microbiota and Faecal and Serum Metabolites Presenting Temporal Changes during Pregnancy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, H.; Guo, R.; Li, S.; Liang, F.; Tian, C.; Zhao, X.; Long, Y.; Liu, F.; Jiang, M.; Zhang, Y.; et al. Systematic analysis of gut microbiota in pregnant women and its correlations with individual heterogeneity. npj Biofilms Microbiomes 2020, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Arany, Z. Maternal cardiac metabolism in pregnancy. Cardiovasc. Res. 2014, 101, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Rossary, A.; Farges, M.-C.; Lamas, B.; Miles, E.A.; Noakes, P.S.; Kremmyda, L.-S.; Vlachava, M.; Diaper, N.D.; Robinson, S.M.; Godfrey, K.M.; et al. Increased consumption of salmon during pregnancy partly prevents the decline of some plasma essential amino acid concentrations in pregnant women. Clin. Nutr. 2014, 33, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Nuriel-Ohayon, M.; Neuman, H.; Koren, O. Microbial changes during pregnancy, birth, and infancy. Front. Microbiol. 2016, 7, 1031. [Google Scholar] [CrossRef] [PubMed]
- Glenwright, A.J.; Pothula, K.R.; Bhamidimarri, S.P.; Chorev, D.S.; Basle, A.; Firbank, S.J.; Zheng, H.; Robinson, C.V.; Winterhalter, M.; Kleinekathoefer, U.; et al. Structural basis for nutrient acquisition by dominant members of the human gut microbiota. Nature 2017, 541, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, A.; Yanez, A.; Price, J.G.; Chow, A.; Merad, M.; Goodridge, H.S.; Mazmanian, S.K. Gut Microbiota Promote Hematopoiesis to Control Bacterial Infection. Cell Host Microbe 2014, 15, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Liu, R. The Role of Diet and Gut Microbiota Interactions in Metabolic Homeostasis. Adv. Biol. 2023, 7, 2300100. [Google Scholar] [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Backhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host Remodeling of the Gut Microbiome and Metabolic Changes during Pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef]
- Sun, Z.; Pan, X.-F.; Li, X.; Jiang, L.; Hu, P.; Wang, Y.; Ye, Y.; Wu, P.; Zhao, B.; Xu, J.; et al. The Gut Microbiome Dynamically Associates with Host Glucose Metabolism throughout Pregnancy: Longitudinal Findings from a Matched Case-Control Study of Gestational Diabetes Mellitus. Adv. Sci. 2023, 10, 2205289. [Google Scholar] [CrossRef]
- DiGiulio, D.B.; Callahan, B.J.; McMurdie, P.J.; Costello, E.K.; Lyell, D.J.; Robaczewska, A.; Sun, C.L.; Goltsman, D.S.A.; Wong, R.J.; Shaw, G.; et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. USA 2015, 112, 11060–11065. [Google Scholar] [CrossRef]
- Pinto, Y.; Frishman, S.; Turjeman, S.; Eshel, A.; Nuriel-Ohayon, M.; Shrossel, O.; Ziv, O.; Walters, W.; Parsonnet, J.; Ley, C.; et al. Gestational diabetes is driven by microbiota-induced inflammation months before diagnosis. Gut 2023, 72, 918–928. [Google Scholar] [CrossRef]
- Needham, B.D.; Kaddurah-Daouk, R.; Mazmanian, S.K. Gut microbial molecules in behavioural and neurodegenerative conditions. Nat. Rev. Neurosci. 2020, 21, 717–731. [Google Scholar] [CrossRef]
- Wang, Q.; Würtz, P.; Auro, K.; Mäkinen, V.-P.; Kangas, A.J.; Soininen, P.; Tiainen, M.; Tynkkynen, T.; Jokelainen, J.; Santalahti, K.; et al. Metabolic profiling of pregnancy: Cross-sectional and longitudinal evidence. BMC Med. 2016, 14, 205. [Google Scholar] [CrossRef]
- Liang, L.; Rasmussen, M.-L.H.; Piening, B.; Shen, X.; Chen, S.; Rost, H.; Snyder, J.K.; Tibshirani, R.; Skotte, L.; Lee, N.C.Y.; et al. Metabolic Dynamics and Prediction of Gestational Age and Time to Delivery in Pregnant Women. Obstet. Gynecol. Surv. 2020, 75, 649–651. [Google Scholar] [CrossRef]
- Cui, M.; Trimigno, A.; Aru, V.; Khakimov, B.; Engelsen, S.B. Human Faecal 1H NMR Metabolomics: Evaluation of Solvent and Sample Processing on Coverage and Reproducibility of Signature Metabolites. Anal. Chem. 2020, 92, 9546–9555. [Google Scholar] [CrossRef]
- Fang, Z.; Pan, T.; Li, L.; Wang, H.; Zhu, J.; Zhang, H.; Zhao, J.; Chen, W.; Lu, W. Bifidobacterium longum mediated tryptophan metabolism to improve atopic dermatitis via the gut-skin axis. Gut Microbes 2022, 14, 2044723. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lu, W.; Wang, L.; Pan, M.; Zhang, H.; Zhao, J.; Chen, W. Assessment of Bifidobacterium Species Using groEL Gene on the Basis of Illumina MiSeq High-Throughput Sequencing. Genes 2017, 8, 336. [Google Scholar] [CrossRef]
- Yang, B.; Ding, M.; Chen, Y.; Han, F.; Yang, C.; Zhao, J.; Malard, P.; Stanton, C.; Ross, R.P.; Zhang, H.; et al. Development of gut microbiota and bifidobacterial communities of neonates in the first 6 weeks and their inheritance from mother. Gut Microbes 2021, 13, 1908100. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Guo, M.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. Integrative Metabolomic Characterization Reveals the Mediating Effect of Bifidobacterium breve on Amino Acid Metabolism in a Mouse Model of Alzheimer’s Disease. Nutrients 2022, 14, 735. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, W.; Chen, Y.; Zhang, X.; Wang, X.; Wang, F.; Qian, Y.; Qiu, J. Identification of markers for tea authenticity assessment: Non-targeted metabolomics of highly similar oolong tea cultivars (Camellia sinensis var. sinensis). Food Control 2022, 142, 109223. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Li, D.; Feng, Y.; Tian, M.; Ji, J.; Hu, X.; Chen, F. Gut microbiota-derived inosine from dietary barley leaf supplementation attenuates colitis through PPARgamma signaling activation. Microbiome 2021, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Li, D.; Ai, C.; Zhao, J.; Zhang, H.; Chen, W. Lactulose differently modulates the composition of luminal and mucosal microbiota in C57BL/6J mice. J. Agric. Food Chem. 2016, 64, 6240–6247. [Google Scholar] [CrossRef] [PubMed]
- Vojinovic, D.; Radjabzadeh, D.; Kurilshikov, A.; Amin, N.; Wijmenga, C.; Franke, L.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Relationship between gut microbiota and circulating metabolites in population-based cohorts. Nat. Commun. 2019, 10, 5813. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Kubota, T.; Nakanishi, Y.; Tsugawa, H.; Suda, W.; Kwon, A.T.-J.; Yazaki, J.; Ikeda, K.; Nemoto, S.; Mochizuki, Y.; et al. Gut microbial carbohydrate metabolism contributes to insulin resistance. Nature 2023, 621, 389–395. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhuo, L.-B.; He, Y.; Fu, Y.; Shen, L.; Xu, F.; Gou, W.; Miao, Z.; Shuai, M.; Liang, Y.; et al. The gut microbiota-bile acid axis links the positive association between chronic insomnia and cardiometabolic diseases. Nat. Commun. 2022, 13, 3002. [Google Scholar] [CrossRef]
- Wang, T.; Ye, Y.; Ji, J.; Zhang, S.; Yang, X.; Xu, J.; Wang, J.-S.; Chen, Z.; Xia, B.; Shen, H.; et al. Astilbin from Smilax glabra Roxb. alleviates high-fat diet-induced metabolic dysfunction. Food Funct. 2022, 13, 5023–5036. [Google Scholar] [CrossRef]
- Du, H.; Wang, Q.; Li, T.; Ren, D.; Yang, X. Grape seed proanthocyanidins reduced the overweight of C57BL/6J mice through modulating adipose thermogenesis and gut microbiota. Food Funct. 2021, 12, 8467–8477. [Google Scholar] [CrossRef]
- Yang, Q.; Liang, Q.; Balakrishnan, B.; Belobrajdic, D.P.; Feng, Q.J.; Zhang, W. Role of dietary nutrients in the modulation of gut microbiota: A narrative review. Nutrients 2020, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Huang, X.; Wu, G.; Zhang, Y.; Xu, H.; Chen, Y.; Yang, H.; Nie, S. Polysaccharides from small black soybean alleviating type 2 diabetes via modulation of gut microbiota and serum metabolism. Food Hydrocoll. 2023, 141, 108670. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Liu, M.; Hu, X.; Zhu, H. Guizhi Fuling Wan, Chinese Herbal Medicine, Ameliorates Insulin Sensitivity in PCOS Model Rats with Insulin Resistance via Remodeling Intestinal Homeostasis. Front. Endocrinol. 2020, 11, 575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, D.; Wu, D.; Gao, X.; Shao, F.; Zhao, M.; Wang, J.; Ma, J.; Wang, W.; Qin, X.; et al. Tissue-resident Lachnospiraceae family bacteria protect against colorectal carcinogenesis by promoting tumor immune surveillance. Cell Host Microbe 2023, 31, 418–432. [Google Scholar] [CrossRef]
- Lima, G.F.; Lopes, R.d.O.; Araujo Mendes, A.B.; Brazao, S.C.; Autran, L.J.; Vieira Motta, N.A.; Brito, F.C.F. Inosine, an endogenous purine nucleoside, avoids early stages of atherosclerosis development associated to eNOS activation and p38 MAPK/NF-kB inhibition in rats. Eur. J. Pharmacol. 2020, 882, 173289. [Google Scholar] [CrossRef]
- Dong, J.; Ping, L.; Meng, Y.; Zhang, K.; Tang, H.; Liu, D.; Li, B.; Huo, G. Bifidobacterium longum BL-10 with Antioxidant Capacity Ameliorates Lipopolysaccharide-Induced Acute Liver Injury in Mice by the Nuclear Factor-κB Pathway. J. Agric. Food Chem. 2022, 70, 8680–8692. [Google Scholar] [CrossRef]
- Xie, Q.; Li, H.; Ma, R.; Ren, M.; Li, Y.; Li, J.; Chen, H.; Chen, Z.; Gong, D.; Wang, J. Effect of Coptis chinensis franch and Magnolia officinalis on intestinal flora and intestinal barrier in a TNBS-induced ulcerative colitis rats model. Phytomedicine 2022, 97, 153927. [Google Scholar] [CrossRef] [PubMed]
- Steck, N.; Hoffmann, M.; Sava, I.; Kim, S.; Hahne, H.; Schemann, M.; Kuster, B.; Sartor, R.B.; Haller, D. Enterococcus faecalis Metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 2011, 141, 959–971. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar]
- Jaffe, R.B. Role of the human fetal adrenal gland in the initiation of parturition. Endocrinol. Parturition 2001, 27, 75–85. [Google Scholar]
- Graham, M.E.; Herbert, W.G.; Song, S.D.; Raman, H.N.; Zhu, J.E.; Gonzalez, P.E.; Walther-Antonio, M.R.S.; Tetel, M.J. Gut and vaginal microbiomes on steroids: Implications for women’s health. Trends Endocrinol. Metab. 2021, 32, 554–565. [Google Scholar] [CrossRef]
- Pernigoni, N.; Zagato, E.; Calcinotto, A.; Troiani, M.; Mestre, R.P.; Cali, B.; Attanasio, G.; Troisi, J.; Minini, M.; Mosole, S.; et al. Commensal bacteria promote endocrine resistance in prostate cancer through androgen biosynthesis. Science 2021, 374, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Nuriel-Ohayon, M.; Neuman, H.; Ziv, O.; Belogolovski, A.; Barsheshet, Y.; Bloch, N.; Uzan, A.; Lahav, R.; Peretz, A.; Frishman, S.; et al. Progesterone Increases Bifidobacterium Relative Abundance during Late Pregnancy. Cell Rep. 2019, 27, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Amaral, W.Z.; Lubach, G.R.; Rendina, D.N.; Phillips, G.J.; Lyte, M.; Coe, C.L.J.M. Significant Microbial Changes Are Evident in the Reproductive Tract of Pregnant Rhesus Monkeys at Mid-Gestation but Their Gut Microbiome Does Not Shift until Late Gestation. Microorganisms 2023, 11, 1481. [Google Scholar] [CrossRef]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Wang, L.; Xu, X.; Chen, Y.; Wang, H.; Wang, G.; Zhao, J.; Chen, W. Crosstalk between the Gut Microbiome and Colonic Motility in Chronic Constipation: Potential Mechanisms and Microbiota Modulation. Nutrients 2022, 14, 3704. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Benarroch, E.E. Acetylcholine in the cerebral cortex Effects and clinical implications. Neurology 2010, 75, 659–665. [Google Scholar] [CrossRef]
- Sorboni, S.G.; Moghaddam, H.S.; Jafarzadeh-Esfehani, R.; Soleimanpour, S. A Comprehensive Review on the Role of the Gut Microbiome in Human Neurological Disorders. Clin. Microbiol. Rev. 2022, 35, e0033820. [Google Scholar] [CrossRef]
- Lana, D.; Giovannini, M.G. The Microbiota-Gut-Brain Axis in Behaviour and Brain Disorders. Int. J. Mol. Sci. 2023, 24, 8460. [Google Scholar] [CrossRef]
- Miyamoto, J.; Igarashi, M.; Watanabe, K.; Karaki, S.-I.; Mukouyama, H.; Kishino, S.; Li, X.; Ichimura, A.; Irie, J.; Sugimoto, Y.; et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat. Commun. 2019, 10, 4007. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-C.; Kuo, K.-L.; Wu, C.-C.; Tarng, D.-C. Indoxyl Sulfate: A Novel Cardiovascular Risk Factor in Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e005022. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.I.; Cnattingius, S.; Dickman, P.W.; Mittleman, M.A.; Ludvigsson, J.F.; Ingelsson, E. Parity and risk of later-life maternal cardiovascular disease. Am. Heart J. 2010, 159, 215–221.e6. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Hu, Y.; Zheng, X.; Ning, Z.; Wu, M.; Xia, F.; Prast-Nielsen, S.; Hu, Y.O.O.; Xu, B. Longitudinal profiling of gut microbiome among tuberculosis patients under anti-tuberculosis treatment in China: Protocol of a prospective cohort study. BMC Pulm. Med. 2019, 19, 211. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Baeza, Y.; Gonzalez, A.; Xu, Z.Z.; Washburne, A.; Herfarth, H.H.; Sartor, R.B.; Knight, R. Guiding longitudinal sampling in IBD cohorts. Gut 2018, 67, 1743–1745. [Google Scholar] [CrossRef]
- Mars, R.A.T.; Yang, Y.; Ward, T.; Houtti, M.; Priya, S.; Lekatz, H.R.; Tang, X.; Sun, Z.; Kalari, K.R.; Korem, T.; et al. Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome. Cell 2020, 182, 1460–1473, Correction in Cell 2020, 183, 1137–1140. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (n = 31) | T1 | T2 | T3 | p Value |
|---|---|---|---|---|
| Gestation weeks | 10.9 ± 0.33 | 23.0 ± 0.47 | 35.5 ± 0.35 | |
| Gestational weight gain (Kg) | 1.39 ± 0.23 | 6.48 ± 0.71 | 12.39 ± 0.87 | p < 0.0001 |
| Cholesterol (mmol/L) | 4.05 ± 0.14 | 5.97 ± 0.43 | 5.87 ± 0.34 | p < 0.0001 |
| Glucose (mmol/L) | 4.49 ± 0.09 | 4.31 ± 0.12 | 4.39 ± 0.11 | 0.27 |
| Triglyceride (mmol/L) | 1.36 ± 0.09 | 3.21 ± 0.33 | 3.42 ± 0.35 | p < 0.0001 |
| High-density lipoprotein Cholesterol (mmol/L) | 1.46 ± 0.07 | 1.69 ± 0.07 | 1.82 ± 0.17 | p < 0.05 |
| Low-density lipoprotein cholesterol (mmol/L) | 2.07 ± 0.09 | 3.02 ± 0.35 | 3.08 ± 0.20 | p < 0.0001 |
| Cystatin C (mg/L) | 0.67 ± 0.02 | 0.83 ± 0.05 | 0.92 ± 0.04 | p < 0.0001 |
| Total Protein (g/L) | 69.24 ± 1.88 | 65.25 ± 0.54 | 63.59 ± 0.66 | p < 0.0001 |
| Alkaline phosphatase (μ/L) | 45.70 ± 2.28 | 76.58 ± 4.51 | 120.80 ± 9.02 | p < 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, R.; Guo, M.; Chen, Y.; Lin, G.; Tian, P.; Wang, L.; Zhao, J.; Chen, W.; Wang, G. Dynamics of the Gut Microbiota and Faecal and Serum Metabolomes during Pregnancy—A Longitudinal Study. Nutrients 2024, 16, 483. https://doi.org/10.3390/nu16040483
Pan R, Guo M, Chen Y, Lin G, Tian P, Wang L, Zhao J, Chen W, Wang G. Dynamics of the Gut Microbiota and Faecal and Serum Metabolomes during Pregnancy—A Longitudinal Study. Nutrients. 2024; 16(4):483. https://doi.org/10.3390/nu16040483
Chicago/Turabian StylePan, Ruili, Min Guo, Ying Chen, Guopeng Lin, Peijun Tian, Linlin Wang, Jianxin Zhao, Wei Chen, and Gang Wang. 2024. "Dynamics of the Gut Microbiota and Faecal and Serum Metabolomes during Pregnancy—A Longitudinal Study" Nutrients 16, no. 4: 483. https://doi.org/10.3390/nu16040483
APA StylePan, R., Guo, M., Chen, Y., Lin, G., Tian, P., Wang, L., Zhao, J., Chen, W., & Wang, G. (2024). Dynamics of the Gut Microbiota and Faecal and Serum Metabolomes during Pregnancy—A Longitudinal Study. Nutrients, 16(4), 483. https://doi.org/10.3390/nu16040483