
Simplification of Dietary Treatment in Pharmacoresistant Epilepsy: Impact of C8 and C10 Fatty Acids on Sirtuins of Neuronal Cells In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Analytical Procedures
2.2.1. Sirtuin Enzyme Activity
2.2.2. Bicinchoninic Assay (BCA)
2.2.3. Protein Expression
2.2.4. Gene Expression
2.2.5. Statistical Analysis
3. Results
3.1. Sirtuin 1 (SIRT1)
3.1.1. Impact of Different Metabolites on SIRT1 Enzyme Activity
3.1.2. Impact of Different Metabolites on Gene Expression of SIRT1
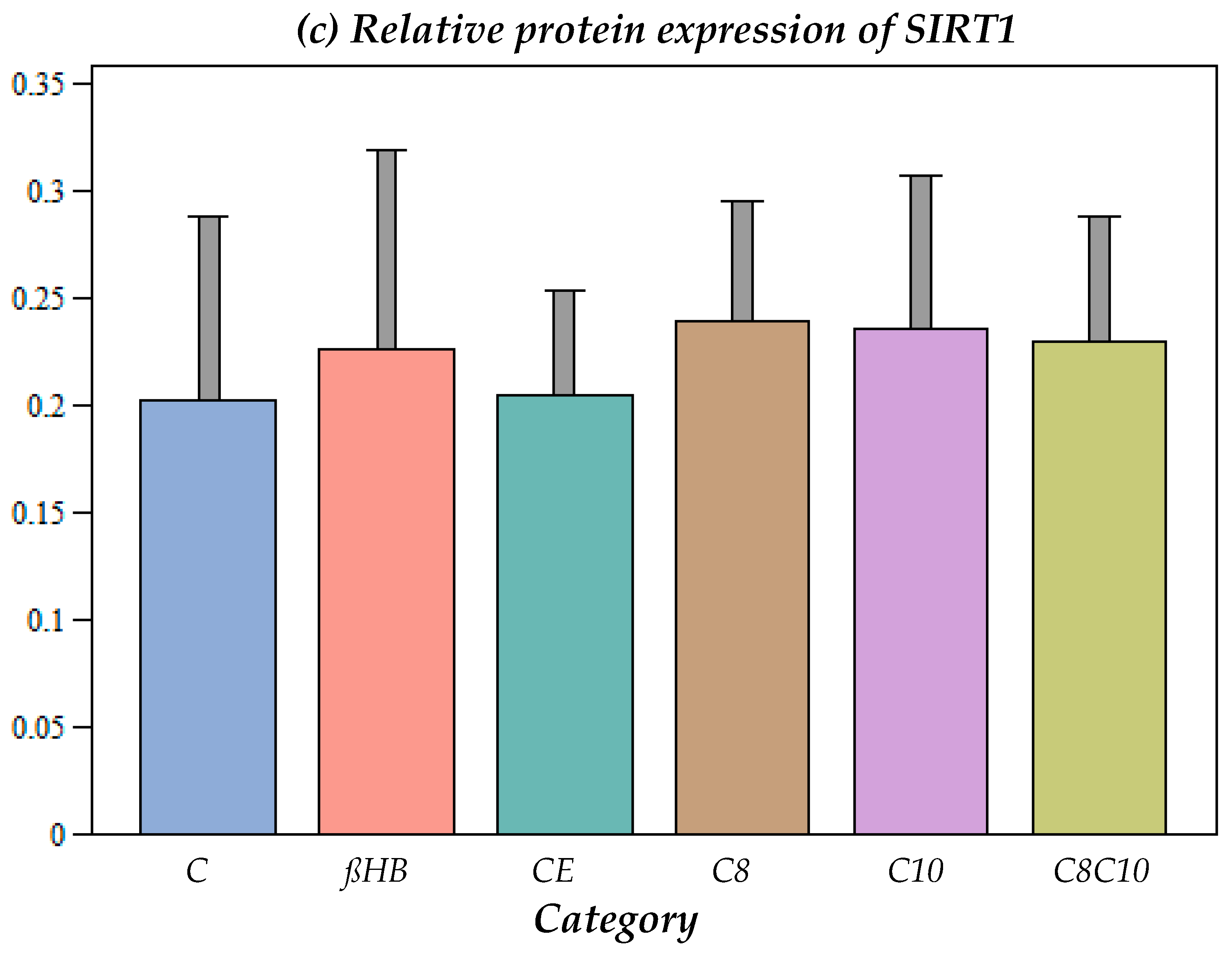
3.1.3. Impact of Different Metabolites on Protein Content of SIRT1
3.1.4. Correlation Analysis between Gene Expression and Enzyme Activity for SIRT1
3.2. Sirtuin 3 (SIRT3)
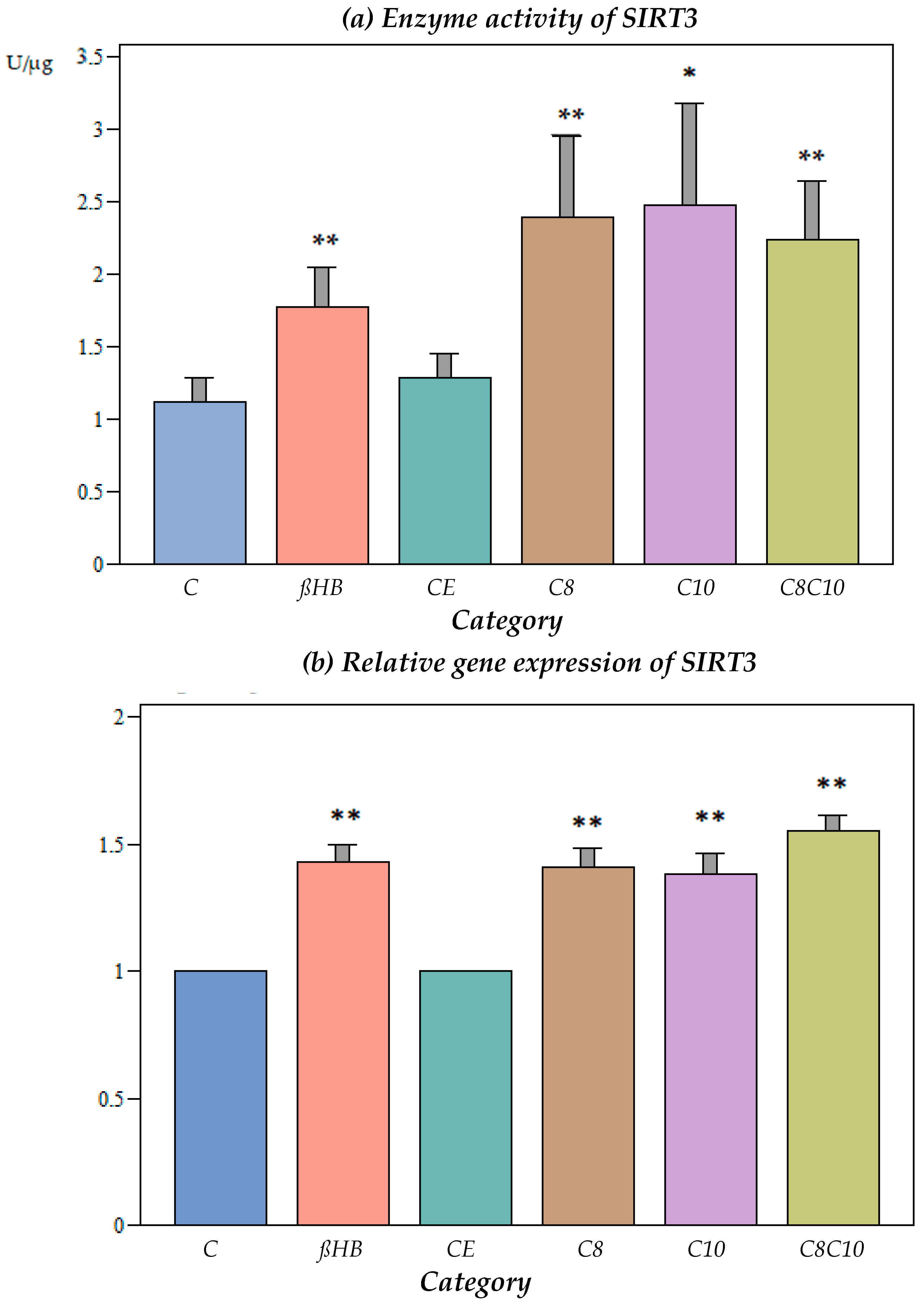
3.2.1. Impact of Different Metabolites on SIRT3 Enzyme Activity
3.2.2. Impact of Different Metabolites on Gene Expression of SIRT3
3.2.3. Impact of Different Metabolites on Protein Content of SIRT3
3.2.4. Correlation Analysis between Gene Expression and Enzyme Activity for SIRT3
3.3. Sirtuin 4 (SIRT4)
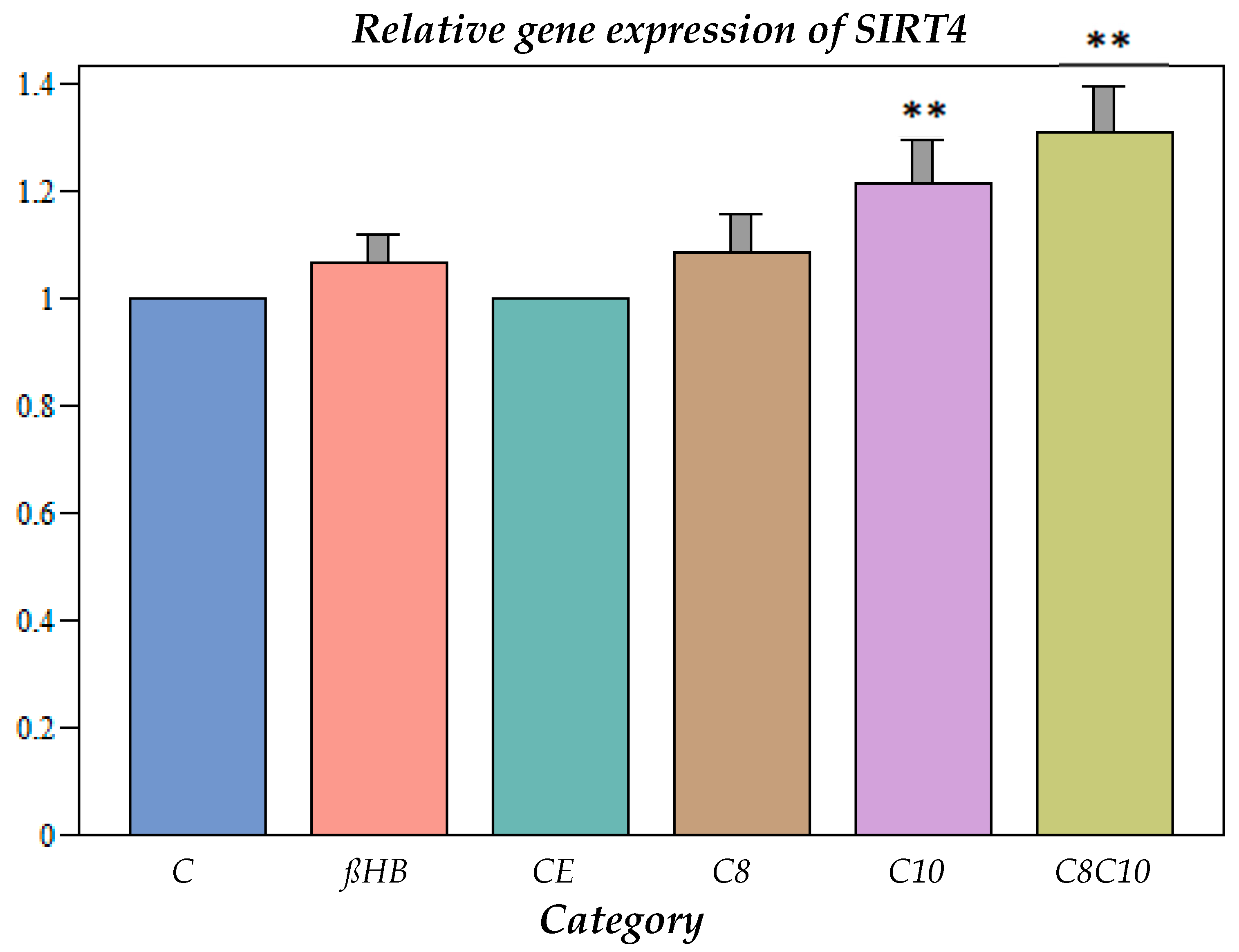
Impact of Different Metabolites on Gene Expression of SIRT4
3.4. Sirtuin 5 (SIRT5)
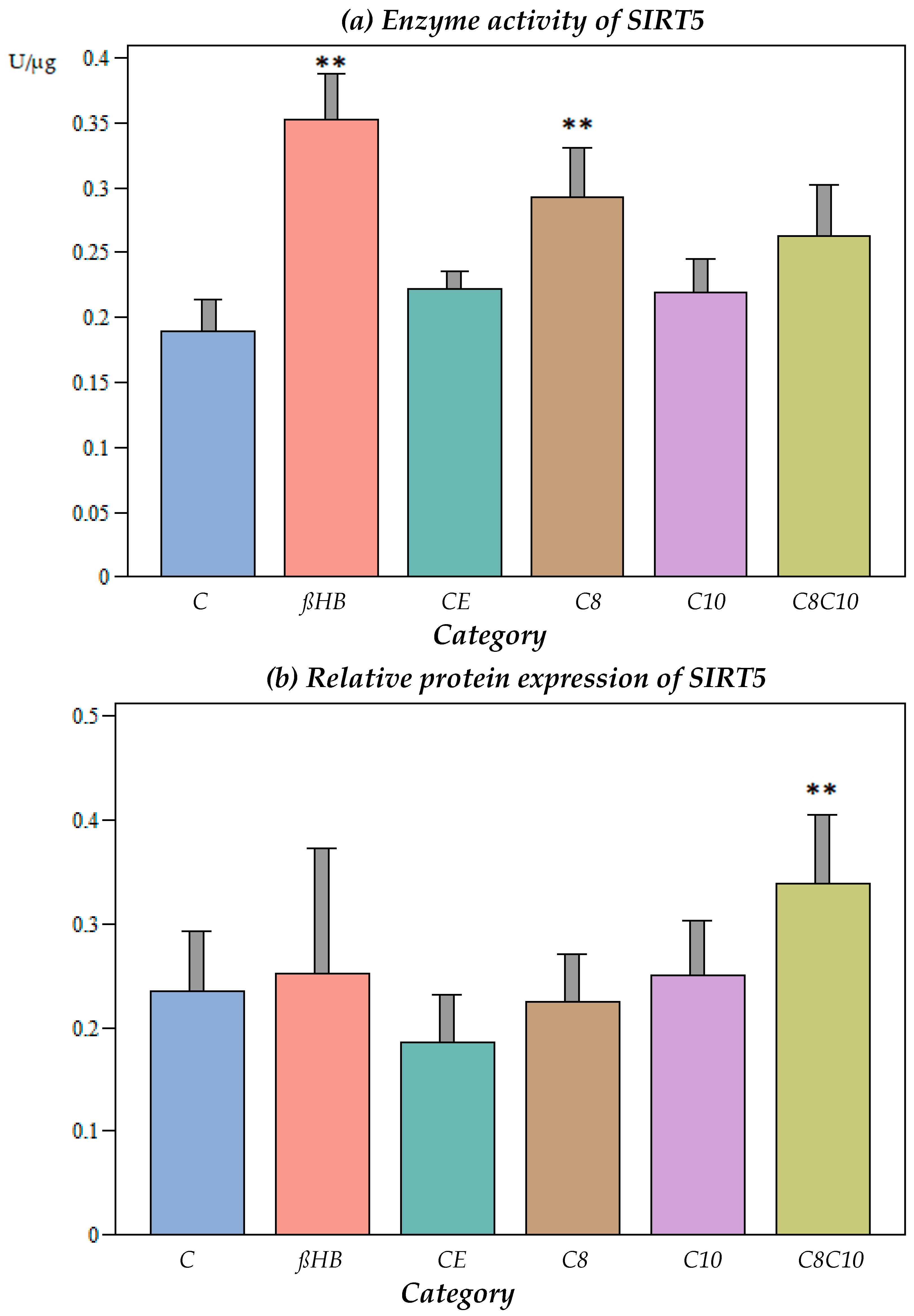
3.4.1. Impact of Different Metabolites on SIRT5 Enzyme Activity
3.4.2. Impact of Different Metabolites on Protein Content of SIRT5
4. Discussion
4.1. Sirtuin 1 (SIRT1)
4.2. Sirtuin 3 (SIRT3)
4.3. Sirtuin 4 (SIRT4)
4.4. Sirtuin 5 (SIRT5)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rho, J.M.; Boison, D. The metabolic basis of epilepsy. Nat. Rev. Neurol. 2022, 18, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Aaberg, K.M.; Gunnes, N.; Bakken, I.J.; Lund Søraas, C.; Berntsen, A.; Magnus, P.; Lossius, M.I.; Stoltenberg, C.; Chin, R.; Surén, P. Incidence and Prevalence of Childhood Epilepsy: A Nationwide Cohort Study. Pediatrics 2017, 139, e20163908. [Google Scholar] [CrossRef]
- Mishra, P.; Mittal, A.K.; Rajput, S.K.; Sinha, J.K. Cognition and memory impairment attenuation via reduction of oxidative stress in acute and chronic mice models of epilepsy using antiepileptogenic Nux vomica. J. Ethnopharmacol. 2021, 267, 113509. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 1971, 21, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Martin-McGill, K.J.; Jackson, C.F.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2018, 11, CD001903. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Augustin, K.; Boddum, K.; Williams, S.; Sun, M.; Terschak, J.A.; Hardege, J.D.; Chen, P.E.; Walker, M.C.; Williams, R.S. Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain 2016, 139, 431–443. [Google Scholar] [CrossRef]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Wang, H.S. Medium-chain triglyceride ketogenic diet, an effective treatment for drug-resistant epilepsy and a comparison with other ketogenic diets. Biomed. J. 2013, 36, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A.; Löscher, W.; Rho, J.M. Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet. Cold Spring Harb. Perspect. Med. 2016, 6, a022780. [Google Scholar] [CrossRef]
- Dabke, P.; Brogden, G.; Naim, H.Y.; Das, A.M. Ketogenic Diet: Impact on Cellular Lipids in Hippocampal Murine Neurons. Nutrients 2020, 12, 3870. [Google Scholar] [CrossRef]
- Dabke, P. Mechanism of Ketogenic Diet: Impact of Beta–Hydroxybutyrate and Decanoic Acid on Sirtuins, Energy Metabolism and Cellular Lipids in a Murine Hippocampal Neuronal Cell Model. Ph.D. Thesis, University of Veterinary Medicine Hannover, Center for Systems Neurosciences, Hannover, Germany, 2020. [Google Scholar]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef]
- Dabke, P.; Das, A.M. Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta-Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons. Nutrients 2020, 12, 2379. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, N.E.; Orford, M.; Vivekananda, U.; Simpson, Z.; Van de Bor, B.; Smith, H.; Balestrini, S.; Rutherford, T.; Brennan, E.; McKenna, J.; et al. A feasibility study of a blend of medium chain triglycerides to manage drug-resistant epilepsy. Brain Commun. 2021, 3, fcab160. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Rho, J.M. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 2007, 48, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovici, C.; Rho, J.M. Metabolic epilepsies amenable to ketogenic therapies: Indications, contraindications, and underlying mechanisms. J. Inherit. Metab. Dis. 2021, 44, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 483–488. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Simeone, T.A.; Simeone, K.A.; Stafstrom, C.E.; Rho, J.M. Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology 2018, 133, 233–241. [Google Scholar] [CrossRef]
- Wlaź, P.; Socała, K.; Nieoczym, D.; Żarnowski, T.; Żarnowska, I.; Czuczwar, S.J.; Gasior, M. Acute anticonvulsant effects of capric acid in seizure tests in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 57, 110–116. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Q.; Zhou, J.; Wu, X.; Zhu, Q. β hydroxybutyrate levels in serum and cerebrospinal fluid under ketone body metabolism in rats. Exp. Anim. 2017, 66, 177–182. [Google Scholar] [CrossRef]
- Min, Z.; Gao, J.; Yu, Y. The Roles of Mitochondrial SIRT4 in Cellular Metabolism. Front Endocrinol 2019, 9, 783. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Och, U.; Klawon, I.; Och, T.; Grüneberg, M.; Fobker, M.; Bordewick-Dell, U.; Marquardt, T. Effect of a Sodium and Calcium DL-β-Hydroxybutyrate Salt in Healthy Adults. J. Nutr. Metab. 2018, 2018, 9812806. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Zhang, X. Nucleus or cytoplasm? The mysterious case of SIRT1’s subcellular localization. Cell Cycle 2016, 15, 3337–3338. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Kuno, A.; Yano, T.; Miura, T.; Hisahara, S.; Ishikawa, S.; Shimamoto, K.; Horio, Y. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 2010, 285, 8375–8382. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Chen, S.D.; Jou, S.B.; Lin, T.K.; Chen, S.F.; Chen, N.C.; Hsu, C.Y. Sirtuin 1 Regulates Mitochondrial Biogenesis and Provides an Endogenous Neuroprotective Mechanism Against Seizure-Induced Neuronal Cell Death in the Hippocampus Following Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 3588. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Denu, J.M. Sirtuin catalysis and regulation. J. Biol. Chem. 2012, 287, 42419–42427. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Deng, X.; Chen, Z.; Ba, X.; Qin, K.; Huang, Y.; Huang, Y.; Li, T.; Yan, J.; Tu, S. SIRT1: A Potential Therapeutic Target in Autoimmune Diseases. Front. Immunol. 2021, 12, 779177. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Sánchez-Gómez, F.J.; Wild, B.; García-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLoS ONE 2013, 8, e73875. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Annu. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012, 100, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Mishra, Y.; Kaundal, R.K. Role of SIRT3 in mitochondrial biology and its therapeutic implications in neurodegenerative disorders. Drug Discov. Today 2023, 28, 103583. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Marcia, C.H.; Deng, C.X.; Finley, L.W.S.; Kim, H.-S.; Gius, D. SIRT3 Is a Mitochondrial Tumor Suppressor: A Scientific Tale That Connects Aberrant Cellular ROS, the Warburg Effect, and Carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef]
- Hirschey, M.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, C.; Levin, B.E. Fatty acid-induced astrocyte ketone production and the control of food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, 1186–1192. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell. 2013, 50, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the Role of Histone Deacetylase and their Inhibitors in Neurodegenerative Disorders: Current Targets and Future Perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.F.; Xu, H.B.; Wang, J.Y.; Lin, Q.; Ruan, Z.; Liu, F.B.; Jin, W.; Huang, H.H.; Chen, X. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem. Biophys. Res. Commun. 2013, 441, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Yang, G.; Chi, X.; Liang, X.; Zhang, Y. Sirtuins: Promising Therapeutic Targets to Treat Ischemic Stroke. Biomolecules 2023, 13, 1210. [Google Scholar] [CrossRef]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L.; et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef]
- Scholl-Bürgi, S.; Höller, A.; Pichler, K.; Michel, M.; Haberlandt, E.; Karall, D. Ketogenic diets in patients with inherited metabolic disorders. J. Inherit. Metab. Dis. 2015, 38, 765–773. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rühling, M.R.; Hartmann, H.; Das, A.M. Simplification of Dietary Treatment in Pharmacoresistant Epilepsy: Impact of C8 and C10 Fatty Acids on Sirtuins of Neuronal Cells In Vitro. Nutrients 2024, 16, 1678. https://doi.org/10.3390/nu16111678
Rühling MR, Hartmann H, Das AM. Simplification of Dietary Treatment in Pharmacoresistant Epilepsy: Impact of C8 and C10 Fatty Acids on Sirtuins of Neuronal Cells In Vitro. Nutrients. 2024; 16(11):1678. https://doi.org/10.3390/nu16111678
Chicago/Turabian StyleRühling, Miriam Rebekka, Hans Hartmann, and Anibh Martin Das. 2024. "Simplification of Dietary Treatment in Pharmacoresistant Epilepsy: Impact of C8 and C10 Fatty Acids on Sirtuins of Neuronal Cells In Vitro" Nutrients 16, no. 11: 1678. https://doi.org/10.3390/nu16111678
APA StyleRühling, M. R., Hartmann, H., & Das, A. M. (2024). Simplification of Dietary Treatment in Pharmacoresistant Epilepsy: Impact of C8 and C10 Fatty Acids on Sirtuins of Neuronal Cells In Vitro. Nutrients, 16(11), 1678. https://doi.org/10.3390/nu16111678