
Implications of Microbiota and Immune System in Development and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease
 , , , ,
, , , ,
Abstract

1. Introduction
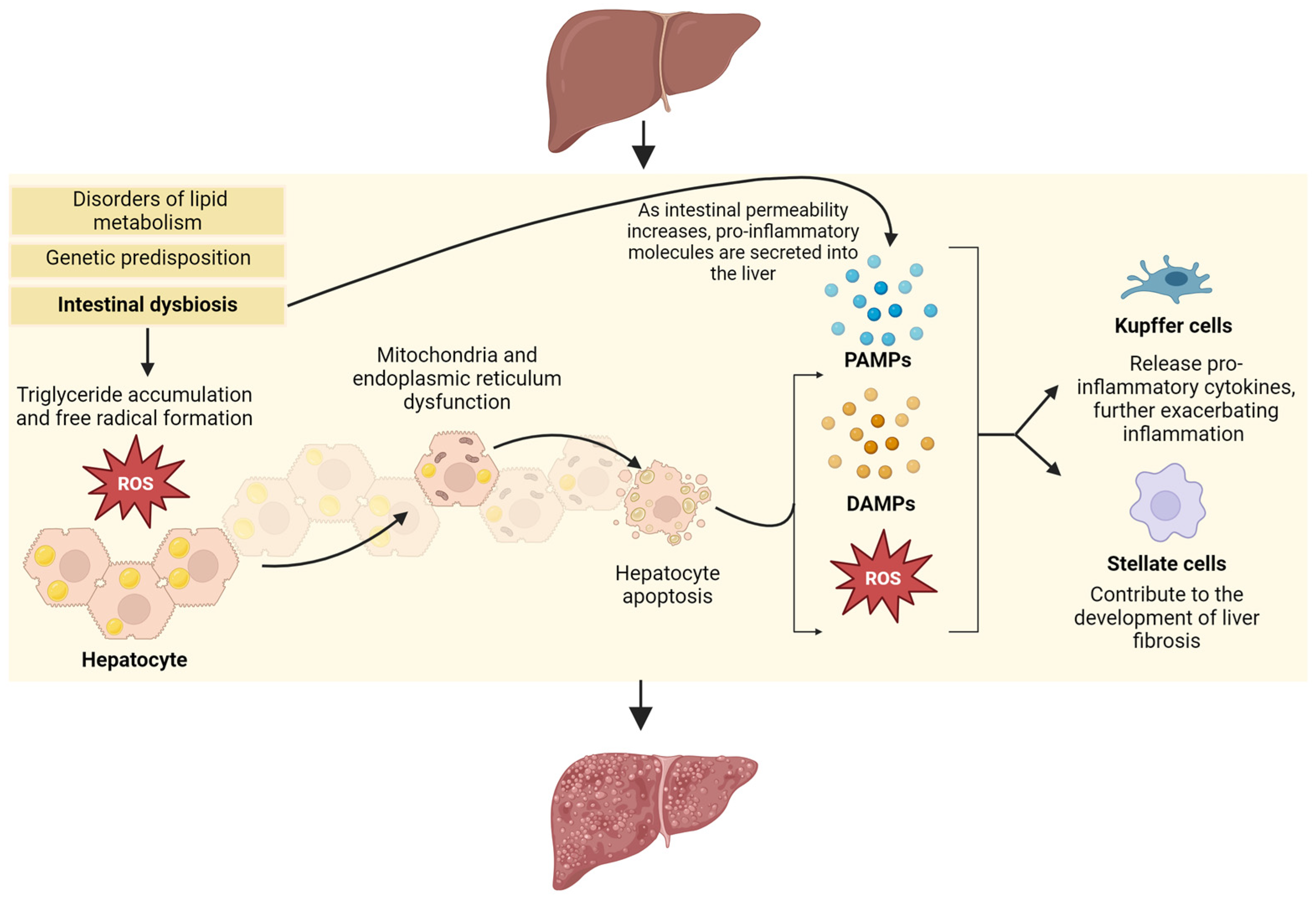
2. Pathophysiology
3. Immunological Implications
3.1. Innate Immune Response
3.2. Adaptive Immune Response
4. The Role of the Microbiome
4.1. Compositional Changes of the Gut Microbiome

{kind=link}
{kind=link}
| HUMAN ADULT STUDIES | ||||
|---|---|---|---|---|
| Author | Population | Samples | Main Effects on Microbiota | Main Effects on Metabolites |
| Adams et al., 2020 [55] | 58 MASLD (minimal–mild fibrosis), 9 MASLD (severe fibrosis), and 55 HCs | Fecal and serum samples | Severe (F3-F4) liver fibrosis:
|
|
| Chen et al., 2020 [56] | 538 MASLD (lean and obese) and 30 lean HCs | Fecal and serum samples | Lean versus obese MASLD patients:
|
|
| Delik et al., 2022 [50] | 20 MASLD and 20 HCs | Intestinal biopsies | ↔ Firmicutes ↑ Gram-negative bacteria ↑ Phylum Proteobacteria ↓ microbial diversity ↓ Bacteroidetes and Actinobacteria | N/A |
| Demir et al., 2022 [57] | 78 obese and non-obese MASLD (40 fibrosis F0-F1 and 38 fibrosis F2-F4), 54 NASH, and 16 HCs | Fecal and serum samples |
|
|
| Hullar et al., 2021 [58] | 511 MASLD and 1033 HCs | Fecal and blood samples | Pooled ethnicities MASLD compared with HCs:
| Pooled ethnicities and genders, MASLD compared with HCs:
|
| 400 Japanese American individuals (178 MASLD and 222 HCs) | Japanese American individuals:
|
| ||
| 257 African American individuals (38 MASLD and 219 HCs) | African American individuals:
| |||
| 316 White individuals (67 MASLD and 249 HCs) | White individuals:
| |||
| 325 Latino individuals (143 MASLD and 182 HCs) | Latino individuals:
|
| ||
| 246 Native Hawaiian individuals (85 MASLD and 161 HCs) | Native Hawaiian individuals:
|
| ||
| Jee et al., 2022 [53] | 16 MASLD (microbiome similar to (PHC-like) and dissimilar (P) from health controls) | Fecal and serum samples |
|
|
| Jiao et al., 2021 [59] | 86 MASLD and 38 HCs | Fecal and serum samples |
|
|
| Lang et al., 2020 [60] | 73 MASLD (37 with F0–F1 fibrosis, 36 with F2–F4 fibrosis, 29 with NAS 0–4, and 44 with NAS 5-8/LCI) and 22 HCs (9 without liver disease and 13 with mild primary biliary cholangitis) | Fecal and serum samples | More advanced disease:
| N/A |
| Leung et al., 2022 [54] | 90 MASLD and 90 HCs | Fecal and serum samples |
|
|
| Mouzaki et al., 2016 [61] | 12 MASL, 16 MASH, and 25 HCs | Fecal and serum samples | ↓ Bacteroidetes in MASH. ↓ Clostridium leptum in MASH. | ↑ CA in MASLD and MASH ↑ CDCA in MASH ↓ LCA in MASLD and MASH ↑ Primary-to-Secondary BA Ratio in MASH ↑ Total BAs in MASH |
| Oh et al., 2021 [62] | 22 MASLD and 44 HCs | Fecal samples | MAFLD compared with HCs:
| N/A |
| Sui et al., 2021 [63] | 59 NHS non-diabetic patients and 32 HCs | Fecal samples | NHS compared with HCs:
|
|
| Wang et al., 2016 [64] | 43 MASLD and 83 HCs | Fecal and serum samples | MASLD compared with HCs:
|
|
| Wang et al., 2021 [65] | 505 MASLD (306 mild, 174 moderate, and 25 severe disease) and 1393 HCs | Breath test and serum samples |
| N/A |
| Yun et al., 2019 [66] | 76 MASLD and 192 HCs | Fecal and blood samples | MASLD compared with HCs:
| N/A |
| HUMAN PEDIATRIC STUDIES | ||||
| Author | Population | Samples | Main Effects on Microbiota | Main Effects on Metabolites |
| Del Chierico et al., 2017 [67] | 61 MASLD (27 MASL, 26 MASH, and 8 obese) and 54 HCs [68] | Fecal and serum samples |
|
|
| Schwimmer et al., 2019 [69] | 87 cases (not-MASH, borderline MASH, and definite MASH) and 37 patients with obesity but without MASLD | Fecal and blood samples |
|
|
| Yu et al., 2021 [51] | 32 MASLD and 36 HCs | Fecal samples |
|
|
4.2. The Intestinal Barrier and the Gut–Liver Axis
5. Metabolic Changes in MASLD
5.1. Short-Chain Fatty Acids
5.2. Bile Acids
5.3. Choline
5.4. Ethanol
6. Treatment
6.1. Antibiotics
6.2. Probiotics
6.3. Fecal Microbiota Transplantation
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Lonardo, A.; Leoni, S.; Alswat, K.A.; Fouad, Y. History of nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2020, 21, 5888. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism 2020, 111, 154170. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Sayiner, M.; Fazel, Y.; Koenig, A.; Henry, L.; Younossi, Z. Current complications and challenges in nonalcoholic steatohepatitis screening and diagnosis. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Shaker, M.; Tabbaa, A.; Albeldawi, M.; Alkhouri, N. Liver transplantation for nonalcoholic fatty liver disease: New challenges and new opportunities. World J. Gastroenterol. 2014, 20, 5320–5330. [Google Scholar] [CrossRef]
- YYilmaz, Y. The heated debate over NAFLD renaming: An ongoing saga. Hepatol. Forum 2023, 4, 89–91. [Google Scholar] [CrossRef]
- Rinella, M.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023, 79, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Day, C.; James, O. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, C.M.; Frühbeck, G.; Escalada, J. Impact of nutritional changes on nonalcoholic fatty liver disease. Nutrients 2019, 11, 677. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yu, C.-H.; Li, X.-J.; Yao, J.-M.; Fang, Z.-Y.; Yoon, S.-H.; Yu, W.-Y. Gut dysbiosis in nonalcoholic fatty liver disease: Pathogenesis, diagnosis, and therapeutic implications. Front. Cell. Infect. Microbiol. 2022, 12, 997018. [Google Scholar] [CrossRef]
- Ore, A.; Akinloye, O.A. Phytotherapy as multi-hit therapy to confront the multiple pathophysiology in non-alcoholic fatty liver disease: A systematic review of experimental interventions. Medicina 2021, 57, 822. [Google Scholar] [CrossRef] [PubMed]
- Huby, T.; Gautier, E.L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 2021, 22, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef]
- Haas, J.T.; Vonghia, L.; Mogilenko, D.A.; Verrijken, A.N.; Molendi-Coste, O.; Fleury, S.; Deprince, A.; Nikitin, A.; Woitrain, E.; Ducrocq-Geoffroy, L.; et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat. Metab. 2019, 1, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; David, E.; Ramadori, P.; Pfister, D.; Safran, M.; Li, B.; Giladi, A.; Jaitin, D.A.; Barboy, O.; Cohen, M.; et al. XCR1+ type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat. Med. 2021, 27, 1043–1054. [Google Scholar] [CrossRef]
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, L.; Chang, N.; Hou, L.; Zhou, X.; Yang, L.; Li, L. Neutrophils undergo switch of apoptosis to NETosis during murine fatty liver injury via S1P receptor 2 signaling. Cell Death Dis. 2020, 11, 379. [Google Scholar] [CrossRef] [PubMed]
- Zang, S.; Wang, L.; Ma, X.; Zhu, G.; Zhuang, Z.; Xun, Y.; Zhao, F.; Yang, W.; Liu, J.; Luo, Y.; et al. Neutrophils play a crucial role in the early stage of nonalcoholic steatohepatitis via neutrophil elastase in mice. Cell Biochem. Biophys. 2015, 73, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Van der Windt, D.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–10966. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Baba, I.; Poupel, L.; Dussaud, S.; Moreau, M.; Gélineau, A.; Marcelin, G.; Magréau-Davy, E.; Ouhachi, M.; Lesnik, P.; et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity 2020, 53, 627–640.e5. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Zhang, Y.; Pan, G.; Xiang, L.-X.; Luo, D.-C.; Shao, J.-Z. Occurrences and functions of Ly6Chi and Ly6Clo macrophages in health and disease. Front. Immunol. 2022, 13, 901672. [Google Scholar] [CrossRef] [PubMed]
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; De Prijck, S.; Vanhockerhout, M.; et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity 2020, 53, 641–657.e14. [Google Scholar]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.-Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef] [PubMed]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef]
- Yuseff, M.-I.; Pierobon, P.; Reversat, A.; Lennon-Duménil, A.-M. How B cells capture, process and present antigens: A crucial role for cell polarity. Nat. Rev. Immunol. 2013, 13, 475–486. [Google Scholar] [CrossRef]
- Fillatreau, S. B cells and their cytokine activities implications in human diseases. Clin. Immunol. 2018, 186, 26–31. [Google Scholar] [CrossRef]
- Barrow, F.; Khan, S.; Fredrickson, G.; Wang, H.; Dietsche, K.; Parthiban, P.; Robert, S.; Kaiser, T.; Winer, S.; Herman, A.; et al. Microbiota-driven activation of intrahepatic B Cells aggravates NASH through innate and adaptive signaling. Hepatology 2021, 74, 704–722. [Google Scholar] [CrossRef] [PubMed]
- Bruzzì, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic. Biol. Med. 2018, 124, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Van Herck, M.A.; Weyler, J.; Kwanten, W.J.; Dirinck, E.L.; De Winter, B.Y.; Francque, S.M.; Vonghia, L. The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Front. Immunol. 2019, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.-K. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Liver Physiol. 2013, 305, G891–G899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shen, J.; Man, K.; Chu, E.S.; Yau, T.O.; Go, M.Y.; Deng, J.; Lu, L.; Wong, V.W.; Sung, J.J.; et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J. Hepatol. 2014, 61, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Fujisawa, T.; Husain, S.; Kioi, M.; Nakajima, A.; Puri, R. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J. Immunol. 2008, 181, 4656–4665. [Google Scholar] [CrossRef] [PubMed]
- Rau, M.; Rehman, A.; Dittrich, M.; Groen, A.K.; Hermanns, H.M.; Seyfried, F.; Beyersdorf, N.; Dandekar, T.; Rosenstiel, P.; Geier, A. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Eur. Gastroenterol. J. 2018, 6, 1496–1507. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Cappelletti, M.; Huppert, S.S.; Iwakura, Y.; Dong, C.; Shanmukhappa, S.K.; Divanovic, S. Regulation of Inflammation by IL-17A and IL-17F Modulates Non-Alcoholic Fatty Liver Disease Pathogenesis. PLoS ONE 2016, 11, e0149783. [Google Scholar] [CrossRef]
- Gomes, A.L.; Teijeiro, A.; Burén, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.-P.; Perna, C.; Djouder, N. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 2016, 30, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Rolla, S.; Alchera, E.; Imarisio, C.; Bardina, V.; Valente, G.; Cappello, P.; Mombello, C.; Follenzi, A.; Novelli, F.; Carini, R. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin. Sci. 2016, 130, 193–203. [Google Scholar] [CrossRef]
- Her, Z.; Tan, J.H.L.; Lim, Y.S.; Tan, S.Y.; Chan, X.Y.; Tan, W.W.S.; Liu, M.; Yong, K.S.M.; Lai, F.; Ceccarello, E.; et al. CD4+ T cells mediate the development of liver fibrosis in high fat diet-Induced NAFLD in humanized mice. Front. Immunol. 2020, 11, 580968. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, G.; Wang, Y.; Li, S.; Zhao, X.; Zhang, C.; Jin, H.; Tian, D.; Liu, K.; Shi, W.; et al. The immunoregulatory effects of CD8 T-cell-derived perforin on diet-induced nonalcoholic steatohepatitis. FASEB J. 2019, 33, 8490–8503. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ye, J.; Shao, C.; Zhong, B. Compositional alterations of gut microbiota in nonalcoholic fatty liver disease patients: A systematic review and meta-analysis. Lipids Health Dis. 2021, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Delik, A.; Dinçer, S.; Ülger, Y.; Akkız, H.; Karaoğullarından, U. Metagenomic identification of gut microbiota distribution on the colonic mucosal biopsy samples in patients with non-alcoholic fatty liver disease. Gene 2022, 833, 146587. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, H.; Chen, L.; Ruan, Y.; Chen, Y.; Liu, Q. Disease-associated gut microbiota reduces the profile of secondary bile acids in pediatric nonalcoholic fatty liver disease. Front. Cell. Infect. Microbiol. 2021, 11, 698852. [Google Scholar] [CrossRef]
- Xiang, H.; Sun, D.; Liu, X.; She, Z.-G.; Chen, Y. The role of the intestinal microbiota in nonalcoholic steatohepatitis. Front. Endocrinol. 2022, 13, 812610. [Google Scholar] [CrossRef] [PubMed]
- Jee, J.J.; Lim, J.; Park, S.; Koh, H.; Lee, H.W. Gut microbial community differentially characterizes patients with nonalcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2022, 37, 1822–1832. [Google Scholar] [CrossRef]
- Leung, H.; Long, X.; Ni, Y.; Qian, L.; Nychas, E.; Siliceo, S.L.; Pohl, D.; Hanhineva, K.; Liu, Y.; Xu, A.; et al. Risk assessment with gut microbiome and metabolite markers in NAFLD development. Sci. Transl. Med. 2022, 14, eabk0855. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Wang, Z.; Liddle, C.; Melton, P.E.; Ariff, A.; Chandraratna, H.; Tan, J.; Ching, H.; Coulter, S.; de Boer, B.; et al. Bile acids associate with specific gut microbiota, low-level alcohol consumption and liver fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2020, 40, 1356–1365. [Google Scholar] [CrossRef]
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A distinct entity shaped by differential metabolic adaptation. Hepatology 2020, 71, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Demir, M.; Lang, S.; Hartmann, P.; Duan, Y.; Martin, A.; Miyamoto, Y.; Bondareva, M.; Zhang, X.; Wang, Y.; Kasper, P.; et al. The fecal mycobiome in non-alcoholic fatty liver disease. J. Hepatol. 2022, 76, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Hullar, M.A.J.; Jenkins, I.C.; Randolph, T.W.; Curtis, K.R.; Monroe, K.R.; Ernst, T.; Shepherd, J.A.; Stram, D.O.; Cheng, I.; Kristal, B.S.; et al. Associations of the gut microbiome with hepatic adiposity in the Multiethnic Cohort Adiposity Phenotype Study. Gut Microbes 2021, 13, 1965463. [Google Scholar] [CrossRef]
- Jiao, N.; Loomba, R.; Yang, Z.-H.; Wu, D.; Fang, S.; Bettencourt, R.; Lan, P.; Zhu, R.; Zhu, L. Alterations in bile acid metabolizing gut microbiota and specific bile acid genes as a precision medicine to subclassify NAFLD. Physiol. Genom. 2021, 53, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal virome signature associated with severity of nonalcoholic fatty liver disease. Gastroenterology 2020, 159, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Lee, J.H.; Cho, M.S.; Kim, H.; Chun, J.; Lee, J.H.; Yoon, Y.; Kang, W. Characterization of gut microbiome in korean patients with metabolic associated fatty liver disease. Nutrients 2021, 13, 1013. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.; Jia, L.; Quan, D.; Zhao, N.; Yang, G. Activation of the gut microbiota-kynurenine-liver axis contributes to the development of nonalcoholic hepatic steatosis in nondiabetic adults. Aging 2021, 13, 21309–21324. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jiang, X.; Cao, M.; Ge, J.; Bao, Q.; Tang, L.; Chen, Y.; Li, L. Altered fecal microbiota correlates with liver biochemistry in nonobese patients with non-alcoholic fatty liver disease. Sci. Rep. 2016, 6, 32002. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dong, F.; Su, H.; Zhu, L.; Shao, S.; Wu, J.; Liu, H.H. pylori is related to NAFLD but only in female: A Cross-sectional Study. Int. J. Med. Sci. 2021, 18, 2303–2311. [Google Scholar] [CrossRef]
- Yun, Y.; Kim, H.-N.; Lee, E.-J.; Ryu, S.; Chang, Y.; Shin, H.; Kim, H.-L.; Kim, T.H.; Yoo, K.; Kim, H.Y. Fecal and blood microbiota profiles and presence of nonalcoholic fatty liver disease in obese versus lean subjects. PLoS ONE 2019, 14, e0213692. [Google Scholar] [CrossRef] [PubMed]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; DeSimone, C.; Song, X.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Johnson, J.S.; Angeles, J.E.; Behling, C.; Belt, P.H.; Borecki, I.; Bross, C.; Durelle, J.; Goyal, N.P.; Hamilton, G.; et al. Microbiome signatures associated with steatohepatitis and moderate to severe fibrosis in children with nonalcoholic fatty liver disease. Gastroenterology 2019, 157, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Bastian, W.; Hasan, I.; Lesmana, C.; Rinaldi, I.; Gani, R. Open access gateway gut microbiota profiles in nonalcoholic fatty liver disease and its possible impact on disease progression evaluated with transient elastography: Lesson learnt from 60 cases. Case Rep. Gastroenterol. 2019, 13, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; De Bandt, J. Fructose and NAFLD: The multifaceted aspects of fructose metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Kwan, S.Y.; Jiao, J.; Qi, J.; Wang, Y.; Wei, P.; McCormick, J.B.; Fisher-Hoch, S.P.; Beretta, L. Bile acid changes associated with liver fibrosis and steatosis in the Mexican-American population of South Texas. Hepatol. Commun. 2020, 4, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Bonfrate, L.; Khalil, M.; De Angelis, M.; Calabrese, F.M.; D’amato, M.; Wang, D.Q.-H.; Di Ciaula, A. Intestinal barrier and permeability in health, obesity and NAFLD. Biomedicines 2021, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cantos, M.V.; Garcia-Morena, D.; Iannone, V.; El-Nezami, H.; Kolehmainen, M.; Kuipers, O.P. Role of microbiota and related metabolites in gastrointestinal tract barrier function in NAFLD. Tissue Barriers 2021, 9, 1879719. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.; Turner, J.R. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.-P.; Zhang, Y.-T.; Zhang, R.-X.; Zhong, H.-J.; He, X.-X. The gut-liver axis in nonalcoholic fatty liver disease: Association of intestinal permeability with disease severity and treatment outcomes. Int. J. Clin. Pract. 2022, 2022, 4797453. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Nieß, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. The role of gut-derived lipopolysaccharides and the intestinal barrier in fatty liver diseases. J. Gastrointest. Surg. 2022, 26, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Rajcic, D.; Brandt, A.; Sánchez, V.; Jung, F.; Staltner, R.; Nier, A.; Trauner, M.; Staufer, K.; Bergheim, I. Alterations of nitric oxide homeostasis as trigger of intestinal barrier dysfunction in non-alcoholic fatty liver disease. J. Cell. Mol. Med. 2022, 26, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.C.; Littlejohn, P.T.; Ayala, V.; Creus-Cuadros, A.; Finlay, B.B. Nonalcoholic fatty liver disease and the gut-liver axis: Exploring an undernutrition perspective. Gastroenterology 2022, 162, 1858–1875.e2. [Google Scholar] [CrossRef]
- Wang, T.; Guo, X.-K.; Xu, H. Disentangling the progression of non-alcoholic fatty liver disease in the human gut microbiota. Front. Microbiol. 2021, 12, 728823. [Google Scholar] [CrossRef]
- Ang, Z.; Ding, J. GPR41 and GPR43 in obesity and inflammation—Protective or causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef]
- Liu, L.; Fu, Q.; Li, T.; Shao, K.; Zhu, X.; Cong, Y.; Zhao, X. Gut microbiota and butyrate contribute to nonalcoholic fatty liver disease in premenopause due to estrogen deficiency. PLoS ONE 2022, 17, e0262855. [Google Scholar] [CrossRef]
- Li, Z.; Yi, C.-X.; Katiraei, S.; Kooijman, S.; Zhou, E.; Chung, C.K.; Gao, Y.; van den Heuvel, J.K.; Meijer, O.C.; Berbée, J.F.P.; et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 2018, 67, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Finnie, I.A.; Dwarakanath, A.D.; Taylor, B.A.; Rhodes, J.M. Colonic mucin synthesis is increased by sodium butyrate. Gut 1995, 36, 93–99. [Google Scholar] [CrossRef]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a g-protein–coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef]
- Xue, R.; Su, L.; Lai, S.; Wang, Y.; Zhao, D.; Fan, J.; Chen, W.; Hylemon, P.B.; Zhou, H. Bile acid receptors and the gut–liver axis in nonalcoholic fatty liver disease. Cells 2021, 10, 2806. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Muthiah, M.D.; Narayan, N.; Siddiqui, M.S.; Puri, P.; Luketic, V.A.; Contos, M.J.; Idowu, M.; Chuang, J.; Billin, A.N.; et al. Metabolic reprogramming of the intestinal microbiome with functional bile acid changes underlie the development of NAFLD. Hepatology 2022, 76, 1811–1824. [Google Scholar] [CrossRef] [PubMed]
- Saga, K.; Iwashita, Y.; Hidano, S.; Aso, Y.; Isaka, K.; Kido, Y.; Tada, K.; Takayama, H.; Masuda, T.; Hirashita, T.; et al. Secondary unconjugated bile acids induce hepatic stellate cell activation. Int. J. Mol. Sci. 2018, 19, 3043. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; Afonso, M.; Simao, A.; Borralho, P.; Rodrigues, C.; Casto, R. Inhibition of NF-κB by deoxycholic acid induces miR-21/PDCD4-dependent hepatocellular apoptosis. Nature 2015, 5, 17528. [Google Scholar]
- Gu, C.; Zhou, Z.; Yu, Z.; He, M.; He, L.; Luo, Z.; Xiao, W.; Yang, Q.; Zhao, F.; Li, W.; et al. Corrigendum: The microbiota and it’s correlation with metabolites in the gut of mice with nonalcoholic fatty liver disease. Front. Cell. Infect. Microbiol. 2022, 12, 972118. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, A.; Li, S.; Lou, P.; Wu, W.; Wang, K.; Yuan, Y.; Xia, J.; Li, B.; Li, L. Longitudinal 16S rRNA sequencing reveals relationships among alterations of gut microbiota and nonalcoholic fatty liver disease progression in mice. Microbiol. Spectr. 2022, 10, e0004722. [Google Scholar] [CrossRef]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.-Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Stofan, M.; Guo, G.L. Bile Acids and FXR: Novel Targets for Liver Diseases. Front. Med. 2020, 7, 544. [Google Scholar] [CrossRef] [PubMed]
- Grzych, G.; Chavez-Talavera, O.; Descat, A.; Thuillier, D.; Verrijken, A.; Kouach, M.; Legry, V.; Verkindt, H.; Raverdy, V.; Legendre, B.; et al. NASH-related increases in plasma bile acid levels depend on insulin resistance. JHEP Rep. 2020, 3, 100222. [Google Scholar] [CrossRef]
- Nogan, A.; Vance, D. A gender-specific role for phosphatidylethanolamine N-methyltransferase-derived phosphatidylcholine in the regulation of plasma high density and very low density lipoproteins in mice. J. Biol. Chem. 2003, 278, 21851–21859. [Google Scholar] [CrossRef] [PubMed]
- Vance, D. Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr. Opin. Lipidol. 2008, 19, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 8, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.L.; Ament, M.E.; Sohel, M.; Dubin, M.; Jenden, D.J.; Roch, M.; Pownall, H.; Farley, W.; Awal, M.; Ahn, C. Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: Proof of a human choline requirement: A placebo-controlled trial. J. Parenter Enter. Nutr. 2001, 25, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Martínez-del Campo, A.; Bodea, S.; Hamer, H.A.; Marks, J.A.; Haiser, H.J.; Turnbaugh, P.J.; Balskus, E.P. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. mBio 2015, 6, e00042-15. [Google Scholar] [CrossRef] [PubMed]
- Idalsoaga, F.; Kulkarni, A.V.; Mousa, O.Y.; Arrese, M.; Arab, J.P. Non-alcoholic fatty liver disease and alcohol-related liver disease: Two intertwined entities. Front. Med. 2020, 7, 448. [Google Scholar] [CrossRef]
- Jeon, S.; Carr, R. Alcohol effects on hepatic lipid metabolism. J. Lipid Res. 2020, 61, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Nagappan, A.; Jung, D.Y.; Kim, J.-H.; Lee, H.; Jung, M.H. Gomisin N alleviates ethanol-induced liver injury through ameliorating lipid metabolism and oxidative stress. Int. J. Mol. Sci. 2018, 19, 2601. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.; Deja, S.; Burgess, S.; Browning, J. Effects of NAFLD on acetyl-CoA partitioning and ketone kinetics in response to a 24-hour fast. Diabetes 2018, 67 (Suppl. S1), 47-OR. [Google Scholar] [CrossRef]
- Rada, P.; González-Rodríguez, A.; García-Monzón, C.; Valverde, A. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7. [Google Scholar] [CrossRef] [PubMed]
- Meijnikman, A.S.; Davids, M.; Herrema, H.; Aydin, O.; Tremaroli, V.; Rios-Morales, M.; Levels, H.; Bruin, S.; de Brauw, M.; Verheij, J.; et al. Microbiome-derived ethanol in nonalcoholic fatty liver disease. Nat. Med. 2022, 28, 2100–2106. [Google Scholar] [CrossRef] [PubMed]
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2015, 91, 1–9. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2012, 57, 601–609. [Google Scholar] [CrossRef]
- Ganguli, S.; DeLeeuw, P.; Satapathy, S.K. A review of current and upcoming treatment modalities in non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Hepatic Med. Evid. Res. 2019, 11, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2014, 62, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Burz, S.D.; Monnoye, M.; Philippe, C.; Farin, W.; Ratziu, V.; Strozzi, F.; Paillarse, J.-M.; Chêne, L.; Blottière, H.M.; Gérard, P. Fecal microbiota transplant from human to mice gives insights into the role of the gut microbiota in non-alcoholic fatty liver disease (NAFLD). Microorganisms 2021, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Madrid, A.; Hurtado, C.; Venegas, M.; Cumsille, F.; Defilippi, C. Long-term treatment with cisapride and antibiotics in liver cirrhosis: Effect on small intestinal motility, bacterial overgrowth, and liver function. Am. J. Gastroenterol. 2001, 96, 1251–1255. [Google Scholar] [CrossRef]
- Fujinaga, Y.; Kawaratani, H.; Kaya, D.; Tsuji, Y.; Ozutsumi, T.; Furukawa, M.; Kitagawa, K.; Sato, S.; Nishimura, N.; Sawada, Y.; et al. Effective combination therapy of angiotensin-ii receptor blocker and rifaximin for hepatic fibrosis in rat model of nonalcoholic steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5589. [Google Scholar] [CrossRef]
- Gangarapu, V.; Ince, A.T.; Baysal, B.; Kayar, Y.; Klç, U.; Gök, Ö.; Uysal, Ö.; Şenturk, H. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razik, A.; Mousa, N.; Shabana, W.; Refaey, M.; Elzehery, R.; Elhelaly, R.; Zalata, K.; Abdelsalam, M.; Eldeeb, A.A.; Awad, M.; et al. Rifaximin in nonalcoholic fatty liver disease: Hit multiple targets with a single shot. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zuo, C.; Huang, W.; Wang, J.; Zhang, Z. Triclosan targeting of gut microbiome ameliorates hepatic steatosis in high fat diet-fed mice. J. Antibiot. 2022, 75, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Degnan, P.H.; Barry, N.A.; Mok, K.C.; Taga, M.E.; Goodman, A.L. Human gut microbes use multiple transporters to distinguish vitamin B12 analogs and compete in the gut. Cell Host Microbe 2014, 15, 47–57. [Google Scholar] [CrossRef]
- Paolella, G.; Mandato, C.; Pierri, L.; Poeta, M.; Di Stasi, M.; Vajro, P. Gut-liver axis and probiotics: Their role in non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15518–15531. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.-S.; Wong, G.L.-H.; Chim, A.M.-L.; Chu, W.C.-W.; Yeung, D.K.-W.; Li, K.C.-T.; Chan, H.L.-Y. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann. Hepatol. 2013, 12, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.; Turnbaugh, P.; Klein, S.; Gordon, J. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.-X.; Ding, W.-J.; Chen, Y.-W.; Fan, J.-G. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 2017, 7, 1529. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Rahman, A.; Parvathy, S.N.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic fecal microbiota transplantation in patients with nonalcoholic fatty liver disease improves abnormal small intestinal permeability: A randomized control trial. Am. J. Gastroenterol. 2020, 115, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popov, J.; Despot, T.; Avelar Rodriguez, D.; Khan, I.; Mech, E.; Khan, M.; Bojadzija, M.; Pai, N. Implications of Microbiota and Immune System in Development and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease. Nutrients 2024, 16, 1668. https://doi.org/10.3390/nu16111668
Popov J, Despot T, Avelar Rodriguez D, Khan I, Mech E, Khan M, Bojadzija M, Pai N. Implications of Microbiota and Immune System in Development and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease. Nutrients. 2024; 16(11):1668. https://doi.org/10.3390/nu16111668
Chicago/Turabian StylePopov, Jelena, Tijana Despot, David Avelar Rodriguez, Irfan Khan, Eugene Mech, Mahrukh Khan, Milan Bojadzija, and Nikhil Pai. 2024. "Implications of Microbiota and Immune System in Development and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease" Nutrients 16, no. 11: 1668. https://doi.org/10.3390/nu16111668
APA StylePopov, J., Despot, T., Avelar Rodriguez, D., Khan, I., Mech, E., Khan, M., Bojadzija, M., & Pai, N. (2024). Implications of Microbiota and Immune System in Development and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease. Nutrients, 16(11), 1668. https://doi.org/10.3390/nu16111668