
Ellagic Acid Prevents α-Synuclein Spread and Mitigates Toxicity by Enhancing Autophagic Flux in an Animal Model of Parkinson’s Disease


Abstract
:1. Introduction
2. Methodology
2.1. PFF Seed Synthesis
2.2. Animals
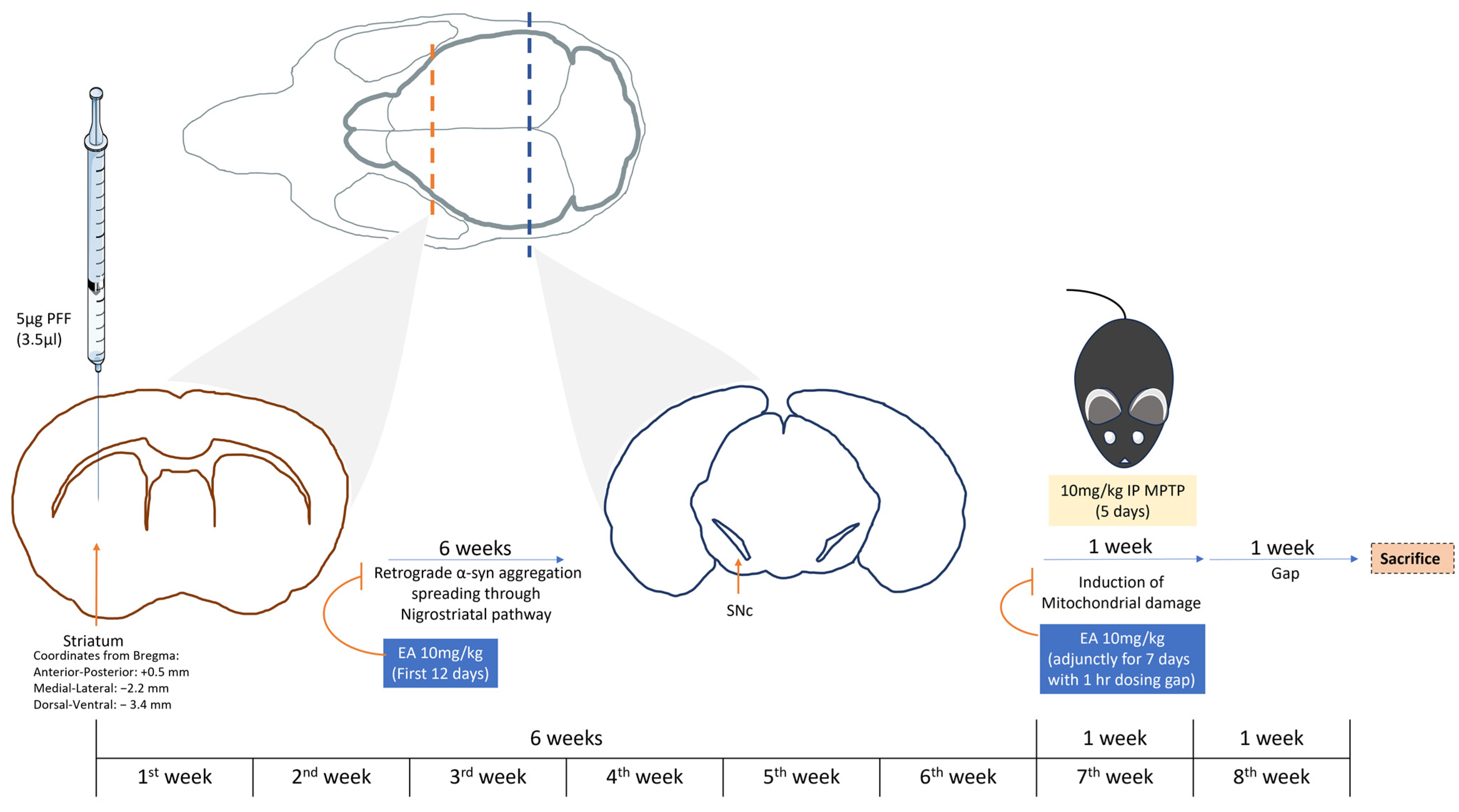
2.3. Study Plan
2.4. Sample Processing
2.4.1. DAB (3,3′-Diaminobenzidine) Stain
2.4.2. Immunofluorescence (IF)
- α-syn species spreading to the dopaminergic neurons in the SNc in phosphorylated form (pSer129) and filamentous form (Proteinase K resistant form).
- Expression of autophagic markers (LC3, p62) in dopaminergic neurons in the SNc.
2.4.3. TUNEL Assay (Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling)
2.4.4. Western Blot Analysis
2.5. Microscopy
2.6. Data Acquisition and Statistical Analysis
3. Results
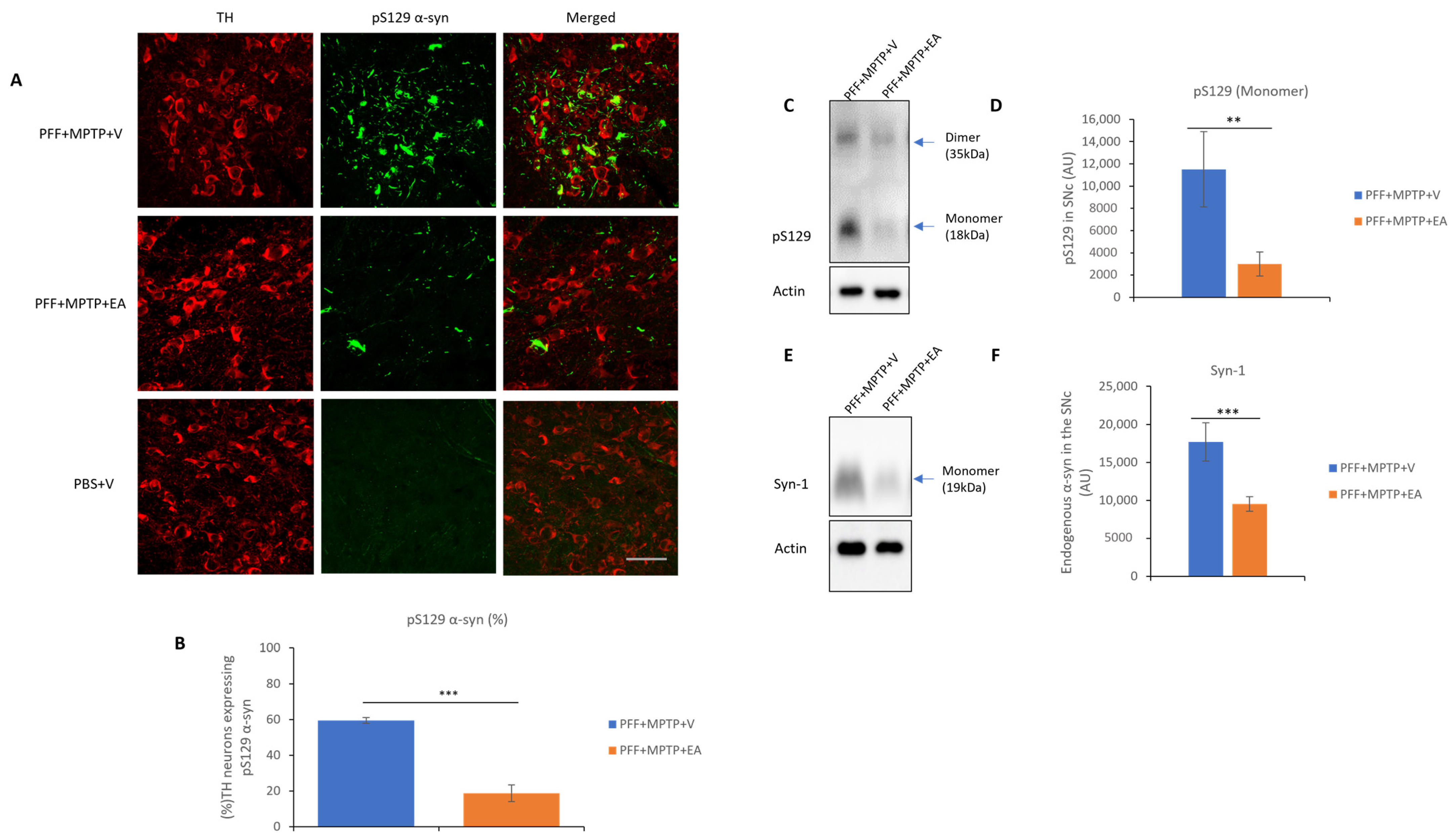
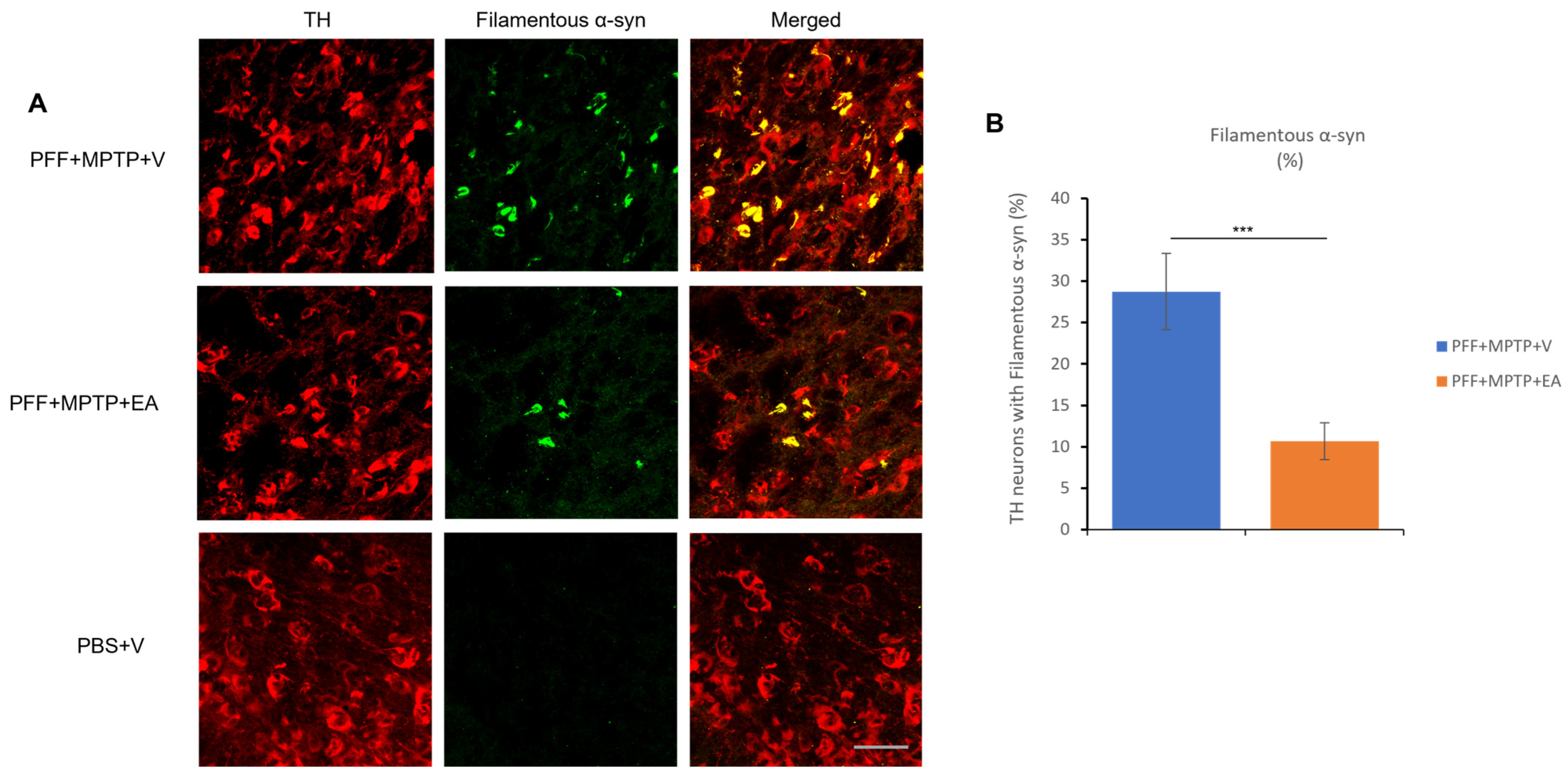
3.1. EA Inhibits MPTP-PFF Induced α-Syn Spreading and Aggregation
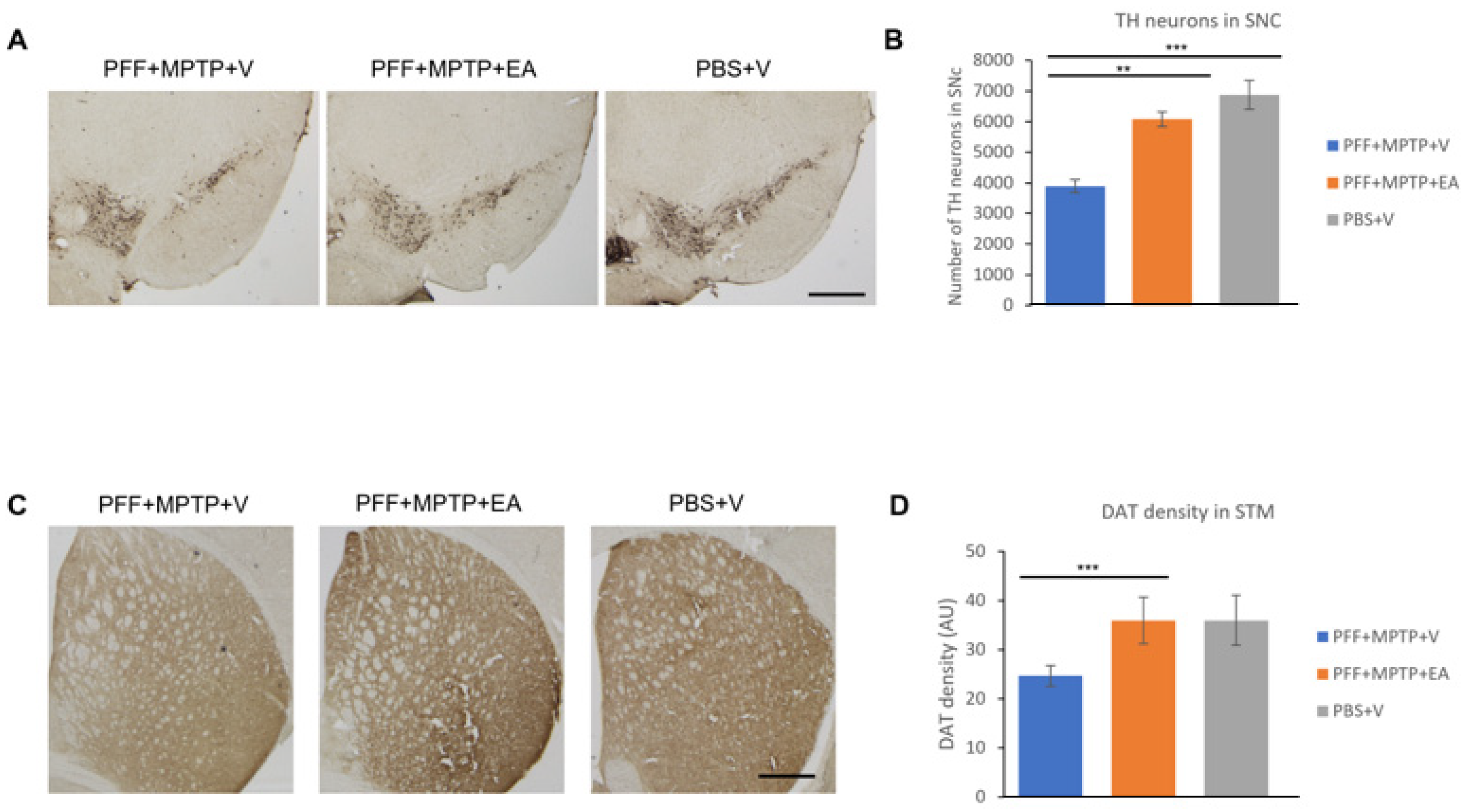
3.2. EA Inhibits Dopaminergic Neuronal Cell Loss and Preserves DAT Terminals
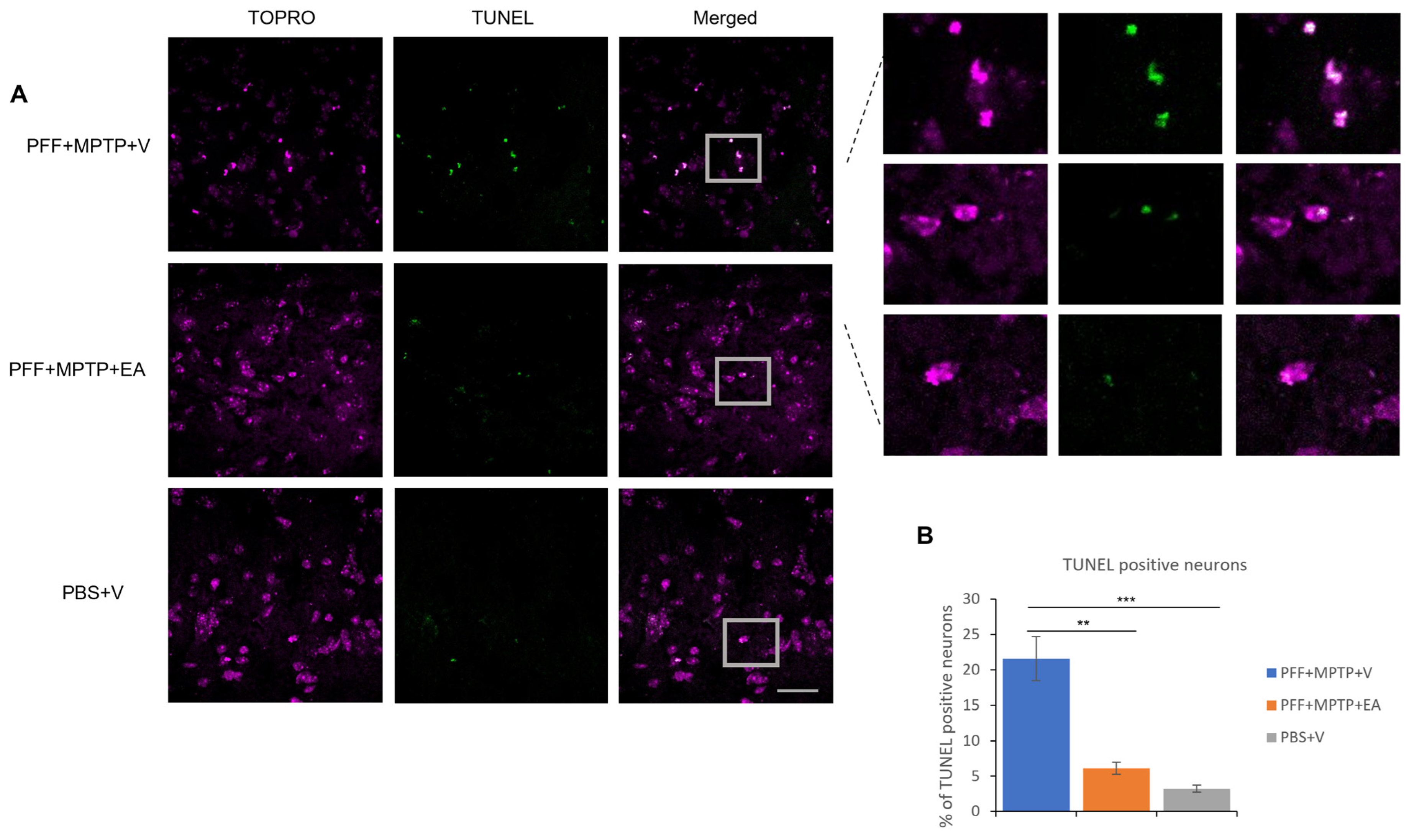
3.3. EA Enhances the Apoptotic Profile of Neurons in the SNc in MPTP-PFF Mouse PD Model
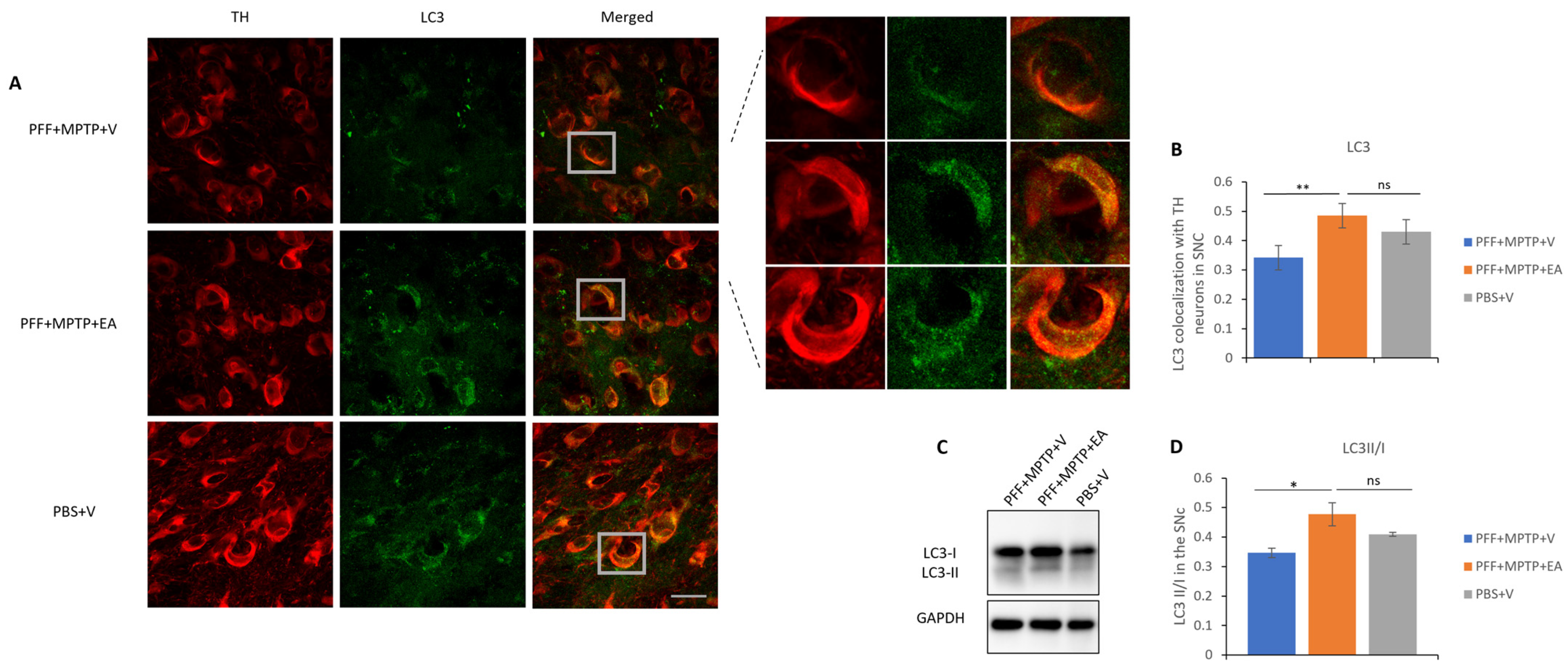
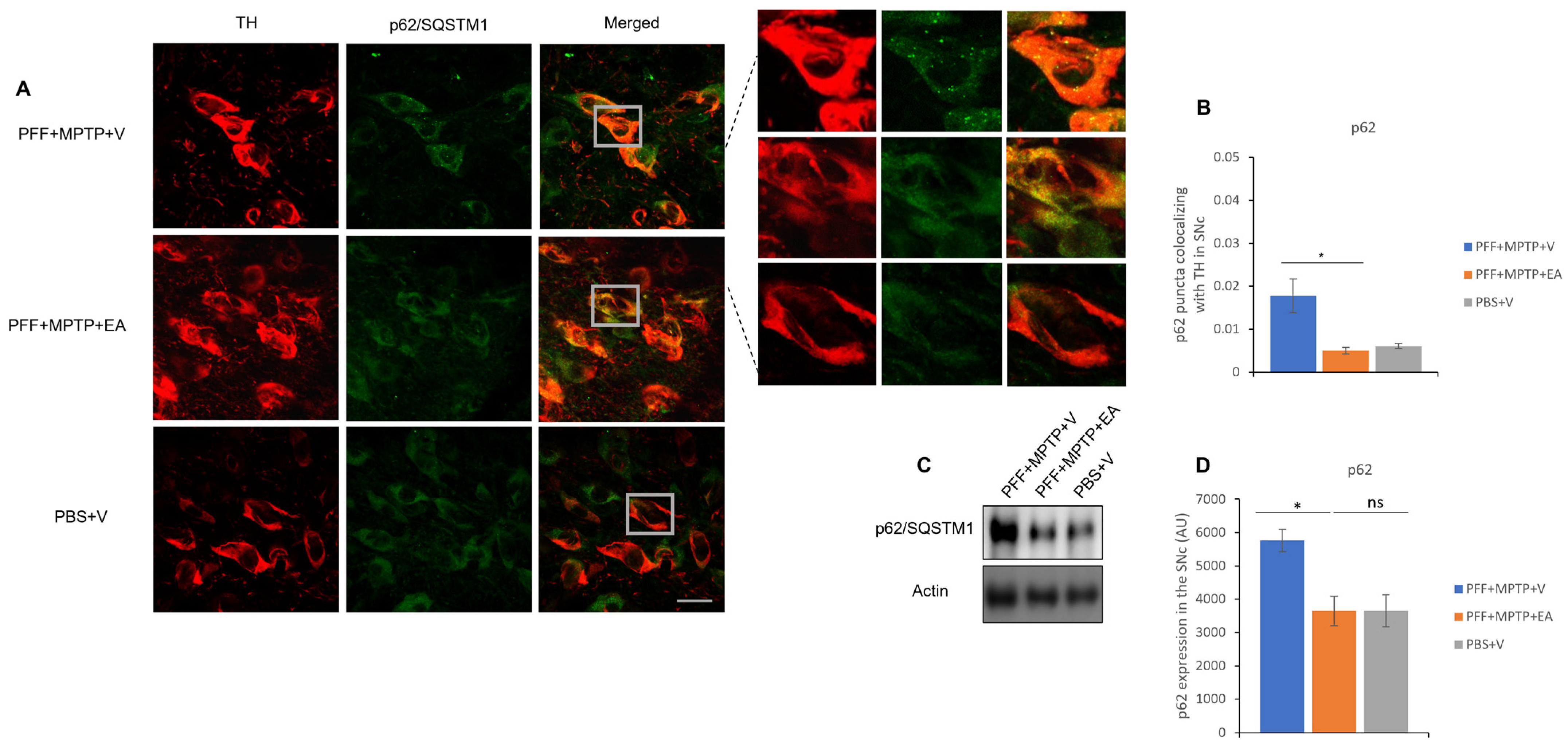
3.4. EA Induces Autophagic Degradation of α-Syn
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rudrapal, M.; Khairnar, S.J.; Khan, J.; Bin Dukhyil, A.; Ansari, M.A.; Alomary, M.N.; Alshabrmi, F.M.; Palai, S.; Deb, P.K.; Devi, R. Dietary Polyphenols and Their Role in Oxidative Stress-Induced Human Diseases: Insights into Protective Effects, Antioxidant Potentials and Mechanism(s) of Action. Front. Pharmacol. 2022, 13, 806470. [Google Scholar] [CrossRef] [PubMed]
- Pereira, Q.C.; Santos, T.W.D.; Fortunato, I.M.; Ribeiro, M.L. The Molecular Mechanism of Polyphenols in the Regulation of Ageing Hallmarks. Int. J. Mol. Sci. 2023, 24, 5508. [Google Scholar] [CrossRef] [PubMed]
- Daniel, E.M.; Krupnick, A.S.; Heur, Y.-H.; Blinzler, J.A.; Nims, R.W.; Stoner, G.D. Extraction, stability, and quantitation of ellagic acid in various fruits and nuts. J. Food Compos. Anal. 1989, 2, 338–349. [Google Scholar] [CrossRef]
- Landete, J.M. Ellagitannins, ellagic acid and their derived metabolites: A review about source, metabolism, functions and health. Food Res. Int. 2011, 44, 1150–1160. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Quispe, C.; Castillo, C.M.S.; Caroca, R.; Lazo-Vélez, M.A.; Antonyak, H.; Polishchuk, A.; Lysiuk, R.; Oliinyk, P.; De Masi, L.; et al. Ellagic Acid: A Review on Its Natural Sources, Chemical Stability, and Therapeutic Potential. Oxid. Med. Cell. Longev. 2022, 2022, 3848084. [Google Scholar] [CrossRef] [PubMed]
- Viladomiu, M.; Hontecillas, R.; Lu, P.; Bassaganya-Riera, J. Preventive and Prophylactic Mechanisms of Action of Pomegranate Bioactive Constituents. Evid.-Based Complement. Altern. Med. 2013, 2013, e789764. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Turrini, F.; Catena, S.; Zunin, P.; Grilli, M.; Pittaluga, A.M.; Boggia, R. Ellagic acid a multi-target bioactive compound for drug discovery in CNS? A narrative review. Eur. J. Med. Chem. 2019, 183, 111724. [Google Scholar] [CrossRef]
- Basu, A.; Penugonda, K. Pomegranate juice: A heart-healthy fruit juice. Nutr. Rev. 2009, 67, 49–56. [Google Scholar] [CrossRef]
- Rosillo, M.A.; Sánchez-Hidalgo, M.; Cárdeno, A.; Aparicio-Soto, M.; Sánchez-Fidalgo, S.; Villegas, I.; de la Lastra, C.A. Dietary supplementation of an ellagic acid-enriched pomegranate extract attenuates chronic colonic inflammation in rats. Pharmacol. Res. 2012, 66, 235–242. [Google Scholar] [CrossRef]
- Mansouri, Z.; Dianat, M.; Radan, M.; Badavi, M. Ellagic Acid Ameliorates Lung Inflammation and Heart Oxidative Stress in Elastase-Induced Emphysema Model in Rat. Inflammation 2020, 43, 1143–1156. [Google Scholar] [CrossRef]
- Aishwarya, V.; Solaipriya, S.; Sivaramakrishnan, V. Role of ellagic acid for the prevention and treatment of liver diseases. Phytother. Res. 2021, 35, 2925–2944. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Choi, Y.J.; Kim, H.-J. Determining the effect of ellagic acid on the proliferation and migration of pancreatic cancer cell lines. Transl. Cancer Res. 2021, 10, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Li, Y.; Gao, H.; Yang, D.; He, X.; Fang, Y.; Zhou, G. Phenolic compound ellagic acid inhibits mitochondrial respiration and tumor growth in lung cancer. Food Funct. 2020, 11, 6332–6339. [Google Scholar] [CrossRef] [PubMed]
- Ceci, C.; Tentori, L.; Atzori, M.G.; Lacal, P.M.; Bonanno, E.; Scimeca, M.; Cicconi, R.; Mattei, M.; De Martino, M.G.; Vespasiani, G.; et al. Ellagic Acid Inhibits Bladder Cancer Invasiveness and In Vivo Tumor Growth. Nutrients 2016, 8, 744. [Google Scholar] [CrossRef] [PubMed]
- Cheshomi, H.; Bahrami, A.R.; Rafatpanah, H.; Matin, M.M. The effects of ellagic acid and other pomegranate (Punica granatum L.) derivatives on human gastric cancer AGS cells. Hum. Exp. Toxicol. 2022, 41, 9603271211064534. [Google Scholar] [CrossRef] [PubMed]
- Yousef, A.I.; El-Masry, O.S.; Yassin, E.H. The anti-oncogenic influence of ellagic acid on colon cancer cells in leptin-enriched microenvironment. Tumor Biol. 2016, 37, 13345–13353. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, M.; Shamsi, A.; Khan, P.; Shahbaaz, M.; AlAjmi, M.F.; Hussain, A.; Hassan, G.M.; Islam, A.; Haque, Q.M.R.; Hassan, I. Ellagic Acid Controls Cell Proliferation and Induces Apoptosis in Breast Cancer Cells via Inhibition of Cyclin-Dependent Kinase 6. Int. J. Mol. Sci. 2020, 21, 3526. [Google Scholar] [CrossRef]
- Engelke, L.H.; Hamacher, A.; Proksch, P.; Kassack, M.U. Ellagic Acid and Resveratrol Prevent the Development of Cisplatin Resistance in the Epithelial Ovarian Cancer Cell Line A2780. J. Cancer 2016, 7, 353–363. [Google Scholar] [CrossRef]
- Goyal, Y.; Koul, A.; Ranawat, P. Ellagic acid modulates cisplatin toxicity in DMH induced colorectal cancer: Studies on membrane alterations. Biochem. Biophys. Rep. 2022, 31, 101319. [Google Scholar] [CrossRef]
- Yakobov, S.; Dhingra, R.; Margulets, V.; Dhingra, A.; Crandall, M.; Kirshenbaum, L.A. Kirshenbaum, Ellagic acid inhibits mitochondrial fission protein Drp-1 and cell proliferation in cancer. Mol. Cell. Biochem. 2023, 478, 2029–2040. [Google Scholar] [CrossRef]
- Rahimi, V.B.; Askari, V.R.; Mousavi, S.H. Ellagic acid reveals promising anti-aging effects against d-galactose-induced aging on human neuroblastoma cell line, SH-SY5Y: A mechanistic study. Biomed. Pharmacother. 2018, 108, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8 (Suppl. 1), S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Vos, T. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 318–324. [Google Scholar] [CrossRef] [PubMed]
- van der Hoek, T.C.; Bus, B.A.; Matui, P.; van der Marck, M.A.; Esselink, R.A.; Tendolkar, I. Prevalence of depression in Parkinson’s disease: Effects of disease stage, motor subtype and gender. J. Neurol. Sci. 2011, 310, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Brás, I.C.; Outeiro, T.F. Alpha-Synuclein: Mechanisms of Release and Pathology Progression in Synucleinopathies. Cells 2021, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef]
- Henderson, M.X.; Trojanowski, J.Q.; Lee, V.M.-Y. α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci. Lett. 2019, 709, 134316. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Recasens, A.; Dehay, B. Alpha-synuclein spreading in Parkinson’s disease. Front. Neuroanat. 2014, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.K.; Ho, H.-A.; Pérez-Acuña, D.; Lee, S.-J. Modeling α-Synuclein Propagation with Preformed Fibril Injections. J. Mov. Disord. 2019, 12, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Li, J.-D. The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed]
- Sonustun, B.; Altay, M.F.; Strand, C.; Ebanks, K.; Hondhamuni, G.; Warner, T.T.; Lashuel, H.A.; Bandopadhyay, R. Pathological Relevance of Post-Translationally Modified Alpha-Synuclein (pSer87, pSer129, nTyr39) in Idiopathic Parkinson’s Disease and Multiple System Atrophy. Cells 2022, 11, 906. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- Kujawska, M.; Jourdes, M.; Kurpik, M.; Szulc, M.; Szaefer, H.; Chmielarz, P.; Kreiner, G.; Krajka-Kuźniak, V.; Mikołajczak, P.; Teissedre, P.-L.; et al. Neuroprotective Effects of Pomegranate Juice against Parkinson’s Disease and Presence of Ellagitannins-Derived Metabolite—Urolithin A—In the Brain. Int. J. Mol. Sci. 2019, 21, 202. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Eid, N.; Kitada, T.; Haque, M.E. Ellagic Acid Prevents α-Synuclein Aggregation and Protects SH-SY5Y Cells from Aggregated α-Synuclein-Induced Toxicity via Suppression of Apoptosis and Activation of Autophagy. Int. J. Mol. Sci. 2021, 22, 13398. [Google Scholar] [CrossRef]
- Ardah, M.T.; Bharathan, G.; Kitada, T.; Haque, M.E. Ellagic Acid Prevents Dopamine Neuron Degeneration from Oxidative Stress and Neuroinflammation in MPTP Model of Parkinson’s Disease. Biomolecules 2020, 10, 1519. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Park. Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- Shen, R.-S.; Abell, C.; Gessner, W.; Brossi, A. Serotonergic conversion of MPTP and dopaminergic accumulation of MPP+. FEBS Lett. 1985, 189, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Merghani, M.M.; Ardah, M.T.; Al Shamsi, M.; Kitada, T.; Haque, M.E. Dose-related biphasic effect of the Parkinson’s disease neurotoxin MPTP, on the spread, accumulation, and toxicity of α-synuclein. Neurotoxicology 2021, 84, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2001; Volume 1, Available online: https://books.google.ae/books?id=tZdjQgAACAAJ (accessed on 1 August 2022).
- Kumar, S.; Kumar, R.; Kumari, M.; Kumari, R.; Saha, S.; Bhavesh, N.S.; Maiti, T.K. Ellagic Acid Inhibits α-Synuclein Aggregation at Multiple Stages and Reduces Its Cytotoxicity. ACS Chem. Neurosci. 2021, 12, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Baluchnejadmojarad, T.; Rabiee, N.; Zabihnejad, S.; Roghani, M. Ellagic acid exerts protective effect in intrastriatal 6-hydroxydopamine rat model of Parkinson’s disease: Possible involvement of ERβ/Nrf2/HO-1 signaling. Brain Res. 2017, 1662, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef]
- Park, H.-A.; Ellis, A.C. Dietary Antioxidants and Parkinson’s Disease. Antioxidants 2020, 9, 570. [Google Scholar] [CrossRef]
- Romeo, I.; Vallarino, G.; Turrini, F.; Roggeri, A.; Olivero, G.; Boggia, R.; Alcaro, S.; Costa, G.; Pittaluga, A. Presynaptic Release-Regulating Alpha2 Autoreceptors: Potential Molecular Target for Ellagic Acid Nutraceutical Properties. Antioxidants 2021, 10, 1759. [Google Scholar] [CrossRef]
- Amor, A.J.; Gómez-Guerrero, C.; Ortega, E.; Sala-Vila, A.; Lázaro, I. Ellagic Acid as a Tool to Limit the Diabetes Burden: Updated Evidence. Antioxidants 2020, 9, 1226. [Google Scholar] [CrossRef] [PubMed]
- Figueira, I.; Garcia, G.; Pimpão, R.C.; Terrasso, A.P.; Costa, I.; Almeida, A.F.; Tavares, L.; Pais, T.F.; Pinto, P.; Ventura, M.R.; et al. Polyphenols journey through blood-brain barrier towards neuronal protection. Sci. Rep. 2017, 7, 11456. [Google Scholar] [CrossRef]
- Long, J.; Guo, Y.; Yang, J.; Henning, S.M.; Lee, R.-P.; Rasmussen, A.; Zhang, L.; Lu, Q.-Y.; Heber, D.; Li, Z. Bioavailability and bioactivity of free ellagic acid compared to pomegranate juice. Food Funct. 2019, 10, 6582–6588. [Google Scholar] [CrossRef] [PubMed]
- Kasai, K.; Yoshimura, M.; Koga, T.; Arii, M.; Kawasaki, S. Effects of oral administration of ellagic acid-rich pomegranate extract on ultraviolet-induced pigmentation in the human skin. J. Nutr. Sci. Vitaminol. 2006, 52, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.; Lalooha, F.; Nooshabadi, M.R.; Dashti, F.; Kavianpour, M.; Haghighian, H.K. Randomized double blind clinical trial evaluating the Ellagic acid effects on insulin resistance, oxidative stress and sex hormones levels in women with polycystic ovarian syndrome. J. Ovarian Res. 2021, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Hajiluian, G.; Karegar, S.J.; Shidfar, F.; Aryaeian, N.; Salehi, M.; Lotfi, T.; Farhangnia, P.; Heshmati, J.; Delbandi, A.-A. The effects of Ellagic acid supplementation on neurotrophic, inflammation, and oxidative stress factors, and indoleamine 2, 3-dioxygenase gene expression in multiple sclerosis patients with mild to moderate depressive symptoms: A randomized, triple-blind, placebo-controlled trial. Phytomedicine 2023, 121, 155094. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, S.; Wang, F.; Yu, H.; Li, X.; Dong, W.; Lin, R.; Liu, Q. Chronic administration of ellagic acid improved the cognition in middle-aged overweight men. Appl. Physiol. Nutr. Metab. 2018, 43, 266–273. [Google Scholar] [CrossRef]
- González-Sarrías, A.; García-Villalba, R.; Núñez-Sánchez, M.Á.; Tomé-Carneiro, J.; Zafrilla, P.; Mulero, J.; Tomás-Barberán, F.A.; Espín, J.C. Identifying the limits for ellagic acid bioavailability: A crossover pharmacokinetic study in healthy volunteers after consumption of pomegranate extracts. J. Funct. Foods 2015, 19, 225–235. [Google Scholar] [CrossRef]
- Lei, F.; Xing, D.M.; Xiang, L.; Zhao, Y.N.; Wang, W.; Zhang, L.J.; Du, L.J. Pharmacokinetic study of ellagic acid in rat after oral administration of pomegranate leaf extract. J. Chromatogr. B 2003, 796, 189–194. [Google Scholar] [CrossRef]
- Zuccari, G.; Baldassari, S.; Ailuno, G.; Turrini, F.; Alfei, S.; Caviglioli, G. Formulation Strategies to Improve Oral Bioavailability of Ellagic Acid. Appl. Sci. 2020, 10, 3353. [Google Scholar] [CrossRef]
- Machiya, Y.; Hara, S.; Arawaka, S.; Fukushima, S.; Sato, H.; Sakamoto, M.; Koyama, S.; Kato, T. Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J. Biol. Chem. 2010, 285, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.-S.Z.; Sun, Y.; Pearce, A.C.; Eleuteri, S.; Kemp, M.; Luckhurst, C.A.; Williams, R.; Mills, R.; Almond, S.; Burzynski, L.; et al. Knockout or inhibition of USP30 protects dopaminergic neurons in a Parkinson’s disease mouse model. Nat. Commun. 2023, 14, 7295. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Park. Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Dahodwala, N.; Shah, K.; He, Y.; Wu, S.S.; Schmidt, P.; Cubillos, F.; Willis, A.W. Sex disparities in access to caregiving in Parkinson disease. Neurology 2018, 90, e48–e54. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Host Species/Cat. No. | Source | Dilution | Assay |
|---|---|---|---|---|
| Primary Antibodies | ||||
| DAT | Rat/TEMECULA MAB369 | Merck, Burlington, MA, USA | 1:1000 | DAB |
| TH | Mouse/Immuno star 22941 | ImmunoStar, Hudson, Wisonsin, USA | 1:1000 | DAB/IF |
| pS129 α-syn | Rabbit/ab59264 | Abcam, Waltham, MA, USA | 1:1000 | IF |
| Conformational α-syn | Rabbit/ab209538 | Abcam, Waltham, MA, USA | 1:1000 | IF |
| LC3A/B | Rabbit/CST 12741 | Cell Signaling Technology, Inc., Danvers, MA. USA | 1:500 | IF |
| p62 | Mouse/ab56416 | Abcam, Waltham, MA, USA | 1:500 | IF |
| Secondary Antibodies | ||||
| Biotin-sp-conjugated | Donkey anti-Rat/Jackson Immuno Research 712-065-153 | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 1:1000 | DAB |
| Biotin-sp-conjugated | Donkey anti-mouse/Jackson Immuno Research 715-065-150 | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 1:1000 | DAB |
| Alexa Fluor 594 | Goat anti-mouse/Invitrogen; A11032 | Thermo Fisher Scientific Pierce Biotechnology, Rockford, IL, USA | 1:1000 | IF |
| Alexa Fluor 488 | Goat anti-rabbit/Invitrogen; A11034 | Thermo Fisher Scientific Pierce Biotechnology, Rockford, IL, USA | 1:1000 | IF |
| Tertiary Antibody | ||||
| Streptavidin-horseradish Peroxidase conjugate | AntiDonkey/AmershamTM; RPN1231-2ML | Cytiva, Buckinghamshire, UK | 1:200 | DAB |
| Antibodies | Host Species/Cat. No. | Dilution |
|---|---|---|
| TH | Mouse/Immuno star 22941 | 1:1000 |
| pS129 α-syn | Rabbit/ab59264 | 1:1000 |
| LC3A/B | Rabbit/CST 12741 | 1:1000 |
| p62 | Mouse/ab56416 | 1:1000 |
| GAPDH | Rabbit/CST 2118 | 1:1000 |
| Actin | Mouse/MAB1501R | 1:1000 |
| Goat Anti-rabbit | Goat/Jacson Immuno Research 111-035-144 | 1:10,000 |
| Goat Anti-mouse | Goat/Jacson Immuno Research 115-035-166 | 1:10,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radwan, N.; Khan, E.; Ardah, M.T.; Kitada, T.; Haque, M.E. Ellagic Acid Prevents α-Synuclein Spread and Mitigates Toxicity by Enhancing Autophagic Flux in an Animal Model of Parkinson’s Disease. Nutrients 2024, 16, 85. https://doi.org/10.3390/nu16010085
Radwan N, Khan E, Ardah MT, Kitada T, Haque ME. Ellagic Acid Prevents α-Synuclein Spread and Mitigates Toxicity by Enhancing Autophagic Flux in an Animal Model of Parkinson’s Disease. Nutrients. 2024; 16(1):85. https://doi.org/10.3390/nu16010085
Chicago/Turabian StyleRadwan, Nada, Engila Khan, Mustafa T. Ardah, Tohru Kitada, and M. Emdadul Haque. 2024. "Ellagic Acid Prevents α-Synuclein Spread and Mitigates Toxicity by Enhancing Autophagic Flux in an Animal Model of Parkinson’s Disease" Nutrients 16, no. 1: 85. https://doi.org/10.3390/nu16010085
APA StyleRadwan, N., Khan, E., Ardah, M. T., Kitada, T., & Haque, M. E. (2024). Ellagic Acid Prevents α-Synuclein Spread and Mitigates Toxicity by Enhancing Autophagic Flux in an Animal Model of Parkinson’s Disease. Nutrients, 16(1), 85. https://doi.org/10.3390/nu16010085