
Molecular Mechanism of Tocotrienol-Mediated Anticancer Properties: A Systematic Review of the Involvement of Endoplasmic Reticulum Stress and Unfolded Protein Response



Abstract
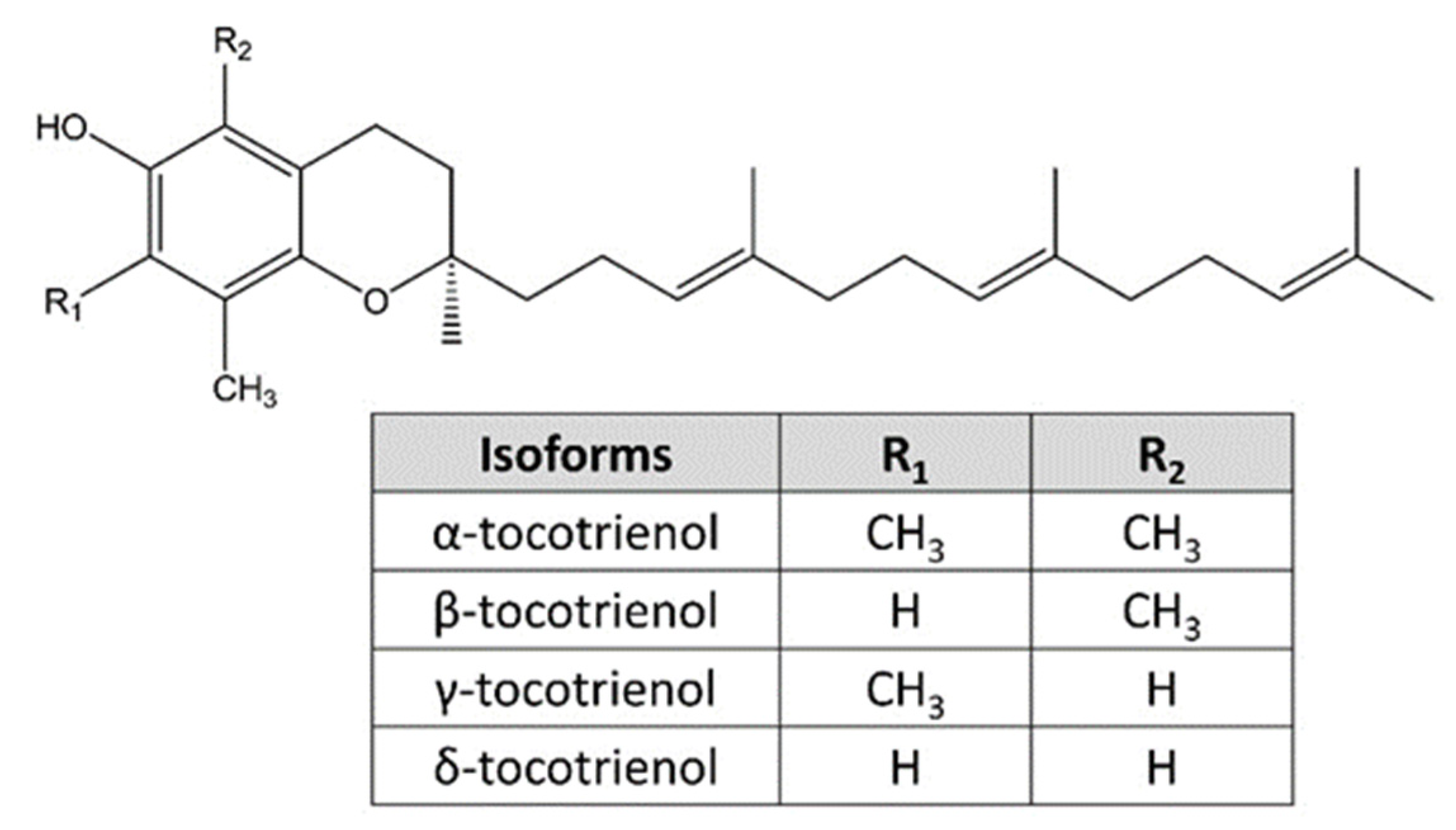
1. Introduction
2. Materials and Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Treatment | Models | Major Findings | OHAT Tier |
|---|---|---|---|---|
| Wali et al. [43] | γ-tocotrienol (0–40 μM, 24 h treatment) | Mammary tumor +SA cells | γ-tocotrienol induced concentration- and time-dependent cell death with the upregulation of ERS response signaling proteins (phosphor-PERK, phosphor-eIF2α, ATF4, ATF6α), cleaved caspase-12 and ERS-related cell death proteins (CHOP & TRB3, but not Grp78). Tocotrienol-mediated ERS was independent of Grp78 and the mevalonate pathway. | 1 |
| Park et al. [44] | α-, δ- & γ-tocotrienols (0–40 μM, 24 h treatment) | Murine mammary tumour 66cl-4-GFP cells, human mammary tumour MCF-7, MDA-MB-231 and MDA-MB-468 cells | δ- & γ-tocotrienols exerted potent anti-cancer activities as compared to α-tocotrienol. γ-tocotrienol induced mammary tumor cell apoptosis in JNK- & p38-mediated CHOP and DR5-dependent manner. ERS was involved in the upstream mechanism of tocotrienol-induced apoptosis as evidenced by the upregulation of ATF4, CHOP, and Grp78 levels; and Xbp-1 mRNA splicing. ERS inhibitor (salubrinal) protected the cells from γ-tocotrienol-induced MAPK activation and apoptosis. | 1 |
| Gopalan et al. [45] | γ-tocotrienol (0–10 μM, 24–72 h treatment) | Human mammary tumor MCF-7 and MDA-MB-435 cells | γ-tocotrienol was more potent than γ-tocopherol. γ-tocotrienol induced mammary tumor cell apoptosis with caspases activation, PARP cleavage, JNK activation, and upregulation of DR5 and CHOP levels. γ-tocotrienol increased the intracellular ceramide and dihydroceramide levels. De novo ceramide synthesis inhibitor protected the cells from tocotrienol-mediated apoptosis, JNK activation, DR5 and CHOP upregulation, and caspases activation. | 1 |
| Patacsil et al. [46] | α- & γ-tocotrienols (0–80 μM, 24–72 h treatment) | Human mammary tumor MCF-7 and MDA-MB-231 cells, and non-cancerous human mammary MCF-10A cells | γ-tocotrienol was more potent than α-tocotrienol. γ-tocotrienol induced mammary tumor cell G1 arrest and apoptosis. Transcriptomic analysis revealed the involvement of ERS response and UPR pathways. γ-tocotrienol upregulated the Grp78, ATF3 and CHOP levels with ERS markers (ATF4, phosphor-PERK, phosphor-IRE1α & eIF2α but not ATF6). | 1 |
| Xiong et al. [47] | γ-tocotrienol (0–20 μM, 24 h treatment) | Human mammary tumor MDA-MB-231 and SUM159 cells | γ-tocotrienol induced mammary tumor cell apoptosis with the upregulation of Grp78, CHOP & DR5 levels. | 1 |
| Tuerdi et al. [48] | γ-tocotrienol (20 μM, 24–48 h treatment) | Human malignant mesothelioma H2052, H28, H242 and MSTO-211H cells | γ-tocotrienol induced malignant mesothelioma cell death with the increase in CHOP, Grp78, and caspase-4 mRNA levels. | 1 |
| Tiwari et al. [49] | γ-tocotrienol (40 μM, 6–24 h treatment) | Human mammary tumour MCF-7 and MDA-MB-231 cells, and non-cancerous human mammary MCF-10A cells | γ-tocotrienol induced mammary tumor cell apoptosis and autophagy with JNK & p38 (but not ERK) activation and early upregulation of Grp78, TRB3, CHOP and ERS markers (IRE1α, phosphor-PERK, phosphor-eIF2α ATF4). | 1 |
| Comitato et al. [50] | TRF, α-, δ- & γ-tocotrienols (5–20 μg/mL, 24–48 h treatment) * 12.6–50.4 μM (δ-tocotrienol) and 12.2–48.7 μM (γ-tocotrienol) | Human cervical tumour HeLa cells and human mammary tumour MCF-7 cells without oestrogen receptor | α-, δ- & γ-tocotrienols (but not TRF) induced the release of endoplasmic reticulum calcium ions into the cytosol. δ- & γ-tocotrienols upregulated the Xbp-1 and CHOP mRNA levels, upregulated Grp78 protein level, and ERβ-independent Xbp-1 alternative splicing and caspase-12 activation. Tocotrienols (especially δ-tocotrienol) induced IRE1α phosphorylation but not ATF6 and PERK phosphorylation. | 1 |
| Marelli et al. [51] | δ-tocotrienol (5–20 μg/mL, 24–48 h treatment) * 12.6–50.4 μM | Human melanoma BLM and A375 cells, and human primary melanocytes Melanoma-xenograft nude mice model was used but no contribution to the mechanistic findings | δ-tocotrienol induced cytotoxicity and apoptosis on melanoma cells but not on non-cancerous melanocytes. δ-tocotrienol activated the caspase 4 and upregulated the ERS markers (Grp78, PERK, phosphor- eIF2α & IRE1α) and ERS-related apoptosis markers (ATF4, CHOP & ERO1α). δ-tocotrienol induced nuclear translocation of CHOP and ATF4 and upregulated the CHOP and IRE1α mRNA. Salubrinal protected the melanoma cells from δ-tocotrienol-induced ERS-mediated apoptosis. | 1 |
| Fontana et al. [52] | δ-tocotrienol (0–20 μg/mL, 24–72 h treatment) * 0–50.4 μM | Human prostate tumour DU145 and PC3 cells, and non-cancerous human prostate epithelial RWPE-1 cells | δ-tocotrienol induced cytotoxicity, apoptosis and autophagy on prostate cancer cells but not on non-cancerous melanocytes. δ-tocotrienol upregulated ERS markers (Grp78, phosphor-eIF2α & IRE1α) and ERS-related apoptosis markers (ATF4 & CHOP). Salubrinal and 4-phenylbutyrate protected the prostate tumour cells from δ-tocotrienol-induced ERS-mediated apoptosis and autophagy. | 1 |
| Ambra et al. [53] | δ- & γ-tocotrienols (5–20 μg/mL, 24 h treatment) * 12.6–50.4 μM (δ-tocotrienol) and 12.2–48.7 μM (γ-tocotrienol) | Human cervical tumour HeLa cells | γ-tocotrienol significantly upregulated 3 miRNAs including miR-190b, miR-215 and miR-148a. δ- & γ-tocotrienols induced Xbp1 alternative splicing via miR-190b. Anti-miR-190b suppressed while miR-190b overexpression promoted tocotrienol-induced apoptosis. | 1 |
3. Results
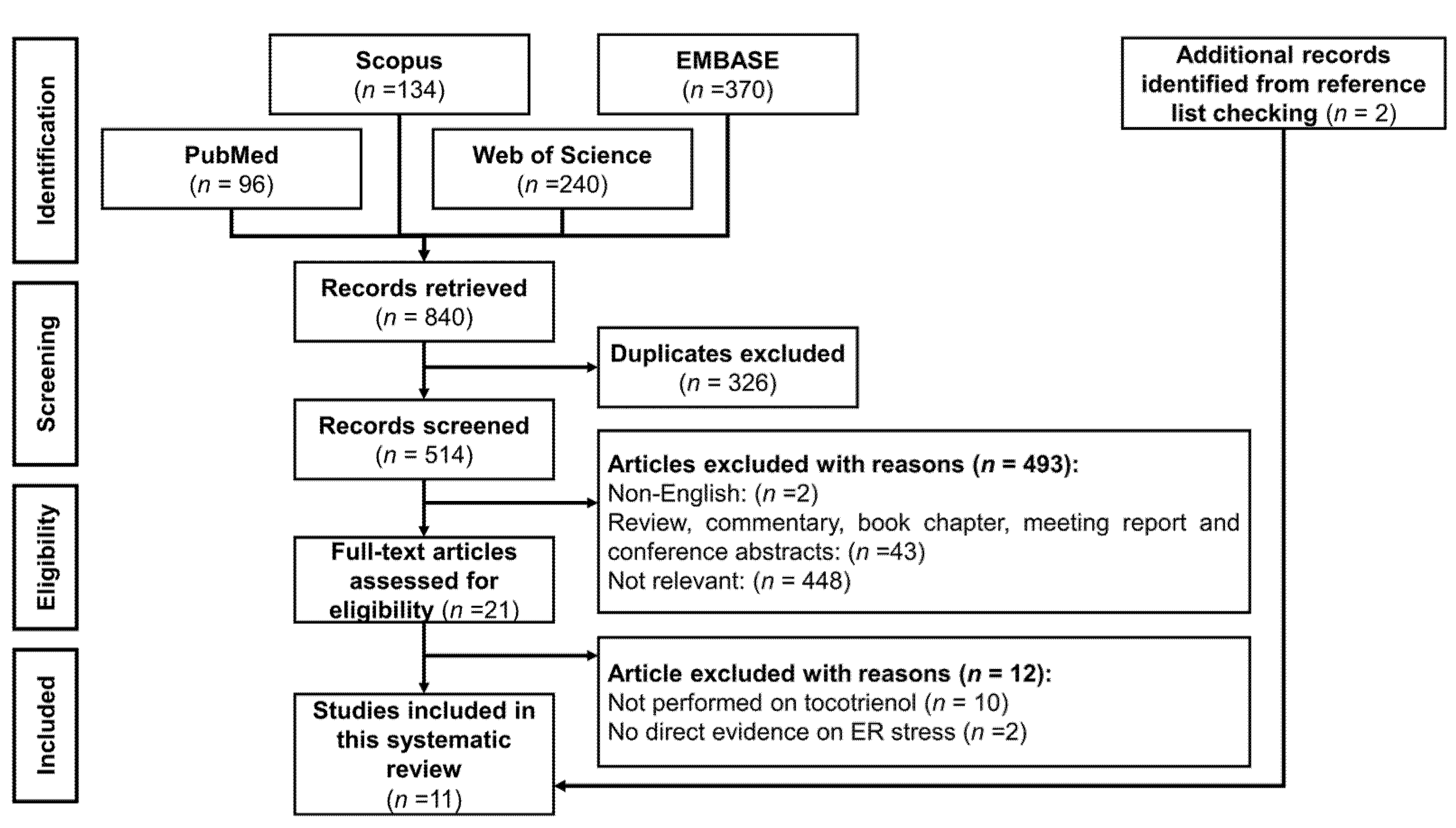
3.1. Selection of Articles
3.2. Study Characteristics
3.3. Anti-Cancer Properties of Tocotrienols
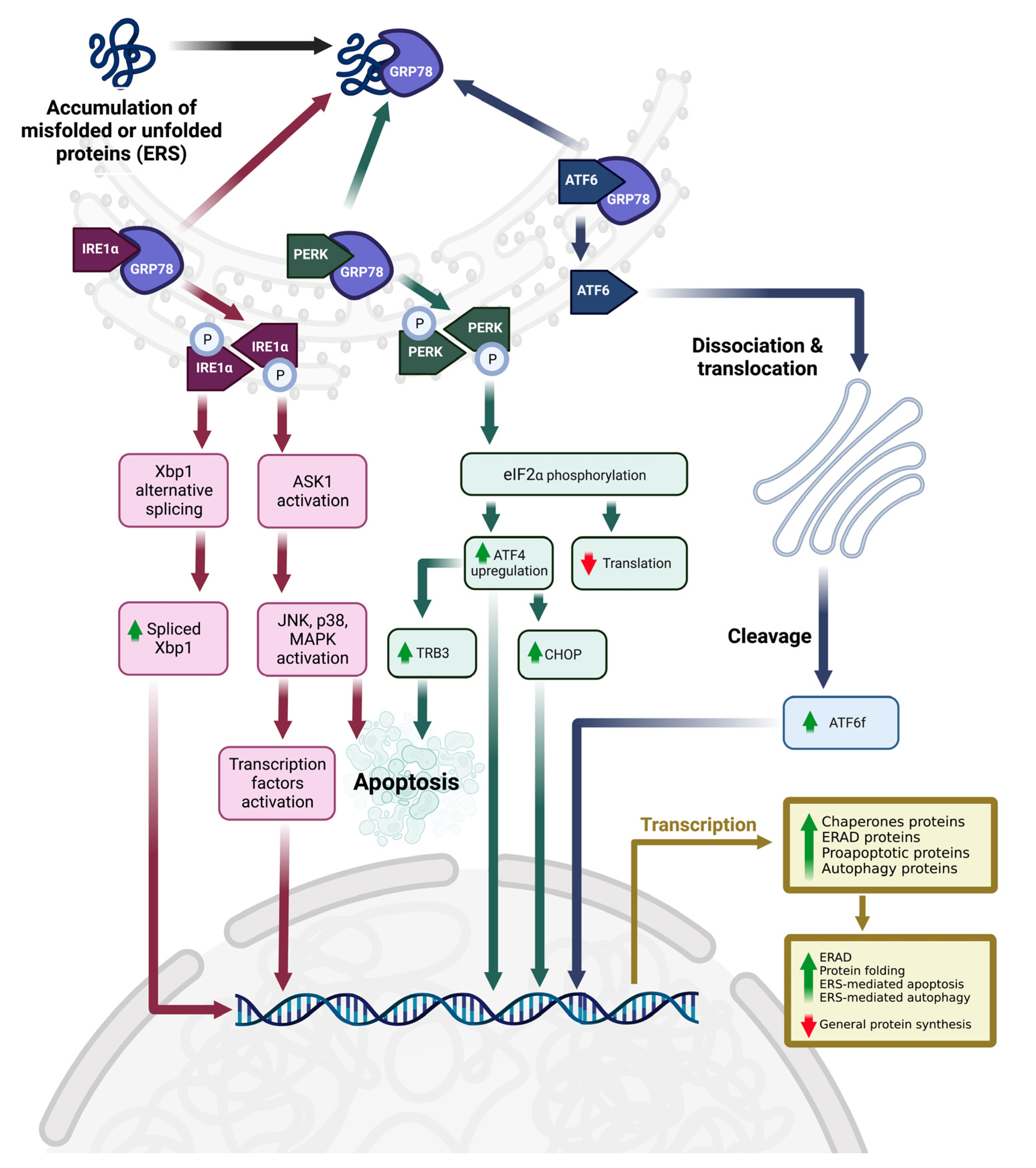
3.4. ERS, UPR, and Upstream Molecular Mechanism
4. Discussion
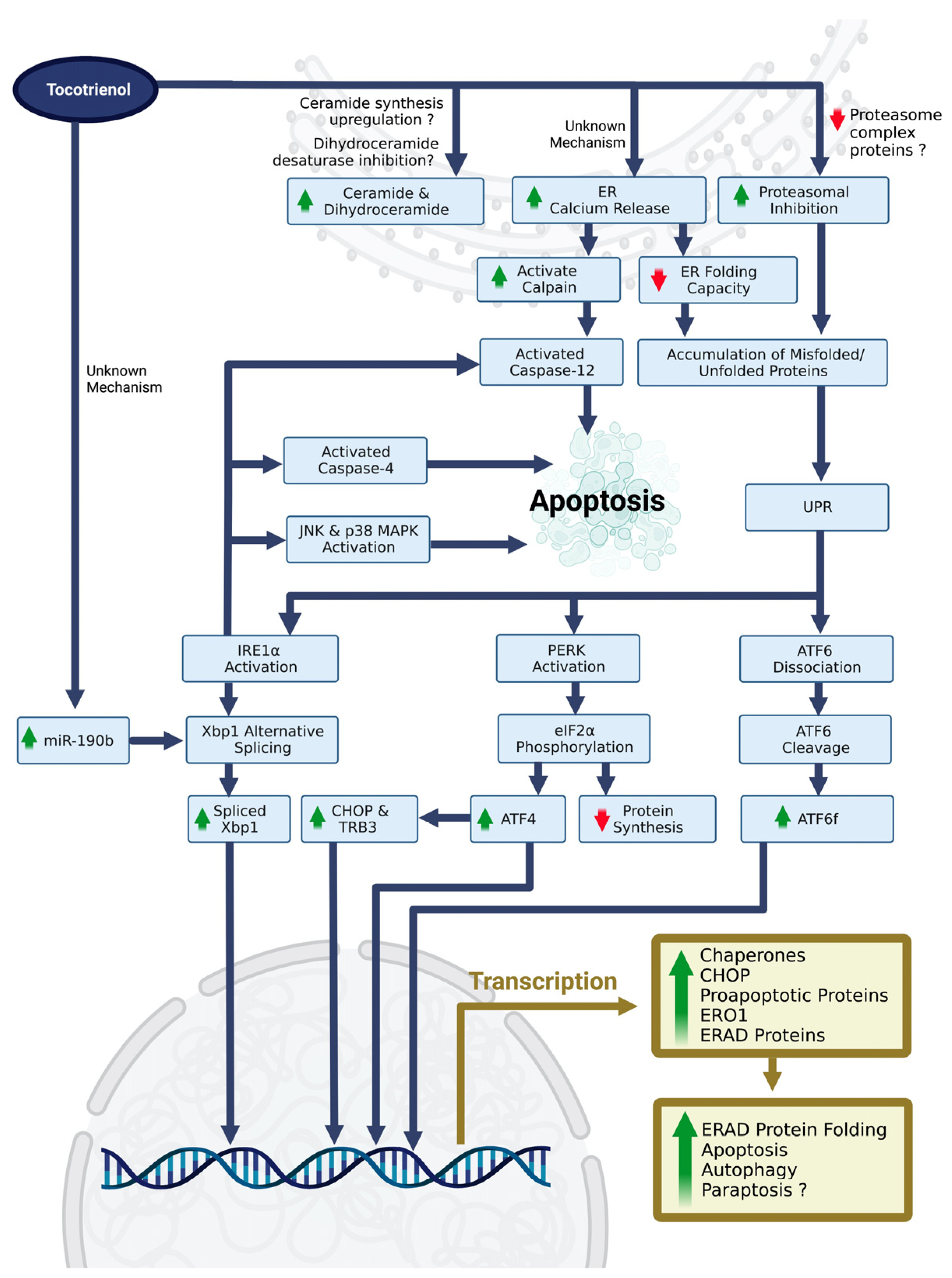
4.1. Tocotrienol-Induced ERS and ERS-Related Cell Death
4.2. Contradicting Findings in Tocotrienol-Mediated ERS
4.3. Novel Approaches in Studying Tocotrienol-Mediated ERS
4.4. Current Understanding of the Upstream Molecular Mechanisms of Tocotrienol-Mediated ERS
4.5. ERS-Inducing Properties of Vitamin E Analogues
4.6. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanvictores, T.; Davis, D.D. Histology, Rough Endoplasmic Reticulum. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mecha, M.F.; Hutchinson, R.B.; Lee, J.H.; Cavagnero, S. Protein folding in vitro and in the cell: From a solitary journey to a team effort. Biophys. Chem. 2022, 287, 106821. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Cohen, R.A.; Tong, X. Endoplasmic Reticulum Stress and Related Pathological Processes. J. Pharmacol. Biomed. Anal. 2013, 1, 1000107. [Google Scholar]
- Sicari, D.; Delaunay-Moisan, A.; Combettes, L.; Chevet, E.; Igbaria, A. A guide to assessing endoplasmic reticulum homeostasis and stress in mammalian systems. FEBS J. 2020, 287, 27–42. [Google Scholar] [CrossRef]
- Enogieru, A.B.; Omoruyi, S.I.; Hiss, D.C.; Ekpo, O.E. GRP78/BIP/HSPA5 as a Therapeutic Target in Models of Parkinson’s Disease: A Mini Review. Adv. Pharmacol. Sci. 2019, 2019, 2706783. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Li, G.; Mongillo, M.; Chin, K.-T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α–mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef]
- Seervi, M.; Sobhan, P.K.; Joseph, J.; Ann Mathew, K.; Santhoshkumar, T.R. ERO1α-dependent endoplasmic reticulum–mitochondrial calcium flux contributes to ER stress and mitochondrial permeabilization by procaspase-activating compound-1 (PAC-1). Cell Death Dis. 2013, 4, e968. [Google Scholar] [CrossRef] [PubMed]
- Avery, J.; Etzion, S.; DeBosch, B.J.; Jin, X.; Lupu, T.S.; Beitinjaneh, B.; Grand, J.; Kovacs, A.; Sambandam, N.; Muslin, A.J. TRB3 Function in Cardiac Endoplasmic Reticulum Stress. Circ. Res. 2010, 106, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, K.; Chojnacka, A.; Górnicka, M. Tocopherols and Tocotrienols—Bioactive Dietary Compounds; What Is Certain, What Is Doubt? Int. J. Mol. Sci. 2021, 22, 6222. [Google Scholar] [CrossRef]
- Elnagar, A.Y.; Wali, V.B.; Sylvester, P.W.; El Sayed, K.A. Design and preliminary structure–activity relationship of redox-silent semisynthetic tocotrienol analogues as inhibitors for breast cancer proliferation and invasion. Bioorg. Med. Chem. 2010, 18, 755–768. [Google Scholar] [CrossRef]
- Smith, L.I.; Ungnade, H.E.; Prichard, W.W. The Chemistry of Vitamin E. I. The Structure and Synthesis of α-Tocopherol. Science 1938, 88, 37–38. [Google Scholar] [CrossRef]
- Mishima, K.; Tanaka, T.; Pu, F.; Egashira, N.; Iwasaki, K.; Hidaka, R.; Matsunaga, K.; Takata, J.; Karube, Y.; Fujiwara, M. Vitamin E isoforms α-tocotrienol and γ-tocopherol prevent cerebral infarction in mice. Neurosci. Lett. 2003, 337, 56–60. [Google Scholar] [CrossRef]
- Ahsan, H.; Ahad, A.; Siddiqui, W.A. A review of characterization of tocotrienols from plant oils and foods. J. Chem. Biol. 2015, 8, 45–59. [Google Scholar] [CrossRef]
- Chun, J.; Lee, J.; Ye, L.; Exler, J.; Eitenmiller, R.R. Tocopherol and tocotrienol contents of raw and processed fruits and vegetables in the United States diet. J. Food Compos. Anal. 2006, 19, 196–204. [Google Scholar] [CrossRef]
- Fu, J.-Y.; Che, H.-L.; Tan, D.M.-Y.; Teng, K.-T. Bioavailability of tocotrienols: Evidence in human studies. Nutr. Metab. 2014, 11, 5. [Google Scholar] [CrossRef]
- Tejpal Singh, H.S.; Aminuddin, A.A.; Pang, K.-L.; Ekeuku, S.O.; Chin, K.-Y. The Role of Tocotrienol in Arthritis Management—A Scoping Review of Literature. Pharmaceuticals 2023, 16, 385. [Google Scholar] [CrossRef]
- Ranasinghe, R.; Mathai, M.; Zulli, A. Revisiting the therapeutic potential of tocotrienol. Biofactors 2022, 48, 813–856. [Google Scholar] [CrossRef]
- Wong, S.K.; Kamisah, Y.; Mohamed, N.; Muhammad, N.; Masbah, N.; Mohd Fahami, N.A.; Mohamed, I.N.; Shuid, A.N.; Mohd Saad, Q.; Abdullah, A.; et al. Potential Role of Tocotrienols on Non-Communicable Diseases: A Review of Current Evidence. Nutrients 2020, 12, 259. [Google Scholar] [CrossRef]
- Pang, K.-L.; Chin, K.-Y. The Role of Tocotrienol in Protecting Against Metabolic Diseases. Molecules 2019, 24, 923. [Google Scholar] [CrossRef]
- Parker, R.A.; Pearce, B.C.; Clark, R.W.; Gordon, D.A.; Wright, J.J. Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 1993, 268, 11230–11238. [Google Scholar] [CrossRef]
- Ramanathan, N.; Tan, E.; Loh, L.; Soh, B.S.; Yap, W.N. Tocotrienol is a cardioprotective agent against ageing-associated cardiovascular disease and its associated morbidities. Nutr. Metab. 2018, 15, 6. [Google Scholar] [CrossRef]
- Wan Hasan, W.N.; Chin, K.Y.; Abd Ghafar, N.; Soelaiman, I.N. Annatto-Derived Tocotrienol Promotes Mineralization of MC3T3-E1 Cells by Enhancing BMP-2 Protein Expression via Inhibiting RhoA Activation and HMG-CoA Reductase Gene Expression. Drug Des. Dev. Ther. 2020, 14, 969–976. [Google Scholar] [CrossRef]
- Aggarwal, V.; Kashyap, D.; Sak, K.; Tuli, H.S.; Jain, A.; Chaudhary, A.; Garg, V.K.; Sethi, G.; Yerer, M.B. Molecular Mechanisms of Action of Tocotrienols in Cancer: Recent Trends and Advancements. Int. J. Mol. Sci. 2019, 20, 656. [Google Scholar] [CrossRef]
- Pang, K.L.; Foong, L.C.; Abd Ghafar, N.; Soelaiman, I.N.; Law, J.X.; Leong, L.M.; Chin, K.Y. Transcriptomic Analysis of the Anticancer Effects of Annatto Tocotrienol, Delta-Tocotrienol and Gamma-Tocotrienol on Chondrosarcoma Cells. Nutrients 2022, 14, 4277. [Google Scholar] [CrossRef]
- Comitato, R.; Ambra, R.; Virgili, F. Tocotrienols: A Family of Molecules with Specific Biological Activities. Antioxidants 2017, 6, 93. [Google Scholar] [CrossRef]
- Nesaretnam, K.; Meganathan, P.; Veerasenan, S.D.; Selvaduray, K.R. Tocotrienols and breast cancer: The evidence to date. Genes Nutr. 2012, 7, 3–9. [Google Scholar] [CrossRef]
- Trujillo, M.; Kharbanda, A.; Corley, C.; Simmons, P.; Allen, A.R. Tocotrienols as an Anti-Breast Cancer Agent. Antioxidants 2021, 10, 1383. [Google Scholar] [CrossRef]
- Zarogoulidis, P.; Cheva, A.; Zarampouka, K.; Huang, H.; Li, C.; Huang, Y.; Katsikogiannis, N.; Zarogoulidis, K. Tocopherols and tocotrienols as anticancer treatment for lung cancer: Future nutrition. J. Thorac. Dis. 2013, 5, 349–352. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sundaram, C.; Prasad, S.; Kannappan, R. Tocotrienols, the vitamin E of the 21st century: Its potential against cancer and other chronic diseases. Biochem. Pharmacol. 2010, 80, 1613–1631. [Google Scholar] [CrossRef]
- Nakatani, Y.; Shimokawa, N.; Urano, Y.; Noguchi, N.; Takagi, M. Suppression of Amyloid-β Adsorption on Endoplasmic Reticulum Stress-Mimicking Membranes by α-Tocopherol and α-Tocotrienol. J. Phys. Chem. Lett. 2022, 13, 11955–11960. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Montagnani Marelli, M. Role of Endoplasmic Reticulum Stress in the Anticancer Activity of Natural Compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef]
- Kim, C.; Kim, B. Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. BMJ 2009, 339, b2535. [Google Scholar] [CrossRef]
- Office of Health Assessment and Translation. Risk of Bias Tool. Available online: https://ntp.niehs.nih.gov/whatwestudy/assessments/noncancer/riskbias/index.html (accessed on 15 March 2023).
- Romeo, S.; Zeni, O.; Sannino, A.; Lagorio, S.; Biffoni, M.; Scarfi, M.R. Genotoxicity of radiofrequency electromagnetic fields: Protocol for a systematic review of in vitro studies. Environ. Int. 2021, 148, 106386. [Google Scholar] [CrossRef]
- Wali, V.B.; Bachawal, S.V.; Sylvester, P.W. Endoplasmic reticulum stress mediates gamma-tocotrienol-induced apoptosis in mammary tumor cells. Apoptosis 2009, 14, 1366–1377. [Google Scholar] [CrossRef]
- Park, S.K.; Sanders, B.G.; Kline, K. Tocotrienols induce apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. Breast Cancer Res. Treat. 2010, 124, 361–375. [Google Scholar] [CrossRef]
- Gopalan, A.; Yu, W.P.; Jiang, Q.; Jang, Y.M.; Sanders, B.G.; Kline, K. Involvement of de novo ceramide synthesis in gamma-tocopherol and gamma-tocotrienol-induced apoptosis in human breast cancer cells. Mol. Nutr. Food Res. 2012, 56, 1803–1811. [Google Scholar] [CrossRef]
- Patacsil, D.; Tran, A.T.; Cho, Y.S.; Suy, S.; Saenz, F.; Malyukova, I.; Ressom, H.; Collins, S.P.; Clarke, R.; Kumar, D. Gamma-tocotrienol induced apoptosis is associated with unfolded protein response in human breast cancer cells. J. Nutr. Biochem. 2012, 23, 93–100. [Google Scholar] [CrossRef]
- Xiong, A.; Yu, W.; Tiwary, R.; Sanders, B.G.; Kline, K. Distinct roles of different forms of vitamin E in DHA-induced apoptosis in triple-negative breast cancer cells. Mol. Nutr. Food Res. 2012, 56, 923–934. [Google Scholar] [CrossRef]
- Tuerdi, G.; Ichinomiya, S.; Sato, H.; Siddig, S.; Suwa, E.; Iwata, H.; Yano, T.; Ueno, K. Synergistic effect of combined treatment with gamma-tocotrienol and statin on human malignant mesothelioma cells. Cancer Lett. 2013, 339, 116–127. [Google Scholar] [CrossRef]
- Tiwari, R.V.; Parajuli, P.; Sylvester, P.W. γ-Tocotrienol-induced endoplasmic reticulum stress and autophagy act concurrently to promote breast cancer cell death. Biochem. Cell Biol. 2015, 93, 306–320. [Google Scholar] [CrossRef]
- Comitato, R.; Guantario, B.; Leoni, G.; Nesaretnam, K.; Ronci, M.B.; Canali, R.; Virgili, F. Tocotrienols induce endoplasmic reticulum stress and apoptosis in cervical cancer cells. Genes Nutr. 2016, 11, 32. [Google Scholar] [CrossRef]
- Marelli, M.M.; Marzagalli, M.; Moretti, R.M.; Beretta, G.; Casati, L.; Comitato, R.; Gravina, G.L.; Festuccia, C.; Limonta, P. Vitamin E delta-tocotrienol triggers endoplasmic reticulum stress-mediated apoptosis in human melanoma cells. Sci. Rep. 2016, 6, 30502. [Google Scholar] [CrossRef]
- Fontana, F.; Moretti, R.M.; Raimondi, M.; Marzagalli, M.; Beretta, G.; Procacci, P.; Sartori, P.; Marelli, M.M.; Limonta, P. δ-Tocotrienol induces apoptosis, involving endoplasmic reticulum stress and autophagy, and paraptosis in prostate cancer cells. Cell Prolif. 2019, 52, e12576. [Google Scholar] [CrossRef]
- Ambra, R.; Manca, S.; Leoni, G.; Guantario, B.; Canali, R.; Comitato, R. Involvement of miR-190b in Xbp1 mRNA Splicing upon Tocotrienol Treatment. Molecules 2020, 26, 163. [Google Scholar] [CrossRef]
- Idriss, M.; Hodroj, M.H.; Fakhoury, R.; Rizk, S. Beta-Tocotrienol Exhibits More Cytotoxic Effects than Gamma-Tocotrienol on Breast Cancer Cells by Promoting Apoptosis via a P53-Independent PI3-Kinase Dependent Pathway. Biomolecules 2020, 10, 577. [Google Scholar] [CrossRef]
- Gopalan, A.; Yu, W.; Sanders, B.; Kline, K. Targeting ceramide and endoplasmic reticulum-stress in vitamin E induced apoptosis in human breast cancer cells. Cancer Res. 2011, 71, 190. [Google Scholar] [CrossRef]
- McAnally, J.A.; Gupta, J.; Sodhani, S.; Bravo, L.; Mo, H. Tocotrienols potentiate lovastatin-mediated growth suppression in vitro and in vivo. Exp. Biol. Med. 2007, 232, 523–531. [Google Scholar]
- Wali, V.B.; Bachawal, S.V.; Sylvester, P.W. Suppression in mevalonate synthesis mediates antitumor effects of combined statin and gamma-tocotrienol treatment. Lipids 2009, 44, 925–934. [Google Scholar] [CrossRef]
- Wali, V.B.; Sylvester, P.W. Synergistic antiproliferative effects of gamma-tocotrienol and statin treatment on mammary tumor cells. Lipids 2007, 42, 1113–1123. [Google Scholar] [CrossRef]
- Elangovan, S.; Hsieh, T.C.; Wu, J.M. Growth inhibition of human MDA-mB-231 breast cancer cells by delta-tocotrienol is associated with loss of cyclin D1/CDK4 expression and accompanying changes in the state of phosphorylation of the retinoblastoma tumor suppressor gene product. Anticancer Res. 2008, 28, 2641–2647. [Google Scholar]
- Nesaretnam, K.; Stephen, R.; Dils, R.; Darbre, P. Tocotrienols inhibit the growth of human breast cancer cells irrespective of estrogen receptor status. Lipids 1998, 33, 461–469. [Google Scholar] [CrossRef]
- Raimondi, M.; Fontana, F.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Ca2+ overload- and ROS-associated mitochondrial dysfunction contributes to δ-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 2021, 26, 277–292. [Google Scholar] [CrossRef]
- Zhang, J.-S.; Li, D.-M.; He, N.; Liu, Y.-H.; Wang, C.-H.; Jiang, S.-Q.; Chen, B.-Q.; Liu, J.-R. A paraptosis-like cell death induced by δ-tocotrienol in human colon carcinoma SW620 cells is associated with the suppression of the Wnt signaling pathway. Toxicology 2011, 285, 8–17. [Google Scholar] [CrossRef]
- Zhang, J.-S.; Li, D.-M.; Ma, Y.; He, N.; Gu, Q.; Wang, F.-S.; Jiang, S.-Q.; Chen, B.-Q.; Liu, J.-R. γ-Tocotrienol Induces Paraptosis-Like Cell Death in Human Colon Carcinoma SW620 Cells. PLoS ONE 2013, 8, e57779. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, Y.; Yang, C.; Huang, R.; Wen, Y.; Zhang, C.; Wu, C.; Zhao, B. Swainsonine Triggers Paraptosis via ER Stress and MAPK Signaling Pathway in Rat Primary Renal Tubular Epithelial Cells. Front. Pharmacol. 2021, 12, 715285. [Google Scholar] [CrossRef] [PubMed]
- Stan, R.C.; Silva, R.L.; de Camargo, M.M. Human GRP78 affinity towards its signaling partners Ire1α and PERK is differently modulated by an unfolded protein client. Biochem. Biophys. Res. Commun. 2017, 487, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rahman, A.; Mokhtar, N.M.; Harun, R.; Jamal, R.; Wan Ngah, W.Z. Transcriptome analysis reveals the molecular mechanisms of combined gamma-tocotrienol and hydroxychavicol in preventing the proliferation of 1321N1, SW1783, and LN18 glioma cancer cells. J. Physiol. Biochem. 2019, 75, 499–517. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Skorczyk-Werner, A.; Chiang, W.C.; Wawrocka, A.; Wicher, K.; Jarmuz-Szymczak, M.; Kostrzewska-Poczekaj, M.; Jamsheer, A.; PLoSki, R.; Rydzanicz, M.; Pojda-Wilczek, D.; et al. Autosomal recessive cone-rod dystrophy can be caused by mutations in the ATF6 gene. Eur. J. Hum. Genet. 2017, 25, 1210–1216. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, S.; Yang, G.; Zou, L.; Huang, X.; Liu, S. The Role of miRNAs during Endoplasmic Reticulum Stress Induced Apoptosis in Digestive Cancer. J. Cancer 2021, 12, 6787–6795. [Google Scholar] [CrossRef]
- Kim, T.; Croce, C.M. MicroRNA and ER stress in cancer. Semin. Cancer Biol. 2021, 75, 3–14. [Google Scholar] [CrossRef]
- Su, S.; Chang, Y.; Andreu-Vieyra, C.; Fang, J.; Yang, Z.; Han, B.; Lee, A.; Liang, G. miR-30d, miR-181a and miR-199a-5p cooperatively suppress the endoplasmic reticulum chaperone and signaling regulator GRP78 in cancer. Oncogene 2013, 32, 4694–4701. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhao, H.; Liu, J.; Zhang, Y.; Wang, X. miRNA-mRNA Regulatory Network Reveals miRNAs in HCT116 in Response to Folic Acid Deficiency via Regulating Vital Genes of Endoplasmic Reticulum Stress Pathway. BioMed Res. Int. 2021, 2021, 6650181. [Google Scholar] [CrossRef]
- Duan, Q.; Wang, X.; Gong, W.; Ni, L.; Chen, C.; He, X.; Chen, F.; Yang, L.; Wang, P.; Wang, D.W. ER stress negatively modulates the expression of the miR-199a/214 cluster to regulates tumor survival and progression in human hepatocellular cancer. PLoS ONE 2012, 7, e31518. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Zou, H.; Liu, X.; He, J.; Zheng, Y.; Xiong, L.; Miao, X. miR-7112-3p targets PERK to regulate the endoplasmic reticulum stress pathway and apoptosis induced by photodynamic therapy in colorectal cancer CX-1 cells. Photodiagn. Photodyn. Ther. 2020, 29, 101663. [Google Scholar] [CrossRef] [PubMed]
- Misiewicz-Krzeminska, I.; Krzeminski, P.; Corchete, L.A.; Quwaider, D.; Rojas, E.A.; Herrero, A.B.; Gutiérrez, N.C. Factors Regulating microRNA Expression and Function in Multiple Myeloma. Noncoding RNA 2019, 5, 9. [Google Scholar] [CrossRef]
- Kalimuthu, K.; Kim, J.H.; Park, Y.S.; Luo, X.; Zhang, L.; Ku, J.-L.; Choudry, M.H.A.; Lee, Y.J. Glucose deprivation-induced endoplasmic reticulum stress response plays a pivotal role in enhancement of TRAIL cytotoxicity. J. Cell. Physiol. 2021, 236, 6666–6677. [Google Scholar] [CrossRef] [PubMed]
- Dronamraju, V.; Ibrahim, B.A.; Briski, K.P.; Sylvester, P.W. γ-Tocotrienol Suppression of the Warburg Effect Is Mediated by AMPK Activation in Human Breast Cancer Cells. Nutr. Cancer 2019, 71, 1214–1228. [Google Scholar] [CrossRef]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef]
- Liu, Z.; Xia, Y.; Li, B.; Xu, H.; Wang, C.; Liu, Y.; Li, Y.; Li, C.; Gao, N.; Li, L. Induction of ER stress-mediated apoptosis by ceramide via disruption of ER Ca homeostasis in human adenoid cystic carcinoma cells. Cell Biosci. 2014, 4, 71. [Google Scholar] [CrossRef]
- Jiang, Q.; Rao, X.; Kim, C.Y.; Freiser, H.; Zhang, Q.; Jiang, Z.; Li, G. Gamma-tocotrienol induces apoptosis and autophagy in prostate cancer cells by increasing intracellular dihydrosphingosine and dihydroceramide. Int. J. Cancer 2012, 130, 685–693. [Google Scholar] [CrossRef]
- Yang, C.; Jiang, Q. Vitamin E δ-tocotrienol inhibits TNF-α-stimulated NF-κB activation by up-regulation of anti-inflammatory A20 via modulation of sphingolipid including elevation of intracellular dihydroceramides. J. Nutr. Biochem. 2019, 64, 101–109. [Google Scholar] [CrossRef]
- Chen, C.L.; Lin, C.F.; Chang, W.T.; Huang, W.C.; Teng, C.F.; Lin, Y.S. Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood 2008, 111, 4365–4374. [Google Scholar] [CrossRef]
- Jang, Y.; Rao, X.; Jiang, Q. Gamma-tocotrienol profoundly alters sphingolipids in cancer cells by inhibition of dihydroceramide desaturase and possibly activation of sphingolipid hydrolysis during prolonged treatment. J. Nutr. Biochem. 2017, 46, 49–56. [Google Scholar] [CrossRef]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Victor, P.; Sarada, D.; Ramkumar, K.M. Crosstalk between endoplasmic reticulum stress and oxidative stress: Focus on protein disulfide isomerase and endoplasmic reticulum oxidase 1. Eur. J. Pharmacol. 2021, 892, 173749. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Abd Manan, N.; Mohamed, N.; Shuid, A.N. Effects of Low-Dose versus High-Dose γ-Tocotrienol on the Bone Cells Exposed to the Hydrogen Peroxide-Induced Oxidative Stress and Apoptosis. Evid. Based Complement. Altern. Med. 2012, 2012, 680834. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Fusegi, M.; Mori, T.; Teshima, K.; Ninomiya, N.; Kohno, K.; Sato, A.; Ishida, T.; Miyakoshi, Y.; Yano, T. A Redox-Silent Analogue of Tocotrienol May Break the Homeostasis of Proteasomes in Human Malignant Mesothelioma Cells by Inhibiting STAT3 and NRF1. Int. J. Mol. Sci. 2022, 23, 2655. [Google Scholar] [CrossRef]
- Ramdas, P.; Radhakrishnan, A.K.; Abdu Sani, A.A.; Kumari, M.; Anandha Rao, J.S.; Abdul-Rahman, P.S. Advancing the Role of Gamma-Tocotrienol as Proteasomes Inhibitor: A Quantitative Proteomic Analysis of MDA-MB-231 Human Breast Cancer Cells. Biomolecules 2019, 10, 19. [Google Scholar] [CrossRef]
- Blair, C.A.; Hu, H.Z.; Huynh, T.; Wu, M.; Yang, C.S.; Zi, X.L. Delta-tocopherol induced endoplasmic reticulum stress causes autophagic degradation of ER and cell death in bladder cancer models. Cancer Res. 2018, 78, 1255. [Google Scholar] [CrossRef]
- Jiang, Q.; Wong, J.; Fyrst, H.; Saba, J.D.; Ames, B.N. gamma-Tocopherol or combinations of vitamin E forms induce cell death in human prostate cancer cells by interrupting sphingolipid synthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 17825–17830. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Z.; Jia, L.; Zhao, Y.; Zhang, X.; Wu, K. Endoplasmic reticulum stress contributes to vitamin E succinate-induced apoptosis in human gastric cancer SGC-7901 cells. Cancer Lett. 2010, 296, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, R.; Yu, W.; Li, J.; Park, S.K.; Sanders, B.G.; Kline, K. Role of endoplasmic reticulum stress in alpha-TEA mediated TRAIL/DR5 death receptor dependent apoptosis. PLoS ONE 2010, 5, e11865. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Bradlow, B.A.; Brace, L.; Manganello, J.; Peterson, D.M.; Pearce, B.C.; Wright, J.J.; Gapor, A.; Elson, C.E. Response of hypercholesterolemic subjects to administration of tocotrienols. Lipids 1995, 30, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.A.; Pearce, B.C.; Nor, R.M.; Gapor, A.; Peterson, D.M.; Elson, C.E. Dietary α-Tocopherol Attenuates the Impact of γ-Tocotrienol on Hepatic 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Activity in Chickens. J. Nutr. 1996, 126, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, C.; Hyatt, J.A.; Vraka, P.S.; Papas, A.; Papas, K.A.; Neophytou, C.; Hadjivassiliou, V.; Constantinou, A.I. Induction of caspase-independent programmed cell death by vitamin E natural homologs and synthetic derivatives. Nutr. Cancer 2009, 61, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Kaneko, S.; Sato, A.; Virgona, N.; Namiki, K.; Yano, T. Combination Effect of δ-Tocotrienol and γ-Tocopherol on Prostate Cancer Cell Growth. J. Nutr. Sci. Vitaminol. 2017, 63, 349–354. [Google Scholar] [CrossRef]
- Jaafar, F.; Abdullah, A.; Makpol, S. Cellular Uptake and Bioavailability of Tocotrienol-Rich Fraction in SIRT1-Inhibited Human Diploid Fibroblasts. Sci. Rep. 2018, 8, 10471. [Google Scholar] [CrossRef]
- Khor, S.C.; Razak, A.M.; Wan Ngah, W.Z.; Mohd Yusof, Y.A.; Abdul Karim, N.; Makpol, S. The Tocotrienol-Rich Fraction Is Superior to Tocopherol in Promoting Myogenic Differentiation in the Prevention of Replicative Senescence of Myoblasts. PLoS ONE 2016, 11, e0149265. [Google Scholar] [CrossRef]
- Khor, S.C.; Wan Ngah, W.Z.; Mohd Yusof, Y.A.; Abdul Karim, N.; Makpol, S. Tocotrienol-Rich Fraction Ameliorates Antioxidant Defense Mechanisms and Improves Replicative Senescence-Associated Oxidative Stress in Human Myoblasts. Oxidative Med. Cell. Longev. 2017, 2017, 3868305. [Google Scholar] [CrossRef]
- Makpol, S.; Durani, L.W.; Chua, K.H.; Mohd Yusof, Y.A.; Ngah, W.Z. Tocotrienol-rich fraction prevents cell cycle arrest and elongates telomere length in senescent human diploid fibroblasts. J. Biomed. Biotechnol. 2011, 2011, 506171. [Google Scholar] [CrossRef]
- Tan, C.M.; Najib, N.A.M.; Suhaimi, N.F.; Halid, N.A.; Cho, V.V.; Abdullah, S.I.; Ismail, M.Z.; Khor, S.C.; Jaafar, F.; Makpol, S. Modulation of Ki67 and myogenic regulatory factor expression by tocotrienol-rich fraction ameliorates myogenic program of senescent human myoblasts. Arch. Med. Sci. 2021, 17, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Then, S.-M.; Sanfeliu, C.; Top, G.M.; Ngah, W.Z.W.; Mazlan, M. γ-Tocotrienol does not substantially protect DS neurons from hydrogen peroxide-induced oxidative injury. Nutr. Metab. 2012, 9, 1. [Google Scholar] [CrossRef]
- Wan Hasan, W.N.; Abd Ghafar, N.; Chin, K.-Y.; Ima-Nirwana, S. Annatto-derived tocotrienol stimulates osteogenic activity in preosteoblastic MC3T3-E1 cells: A temporal sequential study. Drug Des. Dev. Ther. 2018, 12, 1715–1726. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.-L.; Ghafar, N.A.; Soelaiman, I.N.; Chin, K.-Y. Protective Effects of Annatto Tocotrienol and Palm Tocotrienol-Rich Fraction on Chondrocytes Exposed to Monosodium Iodoacetate. Appl. Sci. 2021, 11, 9643. [Google Scholar] [CrossRef]
- Ananthula, S.; Parajuli, P.; Behery, F.A.; Alayoubi, A.Y.; El Sayed, K.A.; Nazzal, S.; Sylvester, P.W. Oxazine Derivatives of γ- and δ-Tocotrienol Display Enhanced Anticancer Activity In Vivo. Anticancer. Res. 2014, 34, 2715. [Google Scholar]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; The PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, K.-L.; Mai, C.-W.; Chin, K.-Y. Molecular Mechanism of Tocotrienol-Mediated Anticancer Properties: A Systematic Review of the Involvement of Endoplasmic Reticulum Stress and Unfolded Protein Response. Nutrients 2023, 15, 1854. https://doi.org/10.3390/nu15081854
Pang K-L, Mai C-W, Chin K-Y. Molecular Mechanism of Tocotrienol-Mediated Anticancer Properties: A Systematic Review of the Involvement of Endoplasmic Reticulum Stress and Unfolded Protein Response. Nutrients. 2023; 15(8):1854. https://doi.org/10.3390/nu15081854
Chicago/Turabian StylePang, Kok-Lun, Chun-Wai Mai, and Kok-Yong Chin. 2023. "Molecular Mechanism of Tocotrienol-Mediated Anticancer Properties: A Systematic Review of the Involvement of Endoplasmic Reticulum Stress and Unfolded Protein Response" Nutrients 15, no. 8: 1854. https://doi.org/10.3390/nu15081854
APA StylePang, K.-L., Mai, C.-W., & Chin, K.-Y. (2023). Molecular Mechanism of Tocotrienol-Mediated Anticancer Properties: A Systematic Review of the Involvement of Endoplasmic Reticulum Stress and Unfolded Protein Response. Nutrients, 15(8), 1854. https://doi.org/10.3390/nu15081854