
Clinical Pathobiochemistry of Vitamin B12 Deficiency: Improving Our Understanding by Exploring Novel Mechanisms with a Focus on Diabetic Neuropathy

 ,
,  ,
, 
Abstract
1. Introduction
2. Biochemistry and (Patho)-Physiology
- Chemistry;
- Vitamin B12 sources, physiological uptake and causes of deficiency;
- Intracellular processing and reduction/oxidation function of B12;
- Physiological functions of Vitamin B12;
- Clinical pathophysiology of B12 deficiency.
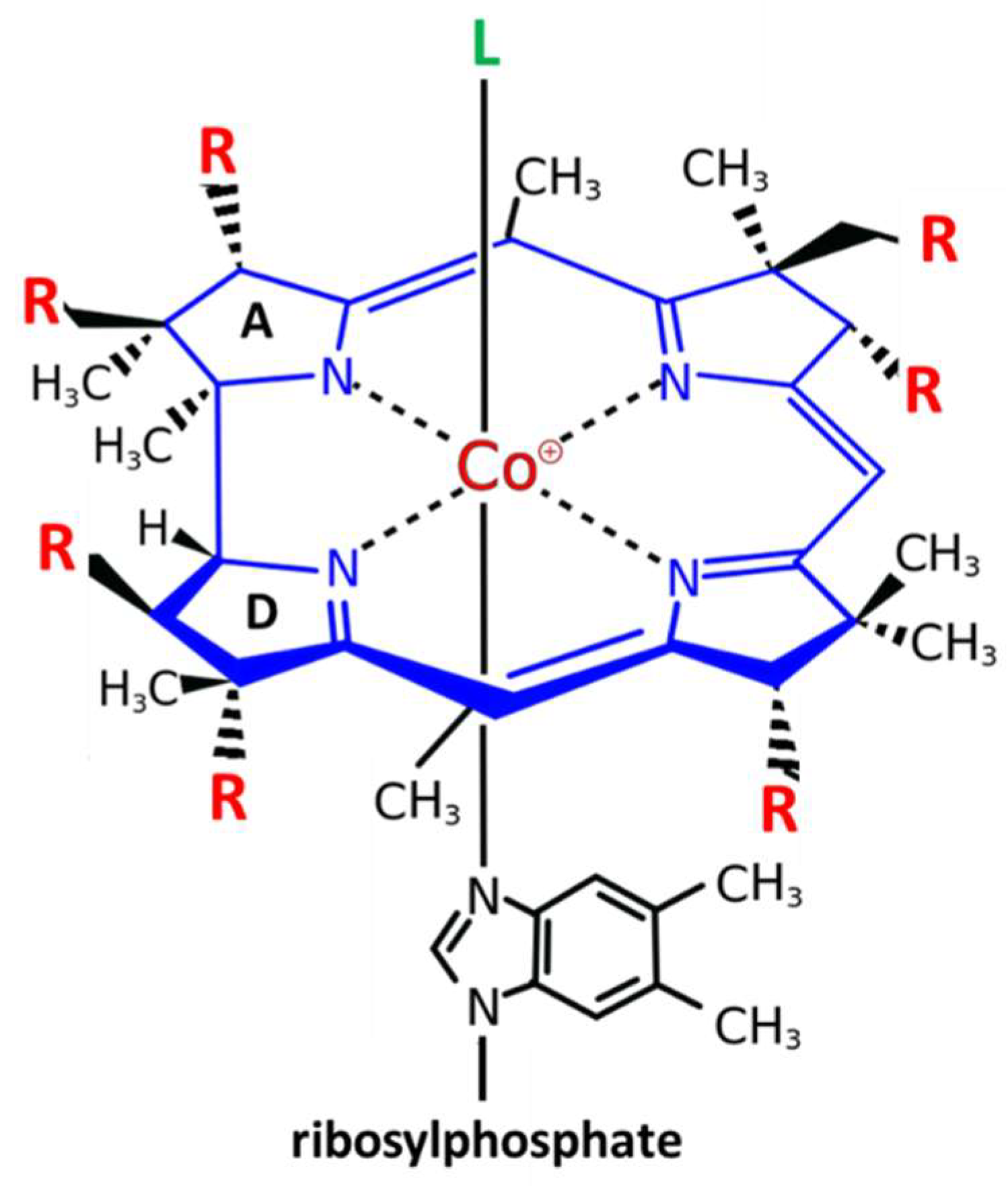
2.1. Chemistry
2.2. Vitamin B12 Sources, Physiological Uptake and Causes of Deficiency
2.3. Intracellular Processing and Reduction/Oxidation Function of B12
2.4. Physiological Functions of Vitamin B12
2.5. Clinical Pathophysiology of B12 Deficiency
3. Involvement of B12 Deficiency in the Development of Diabetic Neuropathy
- Biochemical pathways leading to diabetic neuropathy;
- Common pathophysiological pathways of B12 deficiency-induced neuropathy and diabetic neuropathy;
- B12 may act as an intracellular antioxidant.
3.1. Biochemical Pathways Leading to Diabetic Neuropathy
3.2. B12 Deficiency-Induced Neuropathy and Diabetic Neuropathy Share Common Pathophysiological Pathways
3.3. Evidence for a Role of B12 Acting as an Intracellular Antioxidant
4. Laboratory Determination of B12 Deficiency
- Biomarkers of B12 deficiency;
- Laboratory biomarkers and decision limits for B12 deficiency in the general population;
- B12 deficiency in the elderly population: special considerations and decision limits.
4.1. Biomarkers of B12 Deficiency
4.2. Laboratory Biomarkers for B12 Deficiency in the General Population and Decision Limits
4.3. B12 Deficiency in the Elderly Population
4.4. Summary of Laboratory Assessment of B12 Deficiency
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andres, E. Vitamin B12 (cobalamin) deficiency in elderly patients. Can. Med. Assoc. J. 2004, 171, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, A.; Bertuzzo, L.; Limitone, A.; D’Antona, G.; Cena, H. Perspective: Practical approach to preventing subclinical B12 deficiency in elderly population. Nutrients 2021, 13, 1913. [Google Scholar] [CrossRef]
- Allen, L.H.; Miller, J.W.; de Groot, L.; Rosenberg, I.H.; Smith, A.D.; Refsum, H.; Raiten, D.J. Biomarkers of nutrition for development (BOND): Vitamin B-12 review. J. Nutr. 2018, 148, 1995S–2027S. [Google Scholar] [CrossRef]
- Lin, Q.; Li, K.; Chen, Y.; Xie, J.; Wu, C.; Cui, C.; Deng, B. Oxidative stress in diabetic peripheral neuropathy: Pathway and mechanism-based treatment. Mol. Neurobiol. 2023, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Prim. 2019, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- DCCT Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann. Intern. Med. 1995, 122, 561. [Google Scholar] [CrossRef]
- Ishibashi, F.; Taniguchi, M.; Kosaka, A.; Uetake, H.; Tavakoli, M. Improvement in neuropathy outcomes with normalizing HbA1c in patients with type 2 diabetes. Diabetes Care 2019, 42, 110–118. [Google Scholar] [CrossRef]
- Laiteerapong, N.; Ham, S.A.; Gao, Y.; Moffet, H.H.; Liu, J.Y.; Huang, E.S.; Karter, A.J. The legacy effect in type 2 diabetes: Impact of early glycemic control on future complications (the diabetes & aging study). Diabetes Care 2019, 42, 416–426. [Google Scholar] [CrossRef]
- Bell, D.S.H. Metformin-induced vitamin B12 deficiency can cause or worsen distal symmetrical, autonomic and cardiac neuropathy in the patient with diabetes. Diabetes Obes. Metab. 2022, 24, 1423–1428. [Google Scholar] [CrossRef]
- Karedath, J.; Batool, S.; Arshad, A.; Khalique, S.; Raja, S.; Lal, B.; Chunchu, V.A.; Hirani, S. The impact of vitamin B12 supplementation on clinical outcomes in patients with diabetic neuropathy: A meta-analysis of randomized controlled trials. Cureus 2022, 14, e31783. [Google Scholar] [CrossRef]
- Solomon, L.R. Functional cobalamin (vitamin B12) deficiency: Role of advanced age and disorders associated with increased oxidative stress. Eur. J. Clin. Nutr. 2015, 69, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Gherasim, C.; Lofgren, M.; Banerjee, R. Navigating the B12 road: Assimilation, delivery, and disorders of cobalamin. J. Biol. Chem. 2013, 288, 13186–13193. [Google Scholar] [CrossRef] [PubMed]
- Office of Dietary Supplements Vitamin B12 Fact Sheet for Health Professionals. Available online: https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/#disc (accessed on 19 April 2023).
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.-L.; Brito, A.; Guéant, J.-L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.-H.; et al. Vitamin B12 deficiency. Nat. Rev. Dis. Prim. 2017, 3, 17040. [Google Scholar] [CrossRef] [PubMed]
- Lahner, E.; Norman, G.L.; Severi, C.; Encabo, S.; Shums, Z.; Vannella, L.; Fave, G.D.; Annibale, B. Reassessment of intrinsic factor and parietal cell autoantibodies in atrophic gastritis with respect to cobalamin deficiency. Am. J. Gastroenterol. 2009, 104, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Hart, H.E.; Kuijs, R.; Kooijman-Buiting, A.M.J.; Rutten, G.E.H.M. Influence of duration and dose of metformin on cobalamin deficiency in type 2 diabetes patients using metformin. Acta Diabetol. 2015, 52, 47–53. [Google Scholar] [CrossRef]
- Chapman, L.E.; Darling, A.L.; Brown, J.E. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Metab. 2016, 42, 316–327. [Google Scholar] [CrossRef]
- Infante, M.; Leoni, M.; Caprio, M.; Fabbri, A. Long-term metformin therapy and vitamin B12 deficiency: An association to bear in mind. WJD 2021, 12, 916–931. [Google Scholar] [CrossRef]
- Kim, J.; Ahn, C.W.; Fang, S.; Lee, H.S.; Park, J.S. Association between metformin dose and vitamin B12 deficiency in patients with type 2 diabetes. Medicine 2019, 98, e17918. [Google Scholar] [CrossRef]
- Lam, J.R.; Schneider, J.L.; Zhao, W.; Corley, D.A. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B 12 deficiency. JAMA 2013, 310, 2435. [Google Scholar] [CrossRef]
- Longo, S.L.; Ryan, J.M.; Sheehan, K.B.; Reid, D.J.; Conley, M.P.; Bouwmeester, C.J. Evaluation of vitamin B12 monitoring in patients on metformin in urban ambulatory care settings. Pharm. Pract. 2019, 17, 1499. [Google Scholar] [CrossRef]
- Miller, J.W. Proton pump inhibitors, H2-receptor antagonists, metformin, and vitamin B-12 deficiency: Clinical implications. Adv. Nutr. 2018, 9, 511S–518S. [Google Scholar] [CrossRef]
- de Jager, J.; Kooy, A.; Lehert, P.; Wulffele, M.G.; van der Kolk, J.; Bets, D.; Verburg, J.; Donker, A.J.M.; Stehouwer, C.D.A. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: Randomised placebo controlled trial. BMJ 2010, 340, c2181. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A. Metformin and vitamin B12 deficiency: Where do we stand? J. Pharm. Pharm. Sci. 2016, 19, 382. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.; Muntingh, G.L.; Rheeder, P. Perspectives on peripheral neuropathy as a consequence of metformin-induced vitamin B12 deficiency in T2DM. Int. J. Endocrinol. 2017, 2017, 2452853. [Google Scholar] [CrossRef] [PubMed]
- Pratama, S.; Lauren, B.C.; Wisnu, W. The efficacy of vitamin B12 supplementation for treating vitamin B12 deficiency and peripheral neuropathy in metformin-treated type 2 diabetes mellitus patients: A systematic review. Diabetes Metab. Syndr. Clin. Res. Rev. 2022, 16, 102634. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate, and the methionine remethylation cycle—Biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef]
- Fettelschoss, V.; Burda, P.; Sagné, C.; Coelho, D.; De Laet, C.; Lutz, S.; Suormala, T.; Fowler, B.; Pietrancosta, N.; Gasnier, B.; et al. Clinical or ATPase domain mutations in ABCD4 disrupt the interaction between the vitamin B12-trafficking proteins ABCD4 and LMBD1. J. Biol. Chem. 2017, 292, 11980–11991. [Google Scholar] [CrossRef] [PubMed]
- Padovani, D.; Labunska, T.; Palfey, B.A.; Ballou, D.P.; Banerjee, R. Adenosyltransferase tailors and delivers coenzyme B12. Nat. Chem. Biol. 2008, 4, 194–196. [Google Scholar] [CrossRef]
- Banerjee, R.; Gouda, H.; Pillay, S. Redox-linked coordination chemistry directs vitamin B 12 trafficking. Acc. Chem. Res. 2021, 54, 2003–2013. [Google Scholar] [CrossRef]
- Offringa, A.K.; Bourgonje, A.R.; Schrier, M.S.; Deth, R.C.; van Goor, H. Clinical implications of vitamin B12 as redox-active cofactor. Trends Mol. Med. 2021, 27, 931–934. [Google Scholar] [CrossRef]
- Huemer, M.; Baumgartner, M.R. The clinical presentation of cobalamin-related disorders: From acquired deficiencies to inborn errors of absorption and intracellular pathways. J. Inherit. Metab. Dis. 2019, 42, 686–705. [Google Scholar] [CrossRef]
- Palmer, A.M.; Kamynina, E.; Field, M.S.; Stover, P.J. Folate rescues vitamin B12 depletion-induced inhibition of nuclear thymidylate biosynthesis and genome instability. Proc. Natl. Acad. Sci. USA 2017, 114, E4095–E4102. [Google Scholar] [CrossRef]
- Scott, J. Pathogenesis of subacute combined degeneration: A result of methyl group deficiency. Lancet 1981, 318, 334–337. [Google Scholar] [CrossRef]
- Boachie, J.; Adaikalakoteswari, A.; Samavat, J.; Saravanan, P. Low vitamin B12 and lipid metabolism: Evidence from pre-clinical and clinical studies. Nutrients 2020, 12, 1925. [Google Scholar] [CrossRef]
- Groener, J.B.; Jende, J.M.E.; Kurz, F.T.; Kender, Z.; Treede, R.-D.; Schuh-Hofer, S.; Nawroth, P.P.; Bendszus, M.; Kopf, S. Understanding diabetic neuropathy—From subclinical nerve lesions to severe nerve fiber deficits: A cross-sectional study in patients with type 2 diabetes and healthy control subjects. Diabetes 2020, 69, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.A.; Tesfaye, S.; Newrick, P.G.; Walker, D.; Rajbhandari, S.M.; Siddique, I.; Sharma, A.K.; Boulton, A.J.M.; King, R.H.M.; Thomas, P.K.; et al. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia 2005, 48, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Pasnoor, M.; Dimachkie, M.M.; Kluding, P.; Barohn, R.J. Diabetic neuropathy part 1. Neurol. Clin. 2013, 31, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Zenker, J.; Ziegler, D.; Chrast, R. Novel pathogenic pathways in diabetic neuropathy. Trends Neurosci. 2013, 36, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.G.; Borges, C.G.; Seminotti, B.; Amaral, A.U.; Knebel, L.A.; Eichler, P.; de Oliveira, A.B.; Leipnitz, G.; Wajner, M. Experimental evidence that methylmalonic acid provokes oxidative damage and compromises antioxidant defenses in nerve terminal and striatum of young rats. Cell. Mol. Neurobiol. 2011, 31, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Haslbeck, K.M.; Neundörfer, B.; Schlötzer-Schrehardt, U.; Bierhaus, A.; Schleicher, E.; Pauli, E.; Haslbeck, M.; Hecht, M.; Nawroth, P.; Heuss, D. Activation of the RAGE pathway: A general mechanism in the pathogenesis of polyneuropathies? Neurol. Res. 2007, 29, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Haslbeck, K.-M.; Schleicher, E.; Bierhaus, A.; Nawroth, P.; Haslbeck, M.; Neundörfer, B.; Heuss, D. The AGE/RAGE/NF-ΚB pathway may contribute to the pathogenesis of polyneuropathy in impaired glucose tolerance (IGT). Exp. Clin. Endocrinol. Diabetes 2005, 113, 288–291. [Google Scholar] [CrossRef]
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021. [Google Scholar]
- Pop-Busui, R.; Ang, L.; Boulton, A.; Feldman, E.; Marcus, R.; Mizokami-Stout, K.; Singleton, J.R.; Ziegler, D. Diagnosis and treatment of painful diabetic peripheral neuropathy. Compendia 2022, 2022, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, B.C.; Gallagher, G.; Fridman, V.; Feldman, E.L. Diabetic neuropathy: What does the future hold? Diabetologia 2020, 63, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Román-Pintos, L.M.; Villegas-Rivera, G.; Rodríguez-Carrizalez, A.D.; Miranda-Díaz, A.G.; Cardona-Muñoz, E.G. Diabetic polyneuropathy in type 2 diabetes mellitus: Inflammation, oxidative stress, and mitochondrial function. J. Diabetes Res. 2016, 2016, 3425617. [Google Scholar] [CrossRef]
- Bennett, G.J.; Doyle, T.; Salvemini, D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat. Rev. Neurol. 2014, 10, 326–336. [Google Scholar] [CrossRef]
- Feldman, E.L.; Nave, K.-A.; Jensen, T.S.; Bennett, D.L.H. New horizons in diabetic neuropathy: Mechanisms, bioenergetics, and pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Lian, X.; Liu, H.; Zhang, Y.; Li, Q.; Cai, Y.; Ma, H.; Yu, X. Understanding diabetic neuropathy: Focus on oxidative stress. Oxidative Med. Cell. Longev. 2020, 2020, 9524635. [Google Scholar] [CrossRef] [PubMed]
- Rumora, A.E.; Savelieff, M.G.; Sakowski, S.A.; Feldman, E.L. Disorders of mitochondrial dynamics in peripheral neuropathy: Clues from hereditary neuropathy and diabetes. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 145, pp. 127–176. ISBN 978-0-12-817224-7. [Google Scholar]
- Bierhaus, A.; Haslbeck, K.-M.; Humpert, P.M.; Liliensiek, B.; Dehmer, T.; Morcos, M.; Sayed, A.A.R.; Andrassy, M.; Schiekofer, S.; Schneider, J.G.; et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J. Clin. Investig. 2004, 114, 1741–1751. [Google Scholar] [CrossRef]
- van Zoelen, M.A.; Yang, H.; Florquin, S.; Meijers, J.C.; Akira, S.; Arnold, B.; Nawroth, P.P.; Bierhaus, A.; Tracey, K.J.; van der Poll, T. Role of TOLL-like receptors 2 and 4 and the receptor for advanced glycation end products in high-mobility group box-1- induced inflammation in vivo. Shock 2009, 31, 280–284. [Google Scholar] [CrossRef]
- Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. The RAGE/DIAPH1 signaling axis & implications for the pathogenesis of diabetic complications. Int. J. Mol. Sci. 2022, 23, 4579. [Google Scholar] [CrossRef]
- Thakur, V.; Sadanandan, J.; Chattopadhyay, M. High-mobility group box 1 protein signaling in painful diabetic neuropathy. Int. J. Mol. Sci. 2020, 21, 881. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Sequeira, J.M.; Alarcon, J.M.; Wasek, B.; Arning, E.; Bottiglieri, T.; Quadros, E.V. Neuropathology of vitamin B12 deficiency in the Cd320 −/− mouse. FASEB J. 2019, 33, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.; Muntingh, G.; Rheeder, P. Vitamin B12 deficiency in metformin-treated type-2 diabetes patients, prevalence and association with peripheral neuropathy. BMC Pharmacol. Toxicol. 2016, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, P.R.; Praveen, D.; Aanandhi, M.V. Role of vitamin B12 supplementation on incipient neuropathy in patients with type II diabetes mellitus. Drug Invent. Today 2019, 12, 2536–2539. [Google Scholar]
- Didangelos, T.; Karlafti, E.; Kotzakioulafi, E.; Kontoninas, Z.; Margaritidis, C.; Giannoulaki, P.; Kantartzis, K. Efficacy and safety of the combination of superoxide dismutase, alpha lipoic acid, vitamin b12, and carnitine for 12 months in patients with diabetic neuropathy. Nutrients 2020, 12, 3254. [Google Scholar] [CrossRef]
- Didangelos, T.; Karlafti, E.; Kotzakioulafi, E.; Margariti, E.; Giannoulaki, P.; Batanis, G.; Tesfaye, S.; Kantartzis, K. Vitamin B12 supplementation in diabetic neuropathy: A 1-year, randomized, double-blind, placebo-controlled trial. Nutrients 2021, 13, 395. [Google Scholar] [CrossRef]
- Dizaye, K.F.; Sheet, T.A. Therapeutic effect of pregabalin, vitamin B-groups and their combination on patients with diabetic peripheral poly neuropathy. Middle East J. Fam. Med. 2014, 12. [Google Scholar] [CrossRef]
- Farvid, M.S.; Homayouni, F.; Amiri, Z.; Adelmanesh, F. Improving neuropathy scores in type 2 diabetic patients using micronutrients supplementation. Diabetes Res. Clin. Pract. 2011, 93, 86–94. [Google Scholar] [CrossRef]
- Fonseca, V.A.; Lavery, L.A.; Thethi, T.K.; Daoud, Y.; DeSouza, C.; Ovalle, F.; Denham, D.S.; Bottiglieri, T.; Sheehan, P.; Rosenstock, J. Metanx in type 2 diabetes with peripheral neuropathy: A randomized trial. Am. J. Med. 2013, 126, 141–149. [Google Scholar] [CrossRef]
- Jayabalan, B.; Low, L.L. Vitamin B supplementation for diabetic peripheral neuropathy. Singap. Med. J. 2016, 57, 55–59. [Google Scholar] [CrossRef]
- Jiang, D.-Q.; Xu, L.-C.; Jiang, L.-L.; Li, M.-X.; Wang, Y. Fasudil combined with methylcobalamin or lipoic acid can improve the nerve conduction velocity in patients with diabetic peripheral neuropathy: A meta-analysis. Medicine 2018, 97, e11390. [Google Scholar] [CrossRef]
- Kuwabara, S.; Nakazawa, R.; Azuma, N.; Suzuki, M.; Miyajima, K.; Fukutake, T.; Hattori, T. Intravenous methylcobalamin treatment for uremic and diabetic neuropathy in chronic hemodialysis patients. Intern. Med. 1999, 38, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, X.; Li, Q.; Du, J.; Liu, Z.; Peng, Y.; Xu, M.; Li, Q.; Lei, M.; Wang, C.; et al. Effects of acetyl-L-carnitine and methylcobalamin for diabetic peripheral neuropathy: A multicenter, randomized, double-blind, controlled trial. J. Diabetes Investig. 2016, 7, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Yaqub, B.A.; Siddique, A.; Sulimani, R. Effects of methylcobalamin on diabetic neuropathy. Clin. Neurol. Neurosurg. 1992, 94, 105–111. [Google Scholar] [CrossRef]
- Wang, X.; Yang, W.; Zhu, Y.; Zhang, S.; Jiang, M.; Hu, J.; Zhang, H.-H. Genomic DNA methylation in diabetic chronic complications in patients with type 2 diabetes mellitus. Front. Endocrinol. 2022, 13, 896511. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Elzinga, S.; Eid, S.; Figueroa-Romero, C.; Hinder, L.M.; Pacut, C.; Feldman, E.L.; Hur, J. Genome-wide DNA methylation profiling of human diabetic peripheral neuropathy in subjects with type 2 diabetes mellitus. Epigenetics 2019, 14, 766–779. [Google Scholar] [CrossRef]
- Guo, K.; Eid, S.A.; Elzinga, S.E.; Pacut, C.; Feldman, E.L.; Hur, J. Genome-wide profiling of DNA methylation and gene expression identifies candidate genes for human diabetic neuropathy. Clin. Epigenet. 2020, 12, 123. [Google Scholar] [CrossRef]
- Haslbeck, K.; Schleicher, E.; Friess, U.; Kirchner, A.; Neundörfer, B.; Heuss, D. N ε-carboxymethyllysine in diabetic and non-diabetic polyneuropathies. Acta Neuropathol. 2002, 104, 45–52. [Google Scholar] [CrossRef]
- Luciani, A.; Schumann, A.; Berquez, M.; Chen, Z.; Nieri, D.; Failli, M.; Debaix, H.; Festa, B.P.; Tokonami, N.; Raimondi, A.; et al. Impaired mitophagy links mitochondrial disease to epithelial stress in methylmalonyl-CoA mutase deficiency. Nat Commun. 2020, 11, 970. [Google Scholar] [CrossRef]
- Suarez-Moreira, E.; Yun, J.; Birch, C.S.; Williams, J.H.H.; McCaddon, A.; Brasch, N.E. Vitamin B12 and redox homeostasis: Cob(II)alamin reacts with superoxide at rates approaching superoxide dismutase (SOD). J. Am. Chem. Soc. 2009, 131, 15078–15079. [Google Scholar] [CrossRef]
- Moreira, E.S.; Brasch, N.E.; Yun, J. Vitamin B12 protects against superoxide-induced cell injury in human aortic endothelial cells. Free Radic. Biol. Med. 2011, 51, 876–883. [Google Scholar] [CrossRef]
- Chan, W.; Almasieh, M.; Catrinescu, M.-M.; Levin, L.A. Cobalamin-associated superoxide scavenging in neuronal cells is a potential mechanism for vitamin B12–Deprivation optic neuropathy. Am. J. Pathol. 2018, 188, 160–172. [Google Scholar] [CrossRef]
- Birch, C.S.; Brasch, N.E.; McCaddon, A.; Williams, J.H.H. A novel role for vitamin B12: Cobalamins are intracellular antioxidants in vitro. Free Radic. Biol. Med. 2009, 47, 184–188. [Google Scholar] [CrossRef] [PubMed]
- van de Lagemaat, E.; de Groot, L.; van den Heuvel, E. Vitamin B12 in relation to oxidative stress: A systematic review. Nutrients 2019, 11, 482. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, H.; Ogasawara, S.; Yamagishi, S.-I.; Takahashi, K.; Yagihashi, S. Methylcobalamin effects on diabetic neuropathy and nerve protein kinase C in rats: Methylcobalamin and PKC in diabetic neuropathy. Eur. J. Clin. Investig. 2011, 41, 442–450. [Google Scholar] [CrossRef]
- Wolffenbuttel, B.H.R.; Wouters, H.J.C.M.; Heiner-Fokkema, M.R.; van der Klauw, M.M. The many faces of cobalamin (vitamin B12) deficiency. Mayo Clin. Proc. Innov. Qual. Outcomes 2019, 3, 200–214. [Google Scholar] [CrossRef]
- Nexo, E.; Hoffmann-Lücke, E. Holotranscobalamin, a marker of vitamin B-12 status: Analytical aspects and clinical utility. Am. J. Clin. Nutr. 2011, 94, 359S–365S. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Sherliker, P.; Hin, H.; Nexo, E.; Hvas, A.M.; Schneede, J.; Birks, J.; Ueland, P.M.; Emmens, K.; Scott, J.M.; et al. Detection of vitamin B12 deficiency in older people by measuring vitamin B12 or the active fraction of vitamin B12, holotranscobalamin. Clin. Chem. 2007, 53, 963–970. [Google Scholar] [CrossRef]
- Gwathmey, K.G.; Grogan, J. Nutritional neuropathies. Muscle Nerve 2020, 62, 13–29. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R.; Schorr, H.; Geisel, J. Functional vitamin B12 deficiency and determination of holotranscobalamin in populations at risk. Clin. Chem. Lab. Med. 2003, 41, 1478–1488. [Google Scholar] [CrossRef]
- Golding, P.H. Holotranscobalamin (HoloTC, Active-B12) and herbert’s model for the development of vitamin B12 deficiency: A review and alternative hypothesis. SpringerPlus 2016, 5, 668. [Google Scholar] [CrossRef] [PubMed]
- Risch, M.; Meier, D.W.; Sakem, B.; Escobar, P.M.; Risch, C.; Nydegger, U.; Risch, L. Vitamin B12 and folate levels in healthy swiss senior citizens: A prospective study evaluating reference intervals and decision limits. BMC Geriatr. 2015, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Ugarriza, R.; Palacios, G.; Alder, M.; González-Gross, M. A review of the cut-off points for the diagnosis of vitamin B12 deficiency in the general population. Clin. Chem. Lab. Med. CCLM 2015, 53, 1149–1159. [Google Scholar] [CrossRef]
- Hannibal, L.; Lysne, V.; Bjørke-Monsen, A.-L.; Behringer, S.; Grünert, S.C.; Spiekerkoetter, U.; Jacobsen, D.W.; Blom, H.J. Biomarkers and algorithms for the diagnosis of vitamin B12 deficiency. Front. Mol. Biosci. 2016, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, W.; Obeid, R. Causes and early diagnosis of vitamin B12 deficiency. Dtsch. Ärzteblatt Int. 2008, 105, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, A.; Knudsen, C.S.; Hoejskov, C.S.; Greibe, E.; Parkner, T. Reference intervals for plasma vitamin B12 and plasma/serum methylmalonic acid in Danish children, adults and elderly. Clin. Chim. Acta 2022, 525, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Department of Medicine and Geriatrics, Caritas Medical Centre, Shamshuipo, Hong Kong; Wong, C. Vitamin B12 deficiency in the elderly: Is it worth screening? Hong Kong Med. J. 2015, 21, 155–164. [Google Scholar] [CrossRef]
- Porter, K.M.; Hoey, L.; Hughes, C.F.; Ward, M.; Clements, M.; Strain, J.; Cunningham, C.; Casey, M.C.; Tracey, F.; O’Kane, M.; et al. Associations of atrophic gastritis and proton-pump inhibitor drug use with vitamin B-12 status, and the impact of fortified foods, in older adults. Am. J. Clin. Nutr. 2021, 114, 1286–1294. [Google Scholar] [CrossRef]
- Nardin, R.A.; Amick, A.N.H.; Raynor, E.M. Vitamin B12 and methylmalonic acid levels in patients presenting with polyneuropathy. Muscle Nerve 2007, 36, 532–535. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stages of B12 Metabolism | Defects/Causes of B12 Deficiency |
|---|---|
| 1. Dietary uptake: B12 present in free or protein-bound form is taken up in the diet or as a drug. | Inadequate intake (strict vegan diet, eating disorders etc.). |
| 2. Gastric secretion of intrinsic factor (IF) and HCl: In the stomach, the protein-bound B12 complex is digested by pepsin in the acid milieu, and the free B12 is bound by haptocorrin. This complex and IF (secreted by parietal cells) are transferred to the duodenum. | Impaired secretion or neutralization of HCl • Elderly people • Drugs, e.g., proton pump inhibitors, histamine receptor antagonists Impaired secretion of HCl and IF • Autoimmune gastritis/gastric atrophy (antibodies against parietal cells or IF) • Gastrectomy • Hereditary defects of IF |
| 3. Binding of B12 by IF in the duodenum: In the duodenum, the B12-haptocorrin complex is digested by proteases, and the released B12 is bound by IF with high affinity. | |
| 4. Absorption in ileum: The B12–IF complex is bound to and taken up by specific mucosal receptors in the terminal ileum. | Malabsorption due to, e.g., pancreas insufficiency, surgery, inflammatory bowel disease, or drugs, e.g., biguanides (metformin) |
| 5. Blood-borne transport by transcobalamins: After internalization by the enterocytes, B12 is exported into the blood and subsequently bound to transcobalamins (TC). The complex with holotranscobalamin (HoloTC) carries 10–20% of total B12. HoloTC is the circulating form of B12 which can be taken up by the target cell via the ubiquitous receptor CD 320. The majority of B12 is transported by TC I, but this B12 complex cannot be taken up by peripheral cells. | Congenital defects (very rare in adults) |
| 6. Intracellular transport and lysosomal metabolism: The internalized complex is transported to the lysosomes and degraded, thereby liberating B12. Free intracellular B12 can be metabolized into methylcobalamin or adenosylcobalamin, both being cofactors, the first for methionine synthase and the second for methylmalonyl-CoA mutase. | Congenital defects (very rare in adults) |
| Biomarker | B12 Deficiency | Low/Grey Zone | Adequate Supply |
|---|---|---|---|
| B12 1 (pmol/L) | <131 | 131–315 | ≥316 |
| HoloTC (pmol/L) | <25.8 | 25.8–56.9 | ≥57 |
| MMA 2 (µmol/L) | >0.485 | 0.217–0.485 | ≤0.216 |
| HCys 2,3 (µmol/L) | >26.8 | 9.6–26.8 | ≤9.5 |
| WHO | |||
| B12 1 (pmol/L) | <148 | 148–220 | ≥221 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schleicher, E.; Didangelos, T.; Kotzakioulafi, E.; Cegan, A.; Peter, A.; Kantartzis, K. Clinical Pathobiochemistry of Vitamin B12 Deficiency: Improving Our Understanding by Exploring Novel Mechanisms with a Focus on Diabetic Neuropathy. Nutrients 2023, 15, 2597. https://doi.org/10.3390/nu15112597
Schleicher E, Didangelos T, Kotzakioulafi E, Cegan A, Peter A, Kantartzis K. Clinical Pathobiochemistry of Vitamin B12 Deficiency: Improving Our Understanding by Exploring Novel Mechanisms with a Focus on Diabetic Neuropathy. Nutrients. 2023; 15(11):2597. https://doi.org/10.3390/nu15112597
Chicago/Turabian StyleSchleicher, Erwin, Triantafyllos Didangelos, Evangelia Kotzakioulafi, Alexander Cegan, Andreas Peter, and Konstantinos Kantartzis. 2023. "Clinical Pathobiochemistry of Vitamin B12 Deficiency: Improving Our Understanding by Exploring Novel Mechanisms with a Focus on Diabetic Neuropathy" Nutrients 15, no. 11: 2597. https://doi.org/10.3390/nu15112597
APA StyleSchleicher, E., Didangelos, T., Kotzakioulafi, E., Cegan, A., Peter, A., & Kantartzis, K. (2023). Clinical Pathobiochemistry of Vitamin B12 Deficiency: Improving Our Understanding by Exploring Novel Mechanisms with a Focus on Diabetic Neuropathy. Nutrients, 15(11), 2597. https://doi.org/10.3390/nu15112597