
Branched-Chain Amino Acids and Insulin Resistance, from Protein Supply to Diet-Induced Obesity



Abstract
1. Introduction
2. BCAA Metabolism
2.1. BCAA Inter-Organ Exchanges in the Postprandial Period
2.2. BCAA Celllular Metabolism
2.2.1. BCAA Transport
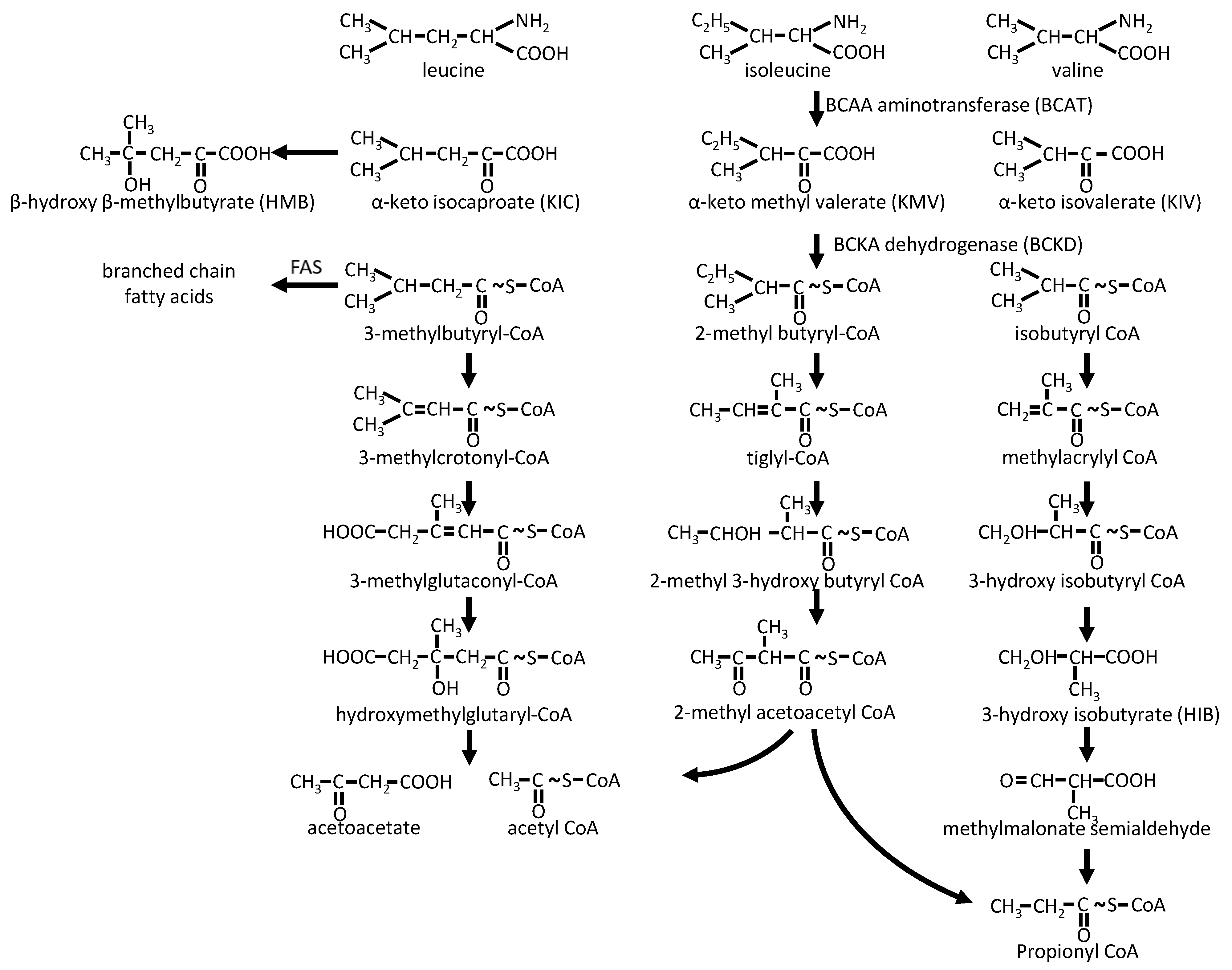
2.2.2. BCAA Catabolism
3. Metabolic Properties of BCAAs/BCKAs
4. Blood BCAAs as Biomarkers of Metabolic Status
4.1. Physiological and Pathological Variations in Blood BCAAs
4.2. Blood BCAAs as Markers of Insulin Resistance
- The decrease in insulin sensitivity affects the metabolism of BCAAs and induces an early increase in their plasma concentrations. This phenomenon is well known for other aspect of insulin resistance, for example in the cardiovascular field where alterations of the vascular function can exist many years before the appearance of clinical manifestations [72];
- The BCAAs or some of their derivatives could at certain doses or in certain situations of energy imbalance promote insulin resistance (e.g., through the increased serine phosphorylation of IRS1).
5. BCAA Metabolism and Insulin Resistance
5.1. BCAA Metabolism in Insulin Resistance Situations
5.1.1. BCAA Metabolism and Insulin Resistance in Humans
5.1.2. BCAA Metabolism in Experimental Models of Insulin Resistance
5.1.3. BCAA Metabolism in Brown/Beige Adipose Tissue
5.2. Do BCAAs Promote Insulin Resistance?
5.2.1. BCAA-Induced Insulin Resistance?

{kind=link}
{kind=link}
| Reference | Diet Type | Lipids | Protides | Carbohydrates |
|---|---|---|---|---|
| [61] | SC | 18% 100% soybean oil | 24% 100% soybean protein | 58.0% 100% wheat/corn |
| [55] | HF | 45% 12.3% soybean oil 87.7% lard | 19% 53% casein 47% free amino acids | 35% 21% corn starch 29% maltodextrin 50% sucrose |
| HF/BCAA | 43% 12.3% soybean oil 87.7% lard | 23% 41% casein 58% free amino acids including BCAA supplement | 34% 21% corn starch 29% maltodextrin 50% sucrose | |
| SC | 10% of kcal 55.5% soybean oil 44.5% lard | 19% of kcal 53% casein 47% free amino acids | 71% of kcal 45% corn starch 5% maltodextrin 50% sucrose | |
| SC/BCAA | 10% 55.5% soybean oil 44.5% lard | 23% 41% casein 58% free amino acids including BCAA supplement | 67% 45% corn starch 5% maltodextrin 50% sucrose | |
| [62] | SC or FD | 16.7% 100% soybean oil | 19.3% 100% casein | 64.0% 100% corn starch or fructose |
| [63] | SC | 8.4% 100% soybean oil | 19.3% 100% casein | 72.4% 66% corn starch 33% maltodextrin |
| FD | 8.4% 100% soybean oil | 19.3% 100% casein | 72.4% 8% corn starch 92% fructose | |
| [64] | HFHS | 45% 20% soybean oil 80% lard | 20% 100% casein | 35% 73% rice starch 27% sucrose |
| +30% fructose in drinking water | ||||
| [65] | SC | 16.8% 100% lard | 26.8% Mixed sources | 56.4% 93% starch 7% sucrose |
| HFHS | 40% 95% milk fat 5% corn oil | 17% 100% casein | 43% 10% corn starch 20% maltodextrin 70% sucrose | |
| HF | 54.8% 94.5% hydrogenated vegetable oil 5.5% corn oil | 21.2% 100% casein | 24% 69.6% corn starch 30.4% sucrose | |
| + BCAAs in drinking water when added | ||||
| [66] | SC | 13.5% 100% lard | 28.5% mixed sources | 58% 84.8% starch 9.8% sucrose 5.5% lactose |
| HFHS | 45% 87.7% lard 12.3% soybean Oil, | 20% 100% casein | 35% 20.8% corn starch 28.6% maltodextrin 50.6% sucrose | |
| [67] | SC | 18% 100% soybean oil | 18% 100% casein | 64% 63.5% wheatstarch 20.7% maltodextrin 15.7% sucrose |
| BCAA adapted by reducing casein and adding free amino acids with or without BCAAs | ||||
| [86] | SC | 13.1% 100% soybean oil | 24.5% mixed sources | 62.4% 91.2% starch 8.8% sucrose |
| HF | 60% 10% soybean oil 90% lard | 20% 100% casein | 20% 64% maltodextrin 36% sucrose | |
| + BCAAs in drinking water when added | ||||
5.2.2. BCAA Metabolites and Insulin Sensitivity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Rossi Fanelli, F.; Cangiano, C.; Capocaccia, L.; Cascino, A.; Ceci, F.; Muscaritoli, M.; Giunchi, G. Use of branched chain amino acids for treating hepatic encephalopathy: Clinical experiences. Gut 1986, 27 (Suppl. 1), 111–115. [Google Scholar] [CrossRef] [PubMed]
- De Bandt, J.P.; Cynober, L. Therapeutic use of branched-chain amino acids in burn, trauma, and sepsis. J. Nutr. 2006, 136 (Suppl. 1), 308S–313S. [Google Scholar] [CrossRef] [PubMed]
- Ten Have, G.A.M.; Jansen, L.; Schooneman, M.G.; Engelen, M.P.K.J.; Deutz, N.E.P. Metabolic flux analysis of branched-chain amino and keto acids (BCAA, BCKA) and β-hydroxy β-methylbutyric acid across multiple organs in the pig. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E629–E640. [Google Scholar] [CrossRef] [PubMed]
- Suryawan, A.; Hawes, J.W.; A Harris, R.; Shimomura, Y.; E Jenkins, A.; Hutson, S.M. A molecular model of human branched-chain amino acid metabolism. Am. J. Clin. Nutr. 1998, 68, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Wahren, J.; Felig, P.; Hagenfeldt, L. Effect of protein ingestion on splanchnic and leg metabolism in normal man and in patients with diabetes mellitus. J. Clin. Investig. 1976, 57, 987–999. [Google Scholar] [CrossRef]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.-X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.-L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef]
- Bröer, S. Amino acid transporters as modulators of glucose homeostasis. Trends Endocrinol. Metab. 2022, 33, 120–135. [Google Scholar] [CrossRef]
- Sperringer, J.E.; Addington, A.; Hutson, S.M. Branched-chain amino acids and brain metabolism. Neurochem. Res. 2017, 42, 1697–1709. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef]
- Van Koevering, M.; Nissen, S. Oxidation of leucine and alpha-ketoisocaproate to beta-hydroxy-beta-methylbutyrate in vivo. Am. J. Physiol. Metab. 1992, 262, E27–E31. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Damuni, Z.; Merryfield, M.L. Regulation of mammalian pyruvate and branched-chain alpha-keto acid dehydrogenase complexes by phosphorylation-dephosphorylation. Curr. Top. Cell. Regul. 1985, 27, 41–49. [Google Scholar] [PubMed]
- Harper, A.E.; Miller, R.H.; Block, K.P. Branched-chain amino acid metabolism. Annu. Rev. Nutr. 1984, 4, 409–454. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Sun, H.; She, P.; Youn, J.-Y.; Warburton, S.; Ping, P.; Vondriska, T.M.; Cai, H.; Lynch, C.J.; Wang, Y. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J. Clin. Investig. 2009, 119, 1678–1687. [Google Scholar] [CrossRef]
- Zhou, M.; Lu, G.; Gao, C.; Wang, Y.; Sun, H. Tissue-specific and nutrient regulation of the branched-chain α-keto acid dehydrogenase phosphatase, protein phosphatase 2Cm (PP2Cm). J. Biol. Chem. 2012, 287, 23397–23406. [Google Scholar] [CrossRef] [PubMed]
- Frick, G.P.; Goodman, H.M. Insulin regulation of the activity and phosphorylation of branched-chain 2-oxo acid dehydrogenase in adipose tissue. Biochem. J. 1989, 258, 229–235. [Google Scholar] [CrossRef]
- Lang, C.H.; Frost, R.A.; Vary, T.C. Regulation of muscle protein synthesis during sepsis and inflammation. Am. J. Physiol. Metab. 2007, 293, E453–E459. [Google Scholar] [CrossRef]
- Kurpad, A.V.; Regan, M.M.; Raj, T.; Gnanou, J.V. Branched-chain amino acid requirements in healthy adult human subjects. J. Nutr. 2006, 136 (Suppl. 1), 256S–263S. [Google Scholar] [CrossRef]
- Wallace, M.; Green, C.R.; Roberts, L.S.; Lee, Y.M.; McCarville, J.L.; Sanchez-Gurmaches, J.; Meurs, N.; Gengatharan, J.M.; Hover, J.D.; Phillips, S.A.; et al. Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat. Chem. Biol. 2018, 14, 1021–1031. [Google Scholar] [CrossRef]
- Neinast, M.; Murashige, D.; Arany, Z. Branched chain amino acids. Annu. Rev. Physiol. 2019, 81, 139–164. [Google Scholar] [CrossRef]
- Tom, A.; Nair, K.S. Assessment of branched-chain amino acid status and potential for biomarkers. J. Nutr. 2006, 136 (Suppl. 1), 324S–330S. [Google Scholar] [CrossRef] [PubMed]
- Choudry, H.A.; Pan, M.; Karinch, A.M.; Souba, W.W. Branched-chain amino acid-enriched nutritional support in surgical and cancer patients. J. Nutr. 2006, 136 (Suppl. 1), 314S–318S. [Google Scholar] [CrossRef] [PubMed]
- Udkoff, M.; Daikhin, Y.; Nissim, I.; Horyn, O.; Luhovyy, B.; Lazarow, A.; Nissim, I. Brain amino acid requirements and toxicity: The example of leucine. J. Nutr. 2005, 135 (Suppl. 6), 1531S–1538S. [Google Scholar] [CrossRef] [PubMed]
- Fernstrom, J.D. Branched-chain amino acids and brain function. J. Nutr. 2005, 135 (Suppl. 6), 1539S–1546S. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.U.; Wajner, M. Pathophysiology of maple syrup urine disease: Focus on the neurotoxic role of the accumulated branched-chain amino acids and branched-chain α-keto acids. Neurochem. Int. 2022, 157, 105360. [Google Scholar] [CrossRef] [PubMed]
- Odessey, R.; Goldberg, A.L. Oxidation of leucine by rat skeletal muscle. Am. J. Physiol. Content 1972, 223, 1376–1383. [Google Scholar] [CrossRef]
- Vary, T.C.; Lynch, C.J. Nutrient signaling components controlling protein synthesis in striated muscle. J. Nutr. 2007, 137, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- De Bandt, J.P. Leucine and mammalian target of rapamycin-dependent activation of muscle protein synthesis in aging. J. Nutr. 2016, 146, 2616S–2624S. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Todorova, P.; Pavlova, N.N.; Tada, Y.; Thompson, C.B.; Finley, L.W.; Overholtzer, M. Leucine retention in lysosomes is regulated by starvation. Proc. Natl. Acad. Sci. USA 2022, 119, e2114912119. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Wang, B.; He, W.; Prosseda, P.P.; Li, L.; Kowal, T.J.; Alvarado, J.A.; Wang, Q.; Hu, Y.; Sun, Y. OCRL regulates lysosome positioning and mTORC1 activity through SSX2IP-mediated microtubule anchoring. EMBO Rep. 2021, 22, e52173. [Google Scholar] [CrossRef] [PubMed]
- Beaumatin, F.; O’Prey, J.; Barthet, V.J.A.; Zunino, B.; Parvy, J.P.; Bachmann, A.M.; O’Prey, M.; Kania, E.; Gonzalez, P.S.; Macintosh, R.; et al. mTORC1 activation requires DRAM-1 by facilitating lysosomal amino acid efflux. Mol. Cell 2019, 76, 163–176. [Google Scholar] [CrossRef]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Heiden, M.G.V.; Sabatini, D.M. mTORC1 Activator SLC38A9 Is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 2017, 171, 642–654.e12. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Jacques, H.; Marette, A. Modulation of insulin action by dietary proteins and amino acids: Role of the mammalian target of rapamycin nutrient sensing pathway. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Krebs, M.; Dombrowski, L.; Brehm, A.; Bernroider, E.; Roth, E.; Nowotny, P.; Waldhäusl, W.; Marette, A.; Roden, M. Overactivation of S6 Kinase 1 as a Cause of Human insulin resistance during increased amino acid availability. Diabetes 2005, 54, 2674–2684. [Google Scholar] [CrossRef]
- Bear, D.E.; Rooyackers, O. HMB and leucine supplementation during critical illness and recovery. Curr. Opin. Clin. Nutr. Metab. Care 2022, 25, 88–92. [Google Scholar] [CrossRef]
- Matthews, D.E. Observations of branched-chain amino acid administration in humans. J. Nutr. 2005, 135 (Suppl. 6), 1580S–1584S. [Google Scholar] [CrossRef]
- Fujita, S.; Dreyer, H.C.; Drummond, M.J.; Glynn, E.L.; Cadenas, J.G.; Yoshizawa, F.; Volpi, E.; Rasmussen, B. Nutrient signalling in the regulation of human muscle protein synthesis. J. Physiol. 2007, 582, 813–823. [Google Scholar] [CrossRef]
- Katsanos, C.S.; Kobayashi, H.; Sheffield-Moore, M.; Aarsland, A.; Wolfe, R.R. A high proportion of leucine is required for optimal stimulation of the rate of muscle protein synthesis by essential amino acids in the elderly. Am. J. Physiol. Metab. 2006, 291, E381–E387. [Google Scholar] [CrossRef]
- Malaisse, W.J. Branched-chain amino and keto acid metabolism in pancreatic islets. Adv. Enzym. Regul. 1986, 25, 203–217. [Google Scholar] [CrossRef]
- Meijer, A.J.; Dubbelhuis, P.F. Amino acid signalling and the integration of metabolism. Biochem. Biophys. Res. Commun. 2004, 313, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Brambilla, E.; Luzi, L.; Landaker, E.J.; Kahn, C.R. Bidirectional modulation of insulin action by amino acids. J. Clin. Investig. 1998, 101, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.; Krssak, M.; Bernroider, E.; Anderwald, C.; Brehm, A.; Meyerspeer, M.; Nowotny, P.; Roth, E.; Waldhäusl, W.; Roden, M. Mechanism of Amino Acid-Induced Skeletal Muscle Insulin Resistance in Humans. Diabetes 2002, 51, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Felig, P.; Owen, O.E.; Wahren, J.; Cahill, G.F. Amino acid metabolism during prolonged starvation. J. Clin. Investig. 1969, 48, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Adibi, S.A. Influence of dietary deprivations on plasma concentration of free amino acids of man. J. Appl. Physiol. 1968, 25, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Wahren, J.; Felig, P.; Cerasi, E.; Luft, R. Splanchnic and peripheral glucose and amino acid metabolism in diabetes mellitus. J. Clin. Investig. 1972, 51, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Felig, P.; Wahren, J. Amino acid metabolism in exercising man. J. Clin. Investig. 1971, 50, 2703–2714. [Google Scholar] [CrossRef]
- Smith, R.; Elia, M. Branched-chain amino acids in catabolic states. Proc. Nutr. Soc. 1983, 42, 473–487. [Google Scholar] [CrossRef]
- Hamaya, R.; Mora, S.; Lawler, P.R.; Cook, N.R.; Ridker, P.M.; Buring, J.E.; Lee, I.-M.; Manson, J.E.; Tobias, D.K. Association of plasma branched-chain amino acid with biomarkers of inflammation and lipid metabolism in women. Circ. Genom. Precis. Med. 2021, 14, 517–525. [Google Scholar] [CrossRef]
- Hamaya, R.; Mora, S.; Lawler, P.R.; Cook, N.R.; E Buring, J.; Lee, I.-M.; E Manson, J.; Tobias, D.K. Association of modifiable lifestyle factors with plasma branched-chain amino acid metabolites in women. J. Nutr. 2022, 152, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Guizar-Heredia, R.; Tovar, A.R.; Granados-Portillo, O.; Pichardo-Ontiveros, E.; Flores-López, A.; González-Salazar, L.E.; Arteaga-Sanchez, L.; Medina-Vera, I.; Orozco-Ruiz, X.; Torres, N.; et al. Serum amino acid concentrations are modified by age, insulin resistance, and BCAT2 rs11548193 and BCKDH rs45500792 polymorphisms in subjects with obesity. Clin. Nutr. 2021, 40, 4209–4215. [Google Scholar] [CrossRef] [PubMed]
- Lotta, L.A.; Scott, R.A.; Sharp, S.J.; Burgess, S.; Luan, J.; Tillin, T.; Schmidt, A.F.; Imamura, F.; Stewart, I.D.; Perry, J.R.; et al. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: A mendelian randomisation analysis. PLoS Med. 2016, 13, e1002179. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M.; Guénard, F.; Garneau, V.; Allam-Ndoul, B.; Lemieux, S.; Pérusse, L.; Vohl, M.-C. Associations between dietary protein sources, plasma BCAA and short-chain acylcarnitine levels in adults. Nutrients 2019, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.D.; Hartle, J.C.; Garrett, R.; Offringa, L.C.; Wasserman, A.S. Maximizing the intersection of human health and the health of the environment with regard to the amount and type of protein produced and consumed in the United States. Nutr. Rev. 2019, 77, 197–215. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Huffman, K.M.; Shah, S.H.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.; Slentz, C.A.; Tanner, C.J.; Kuchibhatla, M.; Houmard, J.A.; Newgard, C.B.; et al. Relationships between circulating metabolic intermediates and insulin action in overweight to obese, inactive men and women. Diabetes Care 2009, 32, 1678–1683. [Google Scholar] [CrossRef]
- Tai, E.S.; Tan, M.L.S.; Stevens, R.D.; Low, Y.L.; Muehlbauer, M.J.; Goh, D.L.M.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Lee, J.J.M.; et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia 2010, 53, 757–767. [Google Scholar] [CrossRef]
- Zhao, X.; Gang, X.; Liu, Y.; Sun, C.; Han, Q.; Wang, G. Using metabolomic profiles as biomarkers for insulin resistance in childhood obesity: A systematic review. J. Diabetes Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Fauman, E.; Erte, I.; Perry, J.R.B.; Kastenmüller, G.; Shin, S.Y.; Petersen, A.K.; Hyde, C.; Psatha, M.; Ward, K.J. Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes 2013, 62, 4270–4276. [Google Scholar] [CrossRef]
- Hamley, S.; Kloosterman, D.; Duthie, T.; Man, C.D.; Visentin, R.; Mason, S.A.; Ang, T.; Selathurai, A.; Kaur, G.; Morales-Scholz, M.G.; et al. Mechanisms of hyperinsulinaemia in apparently healthy non-obese young adults: Role of insulin secretion, clearance and action and associations with plasma amino acids. Diabetologia 2019, 62, 2310–2324. [Google Scholar] [CrossRef]
- She, P.; Van Horn, C.; Reid, T.; Hutson, S.M.; Cooney, R.N.; Lynch, C.J. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Metab. 2007, 293, E1552–E1563. [Google Scholar] [CrossRef] [PubMed]
- David, J.; Dardevet, D.; Mosoni, L.; Savary-Auzeloux, I.; Polakof, S. Impaired skeletal muscle branched-chain amino acids catabolism contributes to their increased circulating levels in a non-obese insulin-resistant fructose-fed rat model. Nutrients 2019, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Ventura, G.; Nubret, E.; Sarfati, G.; Bergheim, I.; De Bandt, J.-P. Citrulline and nonessential amino acids prevent fructose-induced nonalcoholic fatty liver disease in rats. J. Nutr. 2015, 145, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Freese, K.; Waligora-Dupriet, A.-J.; Nubret, E.; Butel, M.-J.; Bergheim, I.; De Bandt, J.-P. Preventive effects of citrulline on Western diet-induced non-alcoholic fatty liver disease in rats. Br. J. Nutr. 2016, 116, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Vijayakumar, A.; White, P.J.; Xu, Y.; Ilkayeva, O.; Lynch, C.J.; Newgard, C.B.; Kahn, B.B. BCAA Supplementation in mice with diet-induced obesity alters the metabolome without impairing glucose homeostasis. Endocrinology 2021, 162, 1–17. [Google Scholar]
- Biswas, D.; Dao, K.T.; Mercer, A.; Cowie, A.M.; Duffley, L.; El Hiani, Y.; Kienesberger, P.C.; Pulinilkunnil, T. Branched-chain ketoacid overload inhibits insulin action in the muscle. J. Biol. Chem. 2020, 295, 15597–15621. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; Cogger, V.C.; Pulpitel, T.; Wahl, D.; Clark, X.; Bagley, E.E.; Gregoriou, G.C.; Senior, A.M.; Wang, Q.-P.; Brandon, A.E.; et al. Branched-chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat. Metab. 2019, 1, 532–545. [Google Scholar] [CrossRef]
- Woo, S.-L.; Yang, J.; Hsu, M.; Yang, A.; Zhang, L.; Lee, R.-P.; Gilbuena, I.; Thames, G.; Huang, J.; Rasmussen, A.; et al. Effects of branched-chain amino acids on glucose metabolism in obese, prediabetic men and women: A randomized, crossover study. Am. J. Clin. Nutr. 2019, 109, 1569–1577. [Google Scholar] [CrossRef]
- Hattersley, J.G.; Pfeiffer, A.F.H.; Roden, M.; Petzke, K.-J.; Hoffmann, D.; Rudovich, N.N.; Randeva, H.S.; Vatish, M.; Osterhoff, M.; Goegebakan, Ö.; et al. Modulation of amino acid metabolic signatures by supplemented isoenergetic diets differing in protein and cereal fiber content. J. Clin. Endocrinol. Metab. 2014, 99, E2599–E2609. [Google Scholar] [CrossRef]
- Flores-Guerrero, J.L.; Osté, M.C.J.; Kieneker, L.M.; Gruppen, E.G.; Wolak-Dinsmore, J.; Otvos, J.D.; Connelly, M.A.; Bakker, S.J.L.; Dullaart, R.P.F. Plasma branched-chain amino acids and risk of incident type 2 diabetes: Results from the PREVEND prospective cohort study. J. Clin. Med. 2018, 7, 513. [Google Scholar] [CrossRef]
- van den Berg, E.H.; Flores-Guerrero, J.L.; Gruppen, E.G.; de Borst, M.H.; Wolak-Dinsmore, J.; Connelly, M.A.; Bakker, S.J. Dullaart RP Non-alcoholic fatty liver disease and risk of incident type 2 diabetes: Role of circulating branched-chain amino acids. Nutrients 2019, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Stern, M.P.; Hazuda, H.P.; Mitchell, B.D.; Patterson, J.K. Cardiovascular risk factors in confirmed prediabetic individuals. Does the clock for coronary heart disease start ticking before the onset of clinical diabetes? JAMA 1990, 263, 2893–2898. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Gulseth, H.L.; Langleite, T.M.; Norheim, F.; Olsen, T.; Refsum, H.; Jensen, J.; Birkeland, K.I.; Drevon, C.A. Branched-chain amino acid metabolism, insulin sensitivity and liver fat response to exercise training in sedentary dysglycaemic and normoglycaemic men. Diabetologia 2021, 64, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Glynn, E.L.; Piner, L.W.; Huffman, K.M.; Slentz, C.A.; Elliot-Penry, L.; Abouassi, H.; White, P.; Bain, J.R.; Muehlbauer, M.J.; Ilkayeva, O.R.; et al. Impact of combined resistance and aerobic exercise training on branched-chain amino acid turnover, glycine metabolism and insulin sensitivity in overweight humans. Diabetologia 2015, 58, 2324–2335. [Google Scholar] [CrossRef]
- Tan, H.C.; Hsu, J.W.; Khoo, C.M.; Tai, E.S.; Yu, S.; Chacko, S.; Lai, O.F.; Jahoor, F. Alterations in branched-chain amino acid kinetics in nonobese but insulin-resistant Asian men. Am. J. Clin. Nutr. 2018, 108, 1220–1228. [Google Scholar] [CrossRef]
- Tan, H.C.; Hsu, J.W.; Kovalik, J.-P.; Eng, A.; Chan, W.H.; Khoo, C.M.; Tai, E.S.; Chacko, S.; Jahoor, F. Branched-chain amino acid oxidation is elevated in adults with morbid obesity and decreases significantly after sleeve gastrectomy. J. Nutr. 2020, 150, 3180–3189. [Google Scholar] [CrossRef]
- Vanweert, F.; de Ligt, M.; Hoeks, J.; Hesselink, M.K.C.; Schrauwen, P.; Phielix, E. Elevated plasma branched-chain amino acid levels correlate with type 2 diabetes–related metabolic disturbances. J. Clin. Endocrinol. Metab. 2020, 106, e1827–e1836. [Google Scholar] [CrossRef]
- Afolabi, P.R.; Scorletti, E.; E Smith, D.; Almehmadi, A.A.; Calder, P.C.; Byrne, C.D. The characterisation of hepatic mitochondrial function in patients with non-alcoholic fatty liver disease (NAFLD) using the 13 C-ketoisocaproate breath test. J. Breath Res. 2018, 12, 046002. [Google Scholar] [CrossRef]
- Grenier-Larouche, T.; Kwee, L.C.; Deleye, Y.; Leon-Mimila, P.; Walejko, J.M.; McGarrah, R.W.; Marceau, S.; Trahan, S.; Racine, C.; Carpentier, A.C.; et al. Altered branched-chain α-keto acid metabolism is a feature of NAFLD in individuals with severe obesity. J. Clin. Investig. 2022, 7, e159204. [Google Scholar] [CrossRef]
- Lackey, D.E.; Lynch, C.J.; Olson, K.C.; Mostaedi, R.; Ali, M.; Smith, W.H.; Karpe, F.; Humphreys, S.; Bedinger, D.H.; Dunn, T.N.; et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1175–E1187. [Google Scholar] [CrossRef]
- White, P.J.; Lapworth, A.L.; An, J.; Wang, L.; McGarrah, R.W.; Stevens, R.D.; Ilkayeva, O.; George, T.; Muehlbauer, M.J.; Bain, J.R.; et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol. Metab. 2016, 5, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Neinast, M.D.; Jang, C.; Hui, S.; Murashige, D.S.; Chu, Q.; Morscher, R.J.; Li, X.; Zhan, L.; White, E.; Anthony, T.G.; et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab. 2019, 29, 417–429.e4. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Wang, Q.; Tajima, K.; Matsushita, M.; Maki, H.; Igarashi, K.; Dai, Z.; White, P.J.; McGarrah, R.W.; Ilkayeva, O.R.; et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 2019, 572, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, V.; Labbé, S.M.; Blondin, D.; Phoenix, S.; Guérin, B.; Haman, F.; Turcotte, E.E.; Richard, D.; Carpentier, A.C. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J. Clin. Investig. 2012, 122, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Yamashita, T.; Osone, T.; Hosooka, T.; Shinohara, M.; Kitahama, S.; Sasaki, K.; Sasaki, D.; Yoneshiro, T.; Suzuki, T.; et al. Bacteroides spp. promotes branched-chain amino acid catabolism in brown fat and inhibits obesity. iScience 2021, 24, 103342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, K.; LeBlanc, R.E.; Loh, D.; Schwartz, G.J.; Yu, Y.-H. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes 2007, 56, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Tappy, L. Effects of amino acids on glucose disposal. Diabetes 1990, 39, 1079–1084. [Google Scholar] [CrossRef]
- Smith, G.I.; Yoshino, J.; Stromsdorfer, K.L.; Klein, S.J.; Magkos, F.; Reeds, D.N.; Klein, S.; Mittendorfer, B. protein ingestion induces muscle insulin resistance independent of leucine-mediated mTOR activation. Diabetes 2015, 64, 1555–1563. [Google Scholar] [CrossRef]
- Harris, L.-A.L.; Smith, G.I.; Patterson, B.W.; Ramaswamy, R.S.; Okunade, A.L.; Kelly, S.C.; Porter, L.C.; Klein, S.; Yoshino, J.; Mittendorfer, B. Alterations in 3-Hydroxyisobutyrate and FGF21 metabolism are associated with protein ingestion–induced insulin resistance. Diabetes 2017, 66, 1871–1878. [Google Scholar] [CrossRef]
- Pal, S.; Ellis, V.; Dhaliwal, S. Effects of whey protein isolate on body composition, lipids, insulin and glucose in overweight and obese individuals. Br. J. Nutr. 2010, 104, 716–723. [Google Scholar] [CrossRef]
- Randolph, A.C.; Markofski, M.M.; Rasmussen, B.B.; Volpi, E. Effect of essential amino acid supplementation and aerobic exercise on insulin sensitivity in healthy older adults: A randomized clinical trial. Clin. Nutr. 2020, 39, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Arentson-Lantz, E.J.; Mikovic, J.; Bhattarai, N.; Fry, C.S.; Lamon, S.; Porter, C.; Paddon-Jones, D. Leucine augments specific skeletal muscle mitochondrial respiratory pathways during recovery following 7 days of physical inactivity in older adults. J. Appl. Physiol. 2021, 130, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Chanet, A.; Verlaan, S.; Salles, J.; Giraudet, C.; Patrac, V.; Pidou, V.; Pouyet, C.; Hafnaoui, N.; Blot, A.; Cano, N.; et al. Supplementing breakfast with a vitamin D and leucine-enriched whey protein medical nutrition drink enhances postprandial muscle protein synthesis and muscle mass in healthy older men. J. Nutr. 2017, 147, 2262–2271. [Google Scholar] [CrossRef] [PubMed]
- Jacob, K.J.; Chevalier, S.; Lamarche, M.; Morais, J.A. Leucine supplementation does not alter insulin sensitivity in prefrail and frail older women following a resistance training protocol. J. Nutr. 2019, 149, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Memelink, R.G.; Pasman, W.J.; Bongers, A.; Tump, A.; van Ginkel, A.; Tromp, W.; Wopereis, S.; Verlaan, S.; de Vogel-van den Bosch, J.; Weijs, P.J.M. Effect of an enriched protein drink on muscle mass and glycemic control during combined lifestyle intervention in older adults with obesity and type 2 diabetes: A double-blind RCT. Nutrients 2020, 13, 64. [Google Scholar] [CrossRef]
- Longo, V.D.; Anderson, R.M. Nutrition, longevity and disease: From molecular mechanisms to interventions. Cell 2022, 185, 1455–1470. [Google Scholar] [CrossRef]
- Szczepańska, E.; Gietka-Czernel, M. FGF21: A Novel Regulator of Glucose and Lipid Metabolism and Whole-Body Energy Balance. Horm. Metab. Res. 2022, 54, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Richardson, N.E.; Green, C.L.; Spicer, A.B.; Murphy, M.E.; Flores, V.; Jang, C.; Kasza, I.; Nikodemova, M.; Wakai, M.H.; et al. The adverse metabolic effects of branched-chain amino acids are mediated by isoleucine and valine. Cell Metab. 2021, 33, 905–922.e6. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Asilmaz, E.; Luca, E.; Friedman, J.M.; Stoffel, M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 2004, 432, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Karusheva, Y.; Koessler, T.; Strassburger, K.; Markgraf, D.; Mastrototaro, L.; Jelenik, T.; Simon, M.-C.; Pesta, D.; Zaharia, O.-P.; Bódis, K.; et al. Short-term dietary reduction of branched-chain amino acids reduces meal-induced insulin secretion and modifies microbiome composition in type 2 diabetes: A randomized controlled crossover trial. Am. J. Clin. Nutr. 2019, 110, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- She, P.; Reid, T.M.; Bronson, S.K.; Vary, T.C.; Hajnal, A.; Lynch, C.J.; Hutson, S.M. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 2007, 6, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.-X.; Zhu, W.-Y.; Lu, X.-C.; Jiang, D.; Xu, F.; Li, J.-T.; Zhang, L.; Wu, Y.-L.; Chen, Z.-J.; Yin, M.; et al. BCAA–BCKA axis regulates WAT browning through acetylation of PRDM16. Nat. Metab. 2022, 4, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Margolis, L.M.; Wilson, M.A.; Whitney, C.C.; Carrigan, C.T.; Murphy, N.E.; Hatch, A.M.; Montain, S.J.; Pasiakos, S.M. Exercising with low muscle glycogen content increases fat oxidation and decreases endogenous, but not exogenous carbohydrate oxidation. Metabolism 2019, 97, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Margolis, L.M.; Karl, J.P.; Wilson, M.A.; Coleman, J.L.; Whitney, C.C.; Pasiakos, S.M. Serum branched-chain amino acid metabolites increase in males when aerobic exercise is initiated with low muscle glycogen. Metabolites 2021, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Howarth, K.R.; Phillips, S.M.; MacDonald, M.J.; Richards, D.; Moreau, N.A.; Gibala, M.J. Effect of glycogen availability on human skeletal muscle protein turnover during exercise and recovery. J. Appl. Physiol. 2010, 109, 431–438. [Google Scholar] [CrossRef]
- Zhou, M.; Shao, J.; Wu, C.-Y.; Shu, L.; Dong, W.; Liu, Y.; Chen, M.; Wynn, R.M.; Wang, J.; Wang, J.; et al. Targeting BCAA Catabolism to treat obesity-associated insulin resistance. Diabetes 2019, 68, 1730–1746. [Google Scholar] [CrossRef]
- Mann, G.; Adegoke, O.A.J. Effects of ketoisocaproic acid and inflammation on glucose transport in muscle cells. Physiol. Rep. 2021, 9, e14673. [Google Scholar] [CrossRef]
- Kitaura, Y.; Shindo, D.; Ogawa, T.; Sato, A.; Shimomura, Y. Antihypertensive drug valsartan as a novel BDK inhibitor. Pharm. Res. 2021, 167, 105518. [Google Scholar] [CrossRef]
- NAVIGATOR Study Group; McMurray, J.J.; Holman, R.R.; Haffner, S.M.; Bethel, M.A.; Holzhauer, B.; Hua, T.A.; Belenkov, Y.; Boolell, M.; Buse, J.B.; et al. Effect of valsartan on the incidence of diabetes and cardiovascular events. N. Engl. J. Med. 2010, 362, 1477–1490. [Google Scholar]
- White, P.J.; McGarrah, R.W.; Grimsrud, P.A.; Tso, S.-C.; Yang, W.-H.; Haldeman, J.M.; Grenier-Larouche, T.; An, J.; Lapworth, A.L.; Astapova, I.; et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 2018, 27, 1281–1293. [Google Scholar] [CrossRef]
- Morrow, M.R.; Batchuluun, B.; Wu, J.; Ahmadi, E.; Leroux, J.M.; Mohammadi-Shemirani, P.; Desjardins, E.M.; Wang, Z.; Tsakiridis, E.E.; Lavoie, D.C.T.; et al. Inhibition of ATP-citrate lyase improves NASH, liver fibrosis, and dyslipidemia. Cell Metab. 2022, 34, 919–936. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; McCoin, C.S.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M.E. Acylcarnitines: Potential implications for skeletal muscle insulin resistance. FASEB J. 2015, 29, 336–345. [Google Scholar] [CrossRef]
- Enooku, K.; Nakagawa, H.; Fujiwara, N.; Kondo, M.; Minami, T.; Hoshida, Y.; Shibahara, J.; Tateishi, R.; Koike, K. Altered serum acylcarnitine profile is associated with the status of nonalcoholic fatty liver disease (NAFLD) and NAFLD-related hepatocellular carcinoma. Sci. Rep. 2019, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Remchak, M.M.E.; Heiston, E.M.; Ballantyne, A.; Dotson, B.L.; Stewart, N.R.; Spaeth, A.M.; Malin, S.K. Insulin sensitivity and metabolic flexibility parallel plasma TCA levels in early chronotype with metabolic syndrome. J. Clin. Endocrinol. Metab. 2022, 107, e3487–e3496. [Google Scholar] [CrossRef] [PubMed]
- Sasarman, F.; Ferdinandusse, S.; Sinasac, D.S.; Fung, E.; Sparkes, R.; Reeves, M.; Rombough, C.; Sass, J.O.; Voit, R.; Ruiter, J.P.N.; et al. 3-Hydroxyisobutyric acid dehydrogenase deficiency: Expanding the clinical spectrum and quantitation of D- and L-3-hydroxyisobutyric acid by an LC-MS/MS method. J. Inherit. Metab. Dis. 2022, 45, 445–455. [Google Scholar] [CrossRef]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Boström, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.-K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. β-Aminoisobutyric acid induces browning of white fat and hepatic β-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef]
- Hatazawa, Y.; Senoo, N.; Tadaishi, M.; Ogawa, Y.; Ezaki, O.; Kamei, Y.; Miura, S. Metabolomic analysis of the skeletal muscle of mice overexpressing PGC-1α. PLoS ONE 2015, 10, e0129084. [Google Scholar] [CrossRef]


| Leu | Val | Ile | BCAAs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Energy a | Protein b | Weight c | Energy a | Protein b | Weight c | Energy a | Protein b | Weight c | Energy a | Protein b | Weight c | ||
| ANIMAL | Chicken | 1.45 | 7.98 | 2.50 | 0.96 | 5.28 | 1.66 | 1.02 | 5.62 | 1.76 | 3.42 | 18.88 | 5.92 |
| Beef | 1.27 | 8.26 | 2.12 | 0.79 | 5.15 | 1.32 | 0.73 | 4.73 | 1.21 | 2.79 | 18.14 | 4.65 | |
| Pork | 1.08 | 8.17 | 2.31 | 0.73 | 5.52 | 1.56 | 0.63 | 4.77 | 1.35 | 2.43 | 18.46 | 5.21 | |
| Salmon | 1.19 | 8.43 | 2.16 | 0.75 | 5.34 | 1.37 | 0.67 | 4.78 | 1.23 | 2.61 | 18.54 | 4.76 | |
| Egg | 0.66 | 8.55 | 1.02 | 0.47 | 6.10 | 0.73 | 0.42 | 5.46 | 0.65 | 1.54 | 20.10 | 2.39 | |
| Whole milk | 0.46 | 8.89 | 0.28 | 0.31 | 6.13 | 0.19 | 0.25 | 4.85 | 0.15 | 1.02 | 19.86 | 0.62 | |
| LEGUMES | Peas | 0.53 | 7.50 | 0.62 | 0.35 | 4.94 | 0.41 | 0.30 | 4.32 | 0.36 | 1.18 | 16.76 | 1.39 |
| Lentils | 0.62 | 7.89 | 0.72 | 0.43 | 5.40 | 0.50 | 0.37 | 4.70 | 0.43 | 1.42 | 17.99 | 1.65 | |
| Soybeans | 0.69 | 7.70 | 1.19 | 0.42 | 4.72 | 0.73 | 0.41 | 4.58 | 0.71 | 1.52 | 17.00 | 2.64 | |
| Black beans | 0.57 | 8.43 | 0.75 | 0.37 | 5.52 | 0.49 | 0.32 | 4.65 | 0.42 | 1.26 | 18.60 | 1.66 | |
| GRAINS | Whole wheat | 0.29 | 6.78 | 0.87 | 0.19 | 4.41 | 0.57 | 0.15 | 3.63 | 0.47 | 0.63 | 14.82 | 1.90 |
| Brown rice | 0.19 | 8.28 | 0.21 | 0.14 | 5.85 | 0.15 | 0.10 | 4.22 | 0.11 | 0.42 | 18.35 | 0.47 | |
| Oats | 0.26 | 7.76 | 0.22 | 0.18 | 5.45 | 0.16 | 0.13 | 3.98 | 0.11 | 0.57 | 17.20 | 0.49 | |
| Quinoa | 0.27 | 7.13 | 0.32 | 0.19 | 5.04 | 0.23 | 0.16 | 4.28 | 0.19 | 0.61 | 16.45 | 0.75 | |
| NUTS | Almonds | 0.24 | 6.53 | 1.42 | 0.14 | 3.79 | 0.82 | 0.12 | 3.33 | 0.72 | 0.51 | 13.64 | 2.96 |
| Peanuts | 0.29 | 6.43 | 1.65 | 0.19 | 4.16 | 1.07 | 0.16 | 3.49 | 0.89 | 0.64 | 14.07 | 3.61 | |
| Pecans | 0.09 | 6.73 | 0.63 | 0.06 | 4.62 | 0.43 | 0.05 | 3.78 | 0.36 | 0.21 | 15.13 | 1.42 | |
| Walnuts | 0.18 | 7.42 | 1.16 | 0.11 | 4.78 | 0.75 | 0.10 | 3.97 | 0.62 | 0.39 | 16.16 | 2.53 | |
| VEGETABLES | Potatoes | 0.12 | 6.01 | 0.10 | 0.11 | 5.60 | 0.09 | 0.08 | 4.08 | 0.07 | 0.30 | 15.69 | 0.26 |
| Carrots | 0.13 | 5.95 | 0.06 | 0.09 | 4.03 | 0.04 | 0.10 | 4.49 | 0.04 | 0.33 | 14.47 | 0.13 | |
| Butternut | 0.15 | 6.88 | 0.06 | 0.12 | 5.26 | 0.05 | 0.10 | 4.72 | 0.04 | 0.37 | 16.87 | 0.15 | |
| Sweet potatoes | 0.14 | 6.19 | 0.12 | 0.13 | 5.77 | 0.12 | 0.08 | 3.67 | 0.07 | 0.35 | 15.63 | 0.32 | |
| Green beans | 0.41 | 7.62 | 0.14 | 0.33 | 6.11 | 0.11 | 0.24 | 4.53 | 0.09 | 0.98 | 18.25 | 0.34 | |
| FRUITS | Apple | 0.03 | 6.47 | 0.02 | 0.03 | 5.97 | 0.02 | 0.02 | 2.98 | 0.01 | 0.08 | 15.42 | 0.04 |
| Banana | 0.10 | 7.97 | 0.09 | 0.07 | 5.51 | 0.06 | 0.04 | 3.28 | 0.04 | 0.21 | 16.76 | 0.18 | |
| Mango | 0.10 | 7.40 | 0.06 | 0.08 | 6.21 | 0.05 | 0.06 | 4.29 | 0.03 | 0.24 | 17.90 | 0.14 | |
| Mushrooms | 0.83 | 6.05 | 0.18 | 1.60 | 11.70 | 0.35 | 0.52 | 3.83 | 0.12 | 2.95 | 21.58 | 0.65 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Bandt, J.-P.; Coumoul, X.; Barouki, R. Branched-Chain Amino Acids and Insulin Resistance, from Protein Supply to Diet-Induced Obesity. Nutrients 2023, 15, 68. https://doi.org/10.3390/nu15010068
De Bandt J-P, Coumoul X, Barouki R. Branched-Chain Amino Acids and Insulin Resistance, from Protein Supply to Diet-Induced Obesity. Nutrients. 2023; 15(1):68. https://doi.org/10.3390/nu15010068
Chicago/Turabian StyleDe Bandt, Jean-Pascal, Xavier Coumoul, and Robert Barouki. 2023. "Branched-Chain Amino Acids and Insulin Resistance, from Protein Supply to Diet-Induced Obesity" Nutrients 15, no. 1: 68. https://doi.org/10.3390/nu15010068
APA StyleDe Bandt, J.-P., Coumoul, X., & Barouki, R. (2023). Branched-Chain Amino Acids and Insulin Resistance, from Protein Supply to Diet-Induced Obesity. Nutrients, 15(1), 68. https://doi.org/10.3390/nu15010068