
The Role and Mechanism of Gut Microbiota in Pulmonary Arterial Hypertension


Abstract
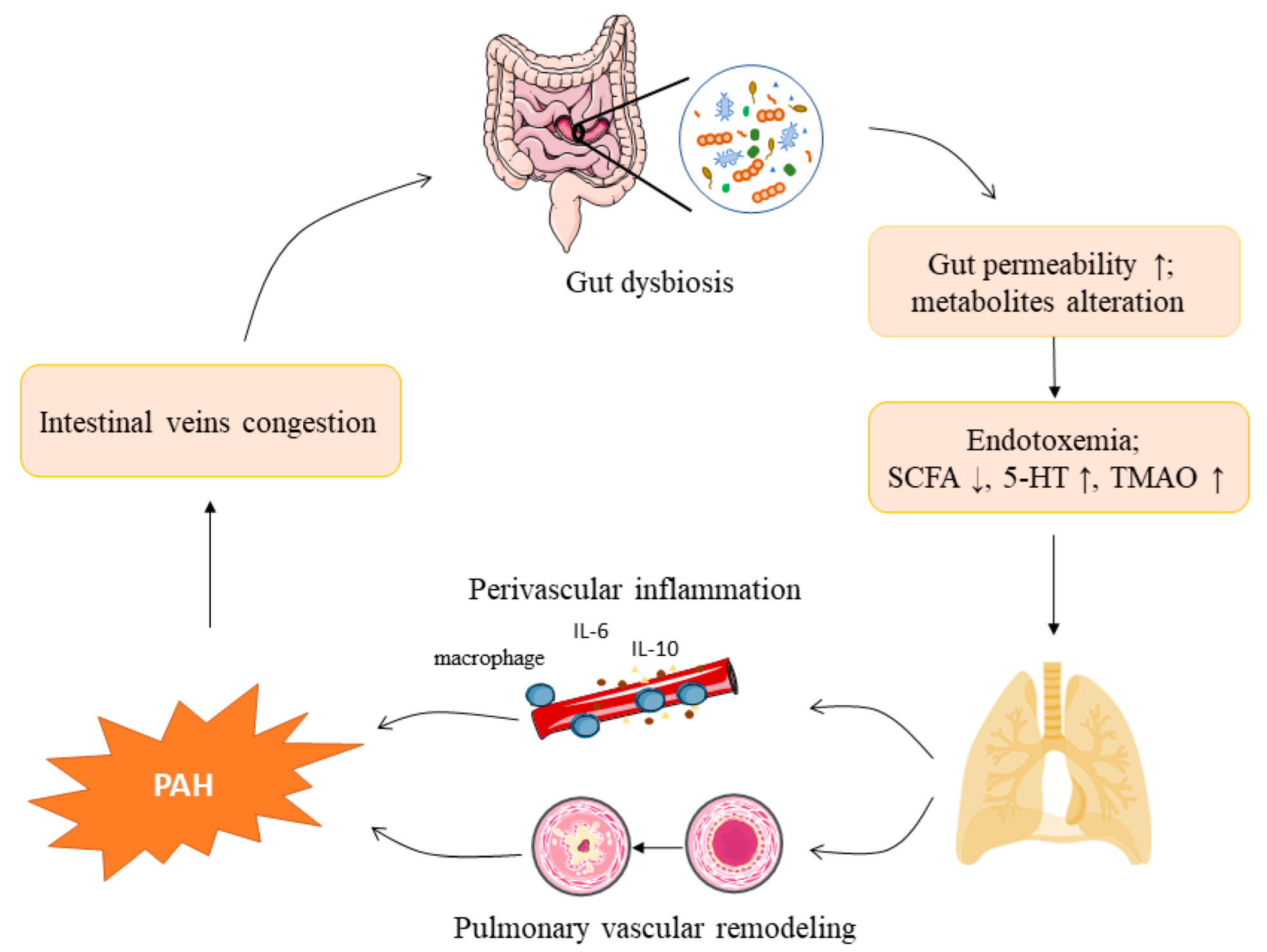
1. Introduction
2. The Gut Microbiota and Pulmonary Arterial Hypertension
2.1. Gut Microbiota Composition
2.2. Shifts in the Gut Microbiota Composition in Pulmonary Arterial Hypertension
2.3. Lipopolysaccharides in Pulmonary Arterial Hypertension
2.4. Gut Microbial Metabolites and Pulmonary Arterial Hypertension
2.5. Short-Chain Fatty Acids
2.6. Trimethylamine N-Oxide
2.7. 5-Hydroxytryptophan
3. Gut Microbiota-Directed Therapies for Pulmonary Arterial Hypertension
3.1. Prebiotics and Probiotics
3.2. Fecal Microbiota Transplantation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McLaughlin, V.V.; McGoon, M.D. Pulmonary Arterial Hypertension. Circulation 2006, 114, 1417–1431. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Gerard, K.; Halloran, K.; Hanhauser, A.; Monacelli, S.; Warford, C.; Thai, P.N.; Chiamvimonvat, N.; Lakshminrusimha, S.; Steinhorn, R.H.; et al. Intestinal Dysbiosis and the Developing Lung: The Role of Toll-Like Receptor 4 in the Gut-Lung Axis. Front. Immunol. 2020, 11, 357. [Google Scholar] [CrossRef] [PubMed]
- Farber, H.W.; Miller, D.P.; Poms, A.D.; Badesch, D.B.; Frost, A.E.; Rouzic, E.M.-L.; Romero, A.J.; Benton, W.W.; Elliott, C.G.; McGoon, M.D.; et al. Five-Year Outcomes of Patients Enrolled in the REVEAL Registry. Chest 2015, 148, 1043–1054. [Google Scholar] [CrossRef]
- Anand, V.; Roy, S.S.; Archer, S.L.; Weir, E.K.; Garg, S.K.; Duval, S.; Thenappan, T. Trends and Outcomes of Pulmonary Arterial Hypertension–Related Hospitalizations in the United States: Analysis of the Nationwide Inpatient Sample Database From 2001 Through 2012. JAMA Cardiol. 2016, 1, 1021. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and Pathobiology of Pulmonary Hypertension: State of the Art and Research Perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Kwant, C.T.; Ruiter, G.; Noordegraaf, A.V. Malnutrition in Pulmonary Arterial Hypertension. Curr. Opin. Pulm. Med. 2019, 25, 405–409. [Google Scholar] [CrossRef]
- Paone, P.; Cani, P.D. Mucus Barrier, Mucins and Gut Microbiota: The Expected Slimy Partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.; Chen, G.Y.; Núñez, G. Role of the Gut Microbiota in Immunity and Inflammatory Disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Clements, S.J.; Carding, S.R. Diet, the Intestinal Microbiota, and Immune Health in Aging. Crit. Rev. Food Sci. 2018, 58, 651–661. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Marinelli, L.; Blottière, H.M.; Larraufie, P.; Lapaque, N. SCFA: Mechanisms and Functional Importance in the Gut. Proc. Nutr. Soc. 2021, 80, 37–49. [Google Scholar] [CrossRef]
- Callejo, M.; Barberá, J.A.; Duarte, J.; Perez-Vizcaino, F. Impact of Nutrition on Pulmonary Arterial Hypertension. Nutrients 2020, 12, 169. [Google Scholar] [CrossRef]
- Brown, J.M.; Hazen, S.L. Microbial Modulation of Cardiovascular Disease. Nat. Rev. Microbiol. 2018, 16, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Shui, Y.; Cui, Y.; Tang, C.; Wang, X.; Qiu, X.; Hu, W.; Fei, L.; Li, Y.; Zhang, S.; et al. Gut Microbiota Dependent Trimethylamine N-Oxide Aggravates Angiotensin II–Induced Hypertension. Redox Biol. 2021, 46, 102115. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal Microbiota Metabolism of l-carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Markó, L.; Höges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G.; et al. Short-Chain Fatty Acid Propionate Protects from Hypertensive Cardiovascular Damage. Circulation 2019, 139, 1407–1421. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.C.; Richards, E.M.; Raizada, M.K. Pulmonary Hypertension: Pathophysiology beyond the Lung. Pharmacol. Res. 2020, 151, 104518. [Google Scholar] [CrossRef]
- Allam-Ndoul, B.; Castonguay-Paradis, S.; Veilleux, A. Gut Microbiota and Intestinal Trans-Epithelial Permeability. Int. J. Mol. Sci. 2020, 21, 6402. [Google Scholar] [CrossRef] [PubMed]
- Hug, H.; Mohajeri, M.H.; La Fata, G. Toll-Like Receptors: Regulators of the Immune Response in the Human Gut. Nutrients 2018, 10, 203. [Google Scholar] [CrossRef]
- Young, K.C.; Hussein, S.M.A.; Dadiz, R.; DeMello, D.; Devia, C.; Hehre, D.; Suguihara, C. Toll-Like Receptor 4–Deficient Mice are Resistant to Chronic Hypoxia-Induced Pulmonary Hypertension. Exp. Lung Res. 2010, 36, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Karoor, V.; Strassheim, D.; Sullivan, T.; Verin, A.; Umapathy, N.S.; Dempsey, E.C.; Frank, D.N.; Stenmark, K.R.; Gerasimovskaya, E. The Short-Chain Fatty Acid Butyrate Attenuates Pulmonary Vascular Remodeling and Inflammation in Hypoxia-Induced Pulmonary Hypertension. Int. J. Mol. Sci. 2021, 22, 9916. [Google Scholar] [CrossRef]
- Huang, Y.; Lin, F.; Tang, R.; Bao, C.; Zhou, Q.; Ye, K.; Shen, Y.; Liu, C.; Hong, C.; Yang, K.; et al. Gut Microbial Metabolite Trimethylamine N-Oxide Aggravates Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2022, 66, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zheng, S.; He, R.; Gui, L.; Lin, M.; Sham, J.S.K. Serotonin and Chronic Hypoxic Pulmonary Hypertension Activate a NADPH Oxidase 4 and TRPM2 Dependent Pathway for Pulmonary Arterial Smooth Muscle Cell Proliferation and Migration. Vasc. Pharmacol. 2021, 138, 106860. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Zmora, N.; Zilberman-Schapira, G.; Suez, J.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Kotler, E.; Zur, M.; Regev-Lehavi, D.; Brik, R.B.; et al. Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics is Associated with Unique Host and Microbiome Features. Cell 2018, 174, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Plé, C.; Breton, J.; Daniel, C.; Foligné, B. Maintaining Gut Ecosystems for Health: Are Transitory Food Bugs Stowaways or Part of the Crew? Int. J. Food Microbiol. 2015, 213, 139–143. [Google Scholar] [CrossRef]
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond gut feelings: How the Gut Microbiota Regulates Blood Pressure. Nat. Rev. Cardiol. 2018, 15, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Jin, J.; Su, X.; Yin, X.; Gao, J.; Wang, X.; Zhang, S.; Bu, P.; Wang, M.; Zhang, Y.; et al. Intestinal Flora Modulates Blood Pressure by Regulating the Synthesis of Intestinal-Derived Corticosterone in High Salt-Induced Hypertension. Circ. Res. 2020, 126, 839–853. [Google Scholar] [CrossRef] [PubMed]
- Gavin, P.G.; Mullaney, J.A.; Loo, D.; Cao, K.L.; Gottlieb, P.A.; Hill, M.M.; Zipris, D.; Hamilton-Williams, E.E. Intestinal Metaproteomics Reveals Host-Microbiota Interactions in Subjects at Risk for Type 1 Diabetes. Diabetes Care 2018, 41, 2178–2186. [Google Scholar] [CrossRef]
- Callejo, M.; Mondejar-Parreño, G.; Barreira, B.; Izquierdo-Garcia, J.L.; Morales-Cano, D.; Esquivel-Ruiz, S.; Moreno, L.; Cogolludo, Á.; Duarte, J.; Perez-Vizcaino, F. Pulmonary Arterial Hypertension Affects the Rat Gut Microbiome. Sci. Rep. 2018, 8, 9681. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Mo, Q.; Wang, L.; Peng, F.; Zhou, Y.; Zou, W.; Sun, R.; Liang, C.; Zheng, M.; Li, H.; et al. Changes in the Gut Microbiome and Metabolome in a Rat Model of Pulmonary Arterial Hypertension. Bioengineered 2021, 12, 5173–5183. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Rigatto, K.; Gazzana, M.B.; Knorst, M.M.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. Altered Gut Microbiome Profile in Patients with Pulmonary Arterial Hypertension. Hypertension 2020, 75, 1063–1071. [Google Scholar] [CrossRef]
- Ikubo, Y.; Sanada, T.J.; Hosomi, K.; Park, J.; Naito, A.; Shoji, H.; Misawa, T.; Suda, R.; Sekine, A.; Sugiura, T.; et al. Altered Gut Microbiota and Its Association with Inflammation in Patients with Chronic Thromboembolic Pulmonary Hypertension: A Single-Center Observational Study in Japan. BMC Pulm. Med. 2022, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Sandek, A.; Bjarnason, I.; Volk, H.; Crane, R.; Meddings, J.B.; Niebauer, J.; Kalra, P.R.; Buhner, S.; Herrmann, R.; Springer, J.; et al. Studies on Bacterial Endotoxin and Intestinal Absorption Function in Patients with Chronic Heart Failure. Int. J. Cardiol. 2012, 157, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Nagatomo, Y.; Tang, W.H.W. Intersections between Microbiome and Heart Failure: Revisiting the Gut Hypothesis. J. Card. Fail. 2015, 21, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Sanada, T.J.; Hosomi, K.; Shoji, H.; Park, J.; Naito, A.; Ikubo, Y.; Yanagisawa, A.; Kobayashi, T.; Miwa, H.; Suda, R.; et al. Gut Microbiota Modification Suppresses the Development of Pulmonary Arterial Hypertension in an SU5416/Hypoxia Rat Model. Pulm. Circ. 2020, 10, 1365052046–1765618565. [Google Scholar] [CrossRef]
- Sharma, R.K.; Oliveira, A.C.; Yang, T.; Karas, M.M.; Li, J.; Lobaton, G.O.; Aquino, V.P.; Robles-Vera, I.; de Kloet, A.D.; Krause, E.G.; et al. Gut Pathology and Its Rescue by ACE2 (Angiotensin-Converting Enzyme 2) in Hypoxia-Induced Pulmonary Hypertension. Hypertension 2020, 76, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Mesmin, L.; Chassaing, B.; Desvaux, M.; De Paepe, K.; Gresse, R.; Sauvaitre, T.; Forano, E.; de Wiele, T.V.; Schuller, S.; Juge, N.; et al. Experimental Models to Study Intestinal Microbes-Mucus Interactions in Health and Disease. Fems Microbiol. Rev. 2019, 43, 457–489. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The Composition of the Gut Microbiota Shapes the Colon Mucus Barrier. Embo Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef]
- Rodriguez-Pineiro, A.M.; Johansson, M.E. The Colonic Mucus Protection Depends on the Microbiota. Gut Microbes 2015, 6, 326–330. [Google Scholar] [CrossRef]
- Sharma, R.K.; Oliveira, A.C.; Yang, T.; Kim, S.; Zubcevic, J.; Aquino, V.; Lobaton, G.O.; Goel, R.; Richards, E.M.; Raizada, M.K. Pulmonary Arterial Hypertension-Associated Changes in Gut Pathology and Microbiota. ERJ Open Res. 2020, 6, 253–2019. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J. Recognition of Lipopolysaccharide Pattern by TLR4 Complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed]
- Bhagwani, A.; Thompson, A.A.R.; Farkas, L. When Innate Immunity Meets Angiogenesis—The Role of Toll-Like Receptors in Endothelial Cells and Pulmonary Hypertension. Front. Med. 2020, 7, 352. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Srivastava, M.; Saqib, U.; Liu, D.; Faisal, S.M.; Sugathan, S.; Bishnoi, S.; Baig, M.S. Potential Therapeutic Targets for Inflammation in Toll-Like Receptor 4 (TLR4)-Mediated Signaling Pathways. Int. Immunopharmacol. 2016, 40, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Jiang, X.; Tamosiuniene, R.; Sung, Y.K.; Qian, J.; Dhillon, G.; Gera, L.; Farkas, L.; Rabinovitch, M.; Zamanian, R.T.; et al. Blocking Macrophage Leukotriene B4 Prevents Endothelial Injury and Reverses Pulmonary Hypertension. Sci. Transl. Med. 2013, 5, 117r–200r. [Google Scholar] [CrossRef] [PubMed]
- Frid, M.G.; Brunetti, J.A.; Burke, D.L.; Carpenter, T.C.; Davie, N.J.; Reeves, J.T.; Roedersheimer, M.T.; van Rooijen, N.; Stenmark, K.R. Hypoxia-Induced Pulmonary Vascular Remodeling Requires Recruitment of Circulating Mesenchymal Precursors of a Monocyte/Macrophage Lineage. Am. J. Pathol. 2006, 168, 659–669. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF- B Pathway in Inflammation. Csh. Perspect. Biol. 2009, 1, a1651. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Crosby, A.; Southwood, M.; Yang, P.; Tajsic, T.; Toshner, M.; Appleby, S.; Shanahan, C.M.; Bloch, K.D.; Pepke-Zaba, J.; et al. Bone Morphogenetic Protein Receptor Type II Deficiency and Increased Inflammatory Cytokine Production. A Gateway to Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 859–872. [Google Scholar] [CrossRef]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 Overexpression Induces Pulmonary Hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J.; Zhao, Y. Endothelial Cells in the Pathogenesis of Pulmonary Arterial Hypertension. Eur. Respir. J. 2021, 58, 2003957. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, R.; Iovene, M.R.; Romano, C.C.; Rizzo, A. Induction of VEGF and MMP-9 Expression by Toll-Like Receptor 2/4 in Human Endothelial Cells Infected with Chlamydia Pneumoniae. Int. J. Immunopathol. Pharmacol. 2012, 25, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-Mesenchymal Transition in Pulmonary Hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef]
- Suzuki, T.; Carrier, E.J.; Talati, M.H.; Rathinasabapathy, A.; Chen, X.; Nishimura, R.; Tada, Y.; Tatsumi, K.; West, J. Isolation and Characterization of Endothelial-to-Mesenchymal Transition Cells in Pulmonary Arterial Hypertension. Am. J. Physiol.-Lung C. 2018, 314, L118–L126. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tada, Y.; Nishimura, R.; Kawasaki, T.; Sekine, A.; Urushibara, T.; Kato, F.; Kinoshita, T.; Ikari, J.; West, J.; et al. Endothelial-to-Mesenchymal Transition in Lipopolysaccharide-Induced Acute Lung Injury Drives a Progenitor Cell-Like Phenotype. Am. J. Physiol.-Lung C. 2016, 310, L1185–L1198. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.J.; Patel, K.D.; Kubes, P. Platelets Express Functional Toll-Like Receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide Stimulates Platelet Secretion and Potentiates Platelet Aggregation via TLR4/MyD88 and the cGMP-Dependent Protein Kinase Pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.M.; Chanthaphavong, R.S.; Sodhi, C.P.; Hackam, D.J.; Billiar, T.R.; Bauer, P.M. Genetic Deletion of Toll-Like Receptor 4 on Platelets Attenuates Experimental Pulmonary Hypertension. Circ. Res. 2014, 114, 1596–1600. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ambalavanan, N.; Liu, H.; Sun, Y.; Jhala, N.; Bradley, W.E.; Dell’Italia, L.J.; Michalek, S.; Wu, H.; Steele, C.; et al. TLR4 Regulates Pulmonary Vascular Homeostasis and Remodeling via Redox Signaling. Front. Biosci. 2016, 21, 397–409. [Google Scholar]
- Chen, L.; He, F.J.; Dong, Y.; Huang, Y.; Wang, C.; Harshfield, G.A.; Zhu, H. Modest Sodium Reduction Increases Circulating Short-Chain Fatty Acids in Untreated Hypertensives. Hypertension 2020, 76, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S. Tryptophan Dietary Impacts Gut Barrier and Metabolic Diseases. Front. Immunol. 2019, 10, 2113. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Su, W.; Rahat-Rozenbloom, S.; Wolever, T.M.S.; Comelli, E.M. Adiposity, Gut Microbiota and Faecal Short Chain Fatty Acids are Linked in Adult Humans. Nutr. Diabetes 2014, 4, e121. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, W.; Liu, Z.; Cong, Y. Microbiota Metabolite Short Chain Fatty Acids, GPCR, and Inflammatory Bowel Diseases. J. Gastroenterol. 2017, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The Gut Microbiota, Bacterial Metabolites and Colorectal Cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of Human Colonic Butyrate-Producing Bacteria Revealed by Analysis of the Butyryl-CoA:Acetate CoA-Transferase Gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef]
- Ze, X.; Duncan, S.H.; Louis, P.; Flint, H.J. Ruminococcus Bromii is a Keystone Species for the Degradation of Resistant Starch in the Human Colon. ISME J. 2012, 6, 1535–1543. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.; Wagenaar, G.; Garssen, J.; Folkerts, G.; Henricks, P. Pro- and Anti-inflammatory Effects of Short Chain Fatty Acids on Immune and Endothelial Cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.A.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The Gut Microbiota and Host Health: A New Clinical Frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-Chain Fatty Acids Activate GPR41 and GPR43 on Intestinal Epithelial cells to Promote Inflammatory Responses in Mice. Gastroenterology 2013, 145, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M.; Kotani, J.; Usami, M. Butyrate and Propionate Induced Activated or Non-Activated Neutrophil Apoptosis via HDAC Inhibitor Activity but without Activating GPR-41/GPR-43 Pathways. Nutrition 2010, 26, 653–661. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.; Park, B.O.; Kwak, Y.S. Perspectives on the Therapeutic Potential of Short-Chain Fatty Acid Receptors. BMB Rep. 2014, 47, 173–178. [Google Scholar] [CrossRef]
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Evidence for the Gut Microbiota Short-Chain Fatty Acids as Key Pathophysiological Molecules Improving Diabetes. Mediat. Inflamm. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of Histone Deacetylases in Epigenetic Regulation: Emerging Paradigms from Studies with Inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef]
- Schilderink, R.; Verseijden, C.; de Jonge, W.J. Dietary Inhibitors of Histone Deacetylases in Intestinal Immunity and Homeostasis. Front. Immunol. 2013, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Das Gupta, K.; Shakespear, M.R.; Iyer, A.; Fairlie, D.P.; Sweet, M.J. Histone Deacetylases in Monocyte/Macrophage Development, Activation and Metabolism: Refining HDAC Targets for Inflammatory and Infectious Diseases. Clin. Transl. Immunol. 2016, 5, e62. [Google Scholar] [CrossRef]
- Tang, J.; Yan, H.; Zhuang, S. Histone Deacetylases as Targets for Treatment of Multiple Diseases. Clin. Sci. 2013, 124, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Leus, N.G.; Zwinderman, M.R.; Dekker, F.J. Histone Deacetylase 3 (HDAC 3) as Emerging Drug Target in NF-κB-Mediated Inflammation. Curr. Opin. Chem. Biol. 2016, 33, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; van Esch, B.C.A.M.; Henricks, P.A.J.; Folkerts, G.; Garssen, J. The Anti-inflammatory Effects of Short Chain Fatty Acids on Lipopolysaccharide- or Tumor Necrosis Factor α-Stimulated Endothelial Cells via Activation of GPR41/43 and Inhibition of HDACs. Front. Pharmacol. 2018, 9, 533. [Google Scholar] [CrossRef]
- Burger-van Paassen, N.; Vincent, A.; Puiman, P.J.; van der Sluis, M.; Bouma, J.; Boehm, G.; van Goudoever, J.B.; van Seuningen, I.; Renes, I.B. The Regulation of Intestinal Mucin MUC2 Expression by Short-Chain Fatty Acids: Implications for Epithelial Protection. Biochem. J. 2009, 420, 211–219. [Google Scholar] [CrossRef]
- Hagihara, M.; Kuroki, Y.; Ariyoshi, T.; Higashi, S.; Fukuda, K.; Yamashita, R.; Matsumoto, A.; Mori, T.; Mimura, K.; Yamaguchi, N.; et al. Clostridium butyricum Modulates the Microbiome to Protect Intestinal Barrier Function in Mice with Antibiotic-Induced Dysbiosis. iScience 2020, 23, 100772. [Google Scholar] [CrossRef]
- Pan, L.L.; Niu, W.; Fang, X.; Liang, W.; Li, H.; Chen, W.; Zhang, H.; Bhatia, M.; Sun, J. Clostridium butyricum Strains Suppress Experimental Acute Pancreatitis by Maintaining Intestinal Homeostasis. Mol. Nutr. Food Res. 2019, 63, e1801419. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, J.; Du, J.; Wang, F.; Fang, R.; Yu, C.; Xiong, J.; Chen, W.; Lu, Z.; Liu, J. Clostridium Butyricum Exerts a Neuroprotective Effect in a Mouse Model of Traumatic Brain Injury via the Gut-Brain Axis. Neurogastroenterol. Motil. 2018, 30, e13260. [Google Scholar] [CrossRef] [PubMed]
- Shang, H.; Sun, J.; Chen, Y.Q. Clostridium Butyricum CGMCC0313.1 Modulates Lipid Profile, Insulin Resistance and Colon Homeostasis in Obese Mice. PLoS ONE 2016, 11, e154373. [Google Scholar] [CrossRef] [PubMed]
- Mackay, C.R.; Artis, D.; Yu, D.; Xavier, R.J.; Ng, A.; Mackay, F.; Sierro, F.; Kranich, J.; Rolph, M.S.; Teixeira, M.M.; et al. Regulation of Inflammatory Responses by Gut Microbiota and Chemoattractant Receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar]
- Yuille, S.; Reichardt, N.; Panda, S.; Dunbar, H.; Mulder, I.E. Human Gut Bacteria as Potent Class I Histone Deacetylase Inhibitors in Vitro through Production of Butyric Acid and Valeric Acid. PLoS ONE 2018, 13, e201073. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M. The Role of Short-Chain Fatty Acids in Health and Disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar]
- Thenappan, T.; Khoruts, A.; Chen, Y.; Weir, E.K. Can Intestinal Microbiota and Circulating Microbial Products Contribute to Pulmonary Arterial Hypertension? Am. J. Physiol.-Heart C 2019, 317, H1093–H1101. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.; et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; Vallim, T.Q.D.A.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-Oxide, a Metabolite Associated with Atherosclerosis, Exhibits Complex Genetic and Dietary Regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal Microbiota Composition Modulates Choline Bioavailability from Diet and Accumulation of the Proatherogenic Metabolite Trimethylamine-N-Oxide. Mbio 2015, 6, e2481. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Ke, B.; Du, J. TMAO: How Gut Microbiota Contributes to Heart Failure. Transl. Res. 2021, 228, 109–125. [Google Scholar] [CrossRef]
- Egermayer, P.; Town, G.I.; Peacock, A.J. Role of Serotonin in the Pathogenesis of Acute and Chronic Pulmonary Hypertension. Thorax 1999, 54, 161–168. [Google Scholar] [CrossRef]
- Ge, X.; Ding, C.; Zhao, W.; Xu, L.; Tian, H.; Gong, J.; Zhu, M.; Li, J.; Li, N. Antibiotics-Induced Depletion of Mice Microbiota Induces Changes in Host Serotonin Biosynthesis and Intestinal Motility. J. Transl. Med. 2017, 15, 13. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Salmonson, C.E.; III, J.F.R.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut Microbes Promote Colonic Serotonin Production through an Effect of Short-Chain Fatty Acids on Enterochromaffin Cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- West, J.D.; Carrier, E.J.; Bloodworth, N.C.; Schroer, A.K.; Chen, P.; Ryzhova, L.M.; Gladson, S.; Shay, S.; Hutcheson, J.D.; Merryman, W.D.; et al. Serotonin 2B Receptor Antagonism Prevents Heritable Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e148657. [Google Scholar] [CrossRef]
- Bloodworth, N.C.; Clark, C.R.; West, J.D.; Snider, J.C.; Gaskill, C.; Shay, S.; Scott, C.; Bastarache, J.; Gladson, S.; Moore, C.; et al. Bone Marrow–Derived Proangiogenic Cells Mediate Pulmonary Arteriole Stiffening via Serotonin 2B Receptor Dependent Mechanism. Circ. Res. 2018, 123, e51–e64. [Google Scholar] [CrossRef]
- Guilluy, C.; Eddahibi, S.; Agard, C.; Guignabert, C.; Izikki, M.; Tu, L.; Savale, L.; Humbert, M.; Fadel, E.; Adnot, S.; et al. RhoA and Rho Kinase Activation in Human Pulmonary Hypertension: Role of 5-HT signaling. Am. J. Respir. Crit. Care Med. 2009, 179, 1151–1158. [Google Scholar] [CrossRef]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ Sensitivity of Smooth Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin Phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef]
- Liu, Y.; Suzuki, Y.J.; Day, R.M.; Fanburg, B.L. Rho Kinase–Induced Nuclear Translocation of ERK1/ERK2 in Smooth Muscle Cell Mitogenesis Caused by Serotonin. Circ. Res. 2004, 95, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.Y.; Mair, K.M.; Harvey, A.P.; Montezano, A.C.; Touyz, R.M.; MacLean, M.R. Serotonin Signaling Through the 5-HT1B Receptor and NADPH Oxidase 1 in Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Eddahibi, S.; Humbert, M.; Fadel, E.; Raffestin, B.; Darmon, M.; Capron, F.; Simonneau, G.; Dartevelle, P.; Hamon, M.; Adnot, S. Serotonin Transporter Overexpression is Responsible for Pulmonary Artery Smooth Muscle Hyperplasia in Primary Pulmonary Hypertension. J. Clin. Investig. 2001, 108, 1141–1150. [Google Scholar] [CrossRef]
- MacLean, M.R.; Deuchar, G.A.; Hicks, M.N.; Morecroft, I.; Shen, S.; Sheward, J.; Colston, J.; Loughlin, L.; Nilsen, M.; Dempsie, Y.; et al. Overexpression of the 5-Hydroxytryptamine Transporter Gene. Circulation 2004, 109, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Marcos, E.; Adnot, S.; Pham, M.H.; Nosjean, A.; Raffestin, B.; Hamon, M.; Eddahibi, S. Serotonin Transporter Inhibitors Protect against Hypoxic Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2003, 168, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Marcos, E.; Fadel, E.; Sanchez, O.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Hamon, M.; Adnot, S.; Eddahibi, S. Serotonin-Induced Smooth Muscle Hyperplasia in Various Forms of Human Pulmonary Hypertension. Circ. Res. 2004, 94, 1263–1270. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Chen, G.; Zhou, Y.; Yao, H.; Tan, H. Upregulation of MiR-361-3p Suppresses Serotonin-Induced Proliferation in Human Pulmonary Artery Smooth Muscle Cells by Targeting SERT. Cell. Mol. Biol. Lett. 2020, 25, 45. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert Consensus Document. The International Scientific Association for Probiotics and Prebiotics Consensus Statement on the Scope and Appropriate Use of the Term Probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Wedgwood, S.; Warford, C.; Agvatisiri, S.R.; Thai, P.N.; Chiamvimonvat, N.; Kalanetra, K.M.; Lakshminrusimha, S.; Steinhorn, R.H.; Mills, D.A.; Underwood, M.A. The Developing Gut–Lung Axis: Postnatal Growth Restriction, Intestinal Dysbiosis, and Pulmonary Hypertension in a Rodent Model. Pediatr. Res. 2020, 87, 472–479. [Google Scholar] [CrossRef]
- Liu, Y.; Fatheree, N.Y.; Mangalat, N.; Rhoads, J.M. Lactobacillus Reuteri Strains Reduce Incidence and Severity of Experimental Necrotizing Enterocolitis via Modulation of TLR4 and NF-κB Signaling in the Intestine. Am. J. Physiol.-Gastr. Liver Physiol. 2012, 302, G608–G617. [Google Scholar]
- Wang, Y.; Gao, L.; Yang, Z.; Chen, F.; Zhang, Y. Effects of Probiotics on Ghrelin and Lungs in Children with Acute Lung Injury: A Double-Blind Randomized, Controlled Trial. Pediatr Pulmonol. 2018, 53, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Blaut, M. Relationship of Prebiotics and Food to Intestinal Microflora. Eur. J. Nutr. 2002, 41, i11–i16. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Byrne, C.S.; Morrison, D.J.; Murphy, K.G.; Preston, T.; Tedford, C.; Garcia-Perez, I.; Fountana, S.; Serrano-Contreras, J.I.; Holmes, E.; et al. Dietary Supplementation with Inulin-Propionate Ester or Inulin Improves Insulin Sensitivity in Adults with Overweight and Obesity with Distinct Effects on the Gut Microbiota, Plasma Metabolome and Systemic Inflammatory Responses: A Randomised Cross-Over Trial. Gut 2019, 68, 1430–1438. [Google Scholar]
- Hiel, S.; Gianfrancesco, M.A.; Rodriguez, J.; Portheault, D.; Leyrolle, Q.; Bindels, L.B.; Gomes Da Silveira Cauduro, C.; Mulders, M.D.G.H.; Zamariola, G.; Azzi, A.; et al. Link between Gut Microbiota and Health Outcomes in Inulin -Treated Obese Patients: Lessons from the Food4Gut Multicenter Randomized Placebo-Controlled Trial. Clin. Nutr. 2020, 39, 3618–3628. [Google Scholar] [CrossRef] [PubMed]
- Nood, V.E.; Vrieze, A.; Nieuwdorp, M.; Fuentes Enriquez De Salamanca, S.; Zoetendal, E.G.; Vos, D.W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal Infusion of Donor Feces for Recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef]
- Moutsoglou, D.M. 2021 American Thoracic Society BEAR Cage Winning Proposal: Microbiome Transplant in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2022, 205, 13–16. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Experimental Models/Populations | Effects and Observation | Preclinical or Clinical | Ref. |
|---|---|---|---|
| SUGEN5416/hypoxia rat model | ↑Firmicutes, Actinobacteria, Cyanobacteria ↓Bacteroides, Akkermansia | Preclinical | Takayuki J. Sanada et al. (2020) [35] |
| MCT-induced rat model | ↑Firmicutes, Proteobacteria, Actinobacteria ↑Allobaculum, Ralstonia, Bifidobacterium ↓Bacteroidota, Spirochaetota ↓Lactobacillus, Romboutsia | Preclinical | Wei Hong et al. (2021) [30] |
| mice with hypoxia | ↑F/B, Proteobacteria, Prevotella, Oscillospira, Ruminococcus ↓Lactobacillus | Preclinical | Ravindra K. Sharma et al. (2020) [36] |
| SUGEN5416/hypoxia rat model | ↓Bacteroidetes, Odoribacteraceae ↑Firmicutes, Peptostreptococcaceae | Preclinical | María Callejo et al. (2018) [29] |
| Patients with type I PAH | ↑S. parasanguinis, R. gnavus, C. aerofaciens ↑Collinsella, Blautia ↓B. crossotus, B. cellulosilyticus, E. siraeum, B. vulgatus, A. muciniphila, ↓B. intestinihominis | Clinical | Seungbum Kim et al. (2020) [31] |
| Patients with CTEPH | ↓Faecalibacterium, Roseburia, Fusicatenibacter | Clinical | Yumiko Ikubo et al. (2022) [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-H.; Yuan, W.; Meng, L.-K.; Zhong, J.-C.; Liu, X.-Y. The Role and Mechanism of Gut Microbiota in Pulmonary Arterial Hypertension. Nutrients 2022, 14, 4278. https://doi.org/10.3390/nu14204278
Chen Y-H, Yuan W, Meng L-K, Zhong J-C, Liu X-Y. The Role and Mechanism of Gut Microbiota in Pulmonary Arterial Hypertension. Nutrients. 2022; 14(20):4278. https://doi.org/10.3390/nu14204278
Chicago/Turabian StyleChen, Yi-Hang, Wen Yuan, Liu-Kun Meng, Jiu-Chang Zhong, and Xiao-Yan Liu. 2022. "The Role and Mechanism of Gut Microbiota in Pulmonary Arterial Hypertension" Nutrients 14, no. 20: 4278. https://doi.org/10.3390/nu14204278
APA StyleChen, Y.-H., Yuan, W., Meng, L.-K., Zhong, J.-C., & Liu, X.-Y. (2022). The Role and Mechanism of Gut Microbiota in Pulmonary Arterial Hypertension. Nutrients, 14(20), 4278. https://doi.org/10.3390/nu14204278