
Effects of 2-Methoxyestradiol, a Main Metabolite of Estradiol on Hepatic ABCA1 Expression in HepG2 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blot Analysis
2.3. Reverse Transcription-Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-qPCR)
2.4. Transfection and Luciferase Reporter Gene Assay
2.5. Cholesterol and Triglyceride Content Assay
2.6. Oil Red O Staining
2.7. Chromatin Immunoprecipitation Assay
2.8. Transfection of Small Interfering RNA (siRNA)
2.9. Statistical Analysis
3. Results
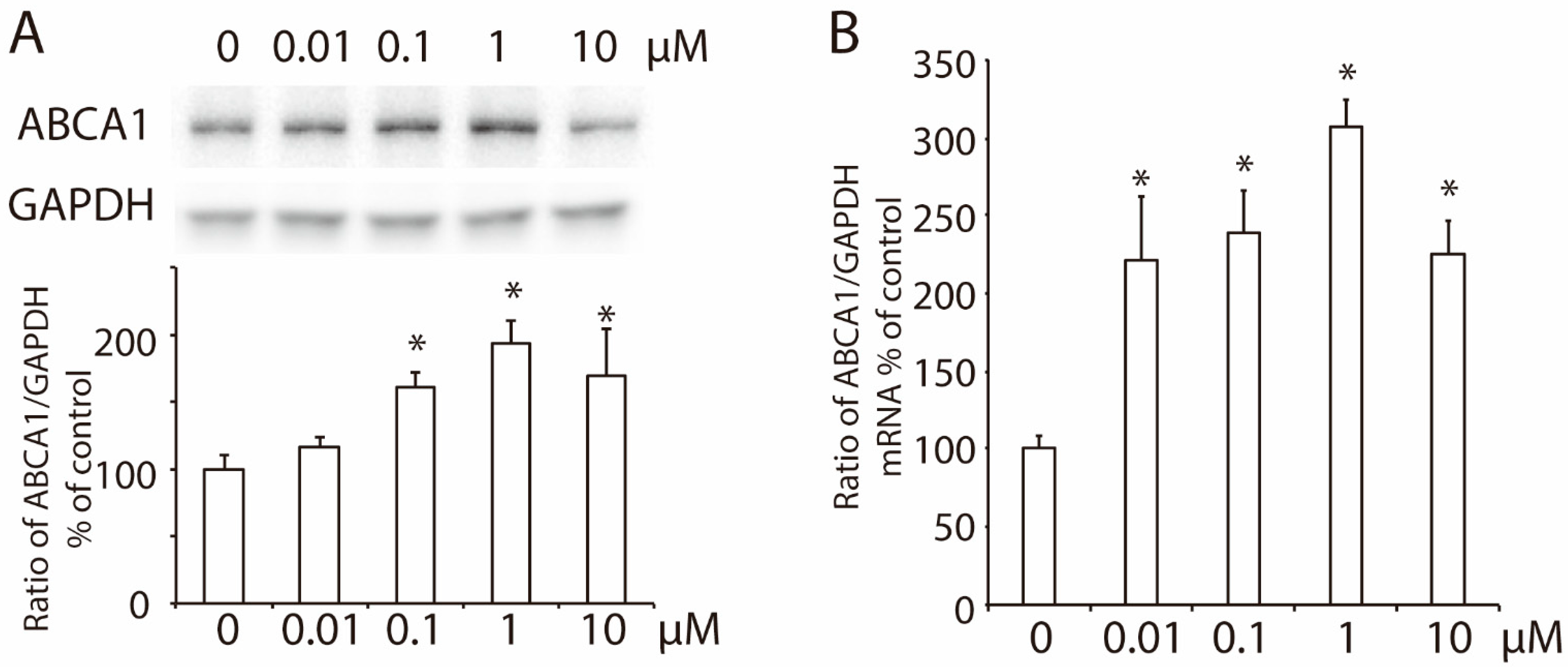
3.1. 2-Methoxyestradiol (2-ME2) Upregulates the Expression of ABCA1 in HepG2 Cells
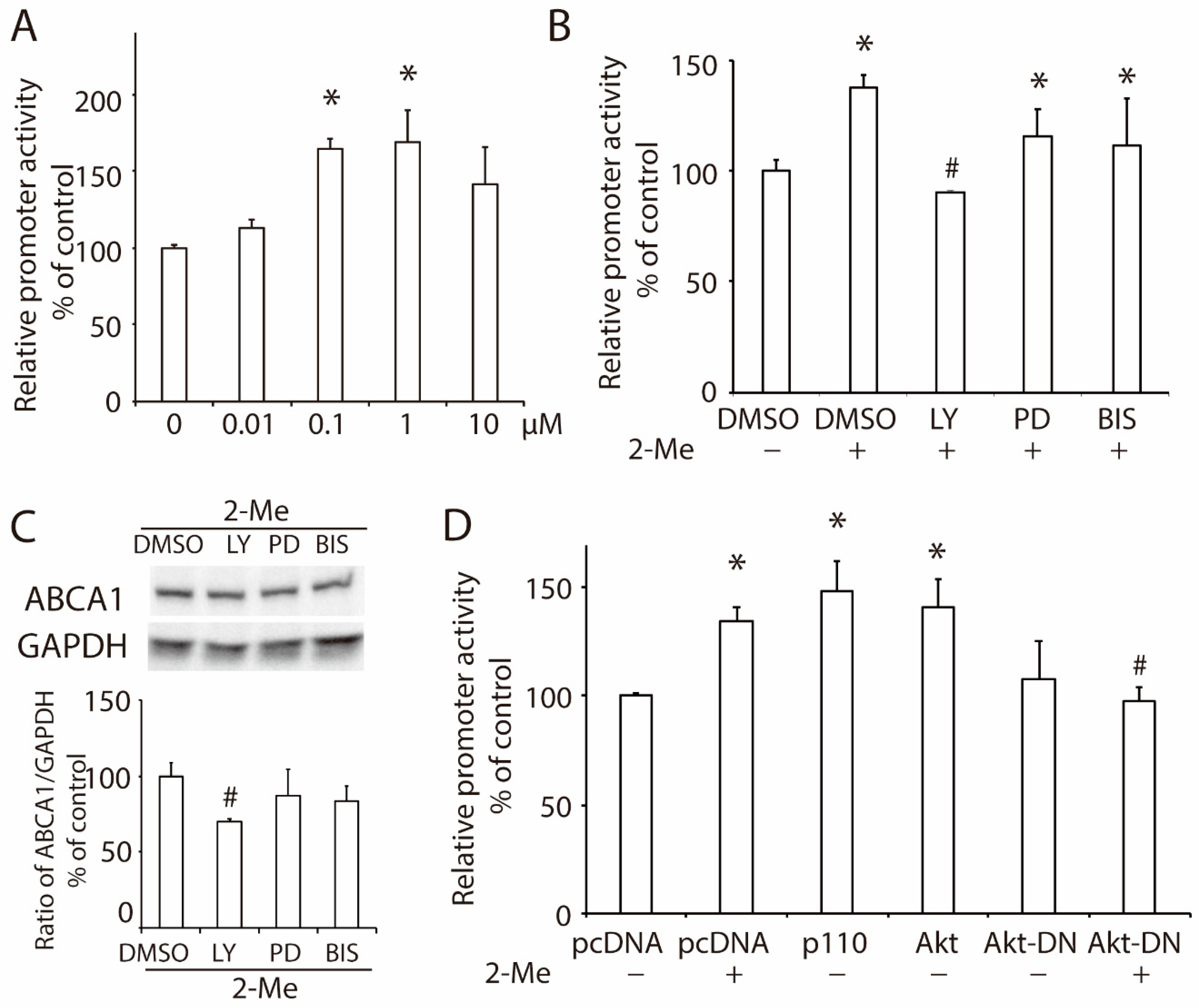
3.2. 2-ME2 Enhances the Promoter Activity of ABCA1 via the PI3K/Akt Pathway in HepG2 Cells
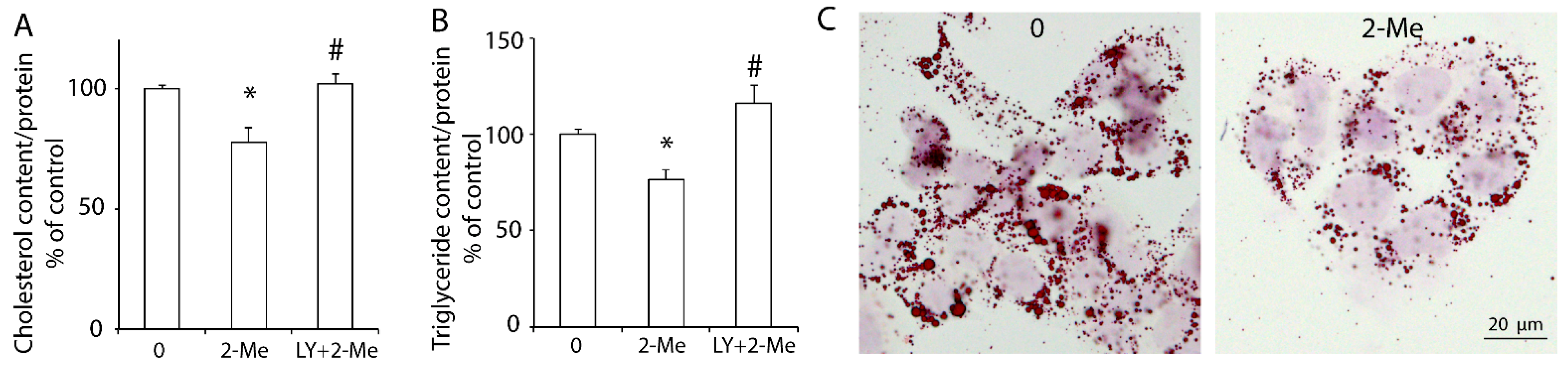
3.3. 2-ME2 Reduces Lipid Content via the PI3K Pathway in HepG2 Cells
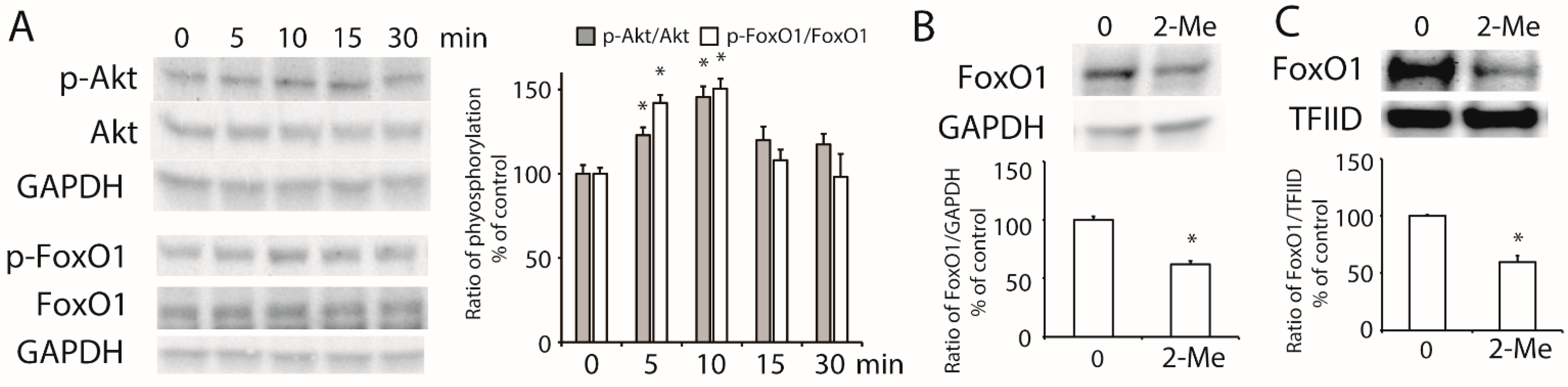
3.4. 2-ME2 Stimulates the Activation of Akt and Transcription Factor FoxO1 in HepG2 Cells
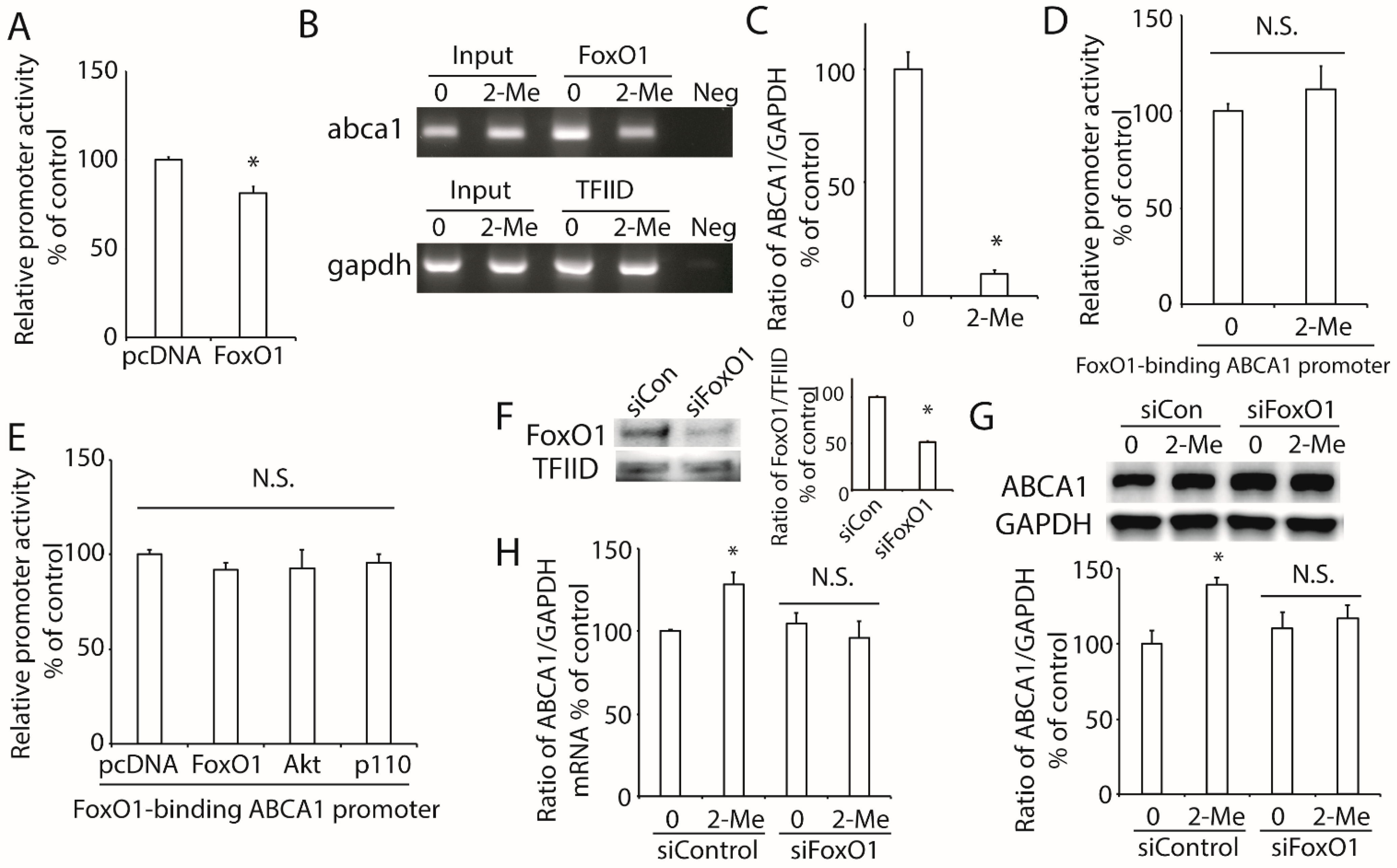
3.5. 2-ME2 Increases ABCA1 Transcription via FoxO1
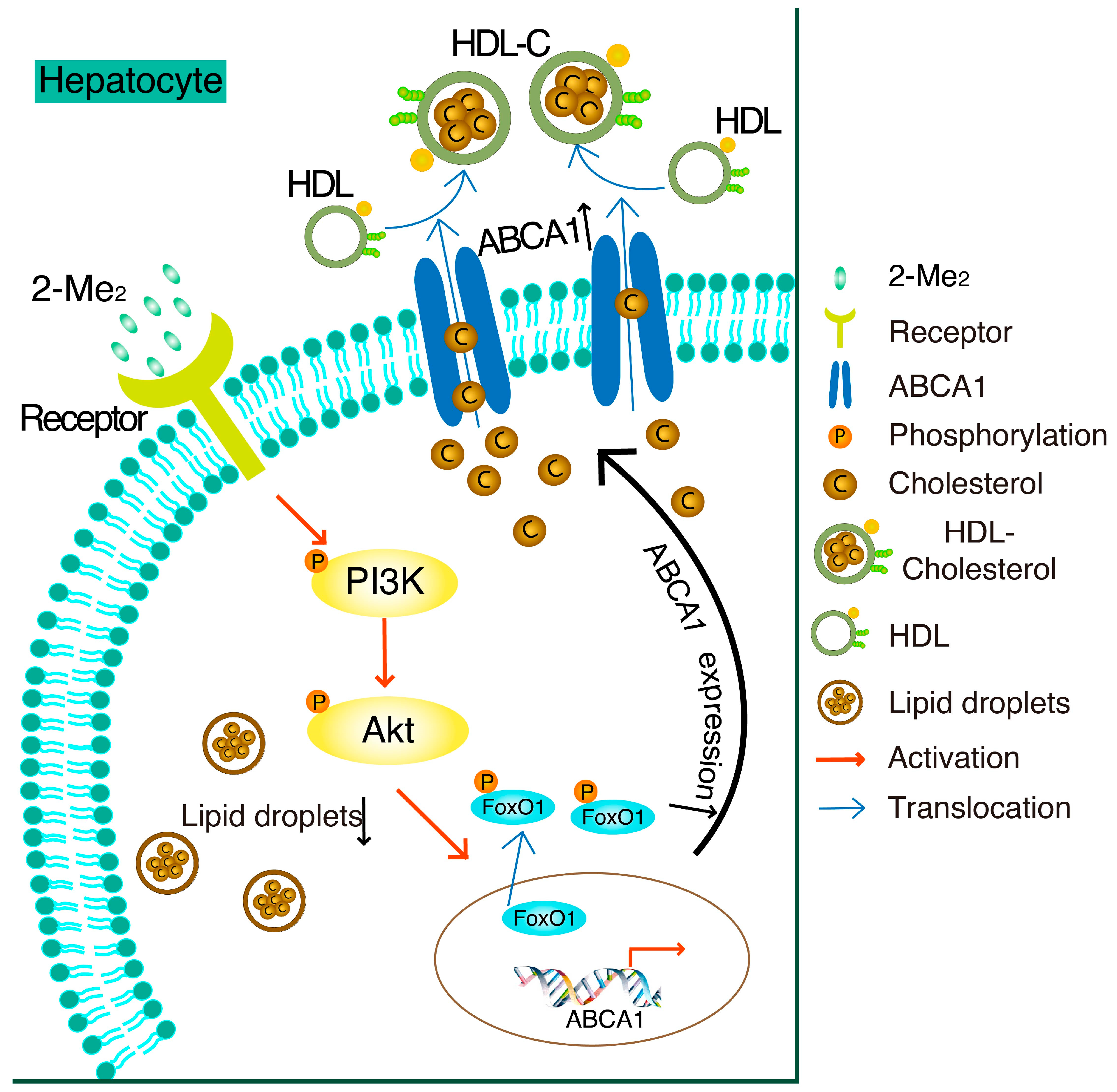
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parada-Bustamante, A.; Valencia, C.; Reuquen, P.; Diaz, P.; Rincion-Rodriguez, R.; Orihuela, P.A. Role of 2-methoxyestradiol, an Endogenous Estrogen Metabolite, in Health and Disease. Mini. Rev. Med. Chem. 2015, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [PubMed]
- Dubey, R.K.; Jackson, E.K. Potential vascular actions of 2-methoxyestradiol. Trends Endocrinol. Metab. 2009, 20, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Mueck, A.O.; Seeger, H. 2-Methoxyestradiol—Biology and mechanism of action. Steroids 2010, 75, 625–631. [Google Scholar] [CrossRef]
- Fielding, C.J.; Fielding, P.E. Molecular physiology of reverse cholesterol transport. J. Lipid Res. 1995, 36, 211–228. [Google Scholar] [CrossRef]
- Oram, J.F. Tangier disease and ABCA1. Biochim. Biophys. Acta 2000, 1529, 321–330. [Google Scholar] [CrossRef]
- Liu, W.; Qin, L.; Yu, H.; Lv, F.; Wang, Y. Apolipoprotein A-I and adenosine triphosphate-binding cassette transporter A1 expression alleviates lipid accumulation in hepatocytes. J. Gastroenterol. Hepatol. 2014, 29, 614–622. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, Y.; Wang, Y.; An, W. Suppression of ABCA1 by unsaturated fatty acids leads to lipid accumulation in HepG2 cells. Biochimie 2010, 92, 958–963. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Cho, H.I.; Seo, M.J.; Lee, S.M. 2-Methoxyestradiol protects against ischemia/reperfusion injury in alcoholic fatty liver by enhancing sirtuin 1-mediated autophagy. Biochem. Pharmacol. 2017, 131, 40–51. [Google Scholar] [CrossRef]
- Murao, K.; Imachi, H.; Yu, X.; Cao, W.M.; Nishiuchi, T.; Chen, K.; Li, J.; Ahmed, R.A.M.; Wong, N.C.W.; Ishida, T. Interferon alpha decreases expression of human scavenger receptor class BI, a possible HCV receptor in hepatocytes. Gut 2008, 57, 664–671. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Ibata, T.; Kobayashi, T.; Dong, T.; Yoshimoto, T.; Yonezaki, K.; Nagata, H.; et al. Angiotensin II induces cholesterol accumulation and impairs insulin secretion by regulating ABCA1 in beta cells. J. Lipid Res. 2018, 59, 1906–1915. [Google Scholar] [CrossRef]
- Murao, K.; Li, J.; Imachi, H.; Muraoka, T.; Masugata, H.; Zhang, G.X.; Kobayashi, R.; Ishida, T.; Tokumitsu, H. Exendin-4 regulates glucokinase expression by CaMKK/CaMKIV pathway in pancreatic beta-cell line. Diabetes Obes. Metab. 2009, 11, 939–946. [Google Scholar] [CrossRef]
- Yu, X.; Murao, K.; Imachi, H.; Li, J.; Nishiuchi, T.; Hosomi, N.; Masugata, H.; Zhang, G.X.; Iwama, H.; Ishida, T. Hyperglycemia suppresses ABCA1 expression in vascular smooth muscle cells. Horm. Metab. Res. 2010, 42, 241–246. [Google Scholar] [CrossRef]
- Sporstol, M.; Tapia, G.; Malerod, L.; Mousavi, S.A.; Berg, T. Pregnane X receptor-agonists down-regulate hepatic ATP-binding cassette transporter A1 and scavenger receptor class B type I. Biochem. Biophys. Res. Commun. 2005, 331, 1533–1541. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Iwama, H.; Zhang, H.; Murao, K. Insulin-like Growth Factor 1 Regulates the Expression of ATP-Binding Cassette Transporter A1 in Pancreatic Beta Cells. Horm. Metab. Res. 2016, 48, 338–344. [Google Scholar] [CrossRef]
- Nishiuchi, Y.; Murao, K.; Imachi, H.; Nishiuchi, T.; Iwama, H.; Ishida, T. Transcriptional factor prolactin regulatory element-binding protein-mediated gene transcription of ABCA1 via 3′,5′-cyclic adenosine-5′-monophosphate. Atherosclerosis 2010, 212, 418–425. [Google Scholar] [CrossRef]
- Murao, K.; Wada, Y.; Nakamura, T.; Taylor, A.H.; Mooradian, A.D.; Wong, N.C. Effects of glucose and insulin on rat apolipoprotein A-I gene expression. J. Biol. Chem. 1998, 273, 18959–18965. [Google Scholar] [CrossRef]
- Fukunaga, K.; Imachi, H.; Lyu, J.; Dong, T.; Sato, S.; Ibata, T.; Kobayashi, T.; Yoshimoto, T.; Yonezaki, K.; Matsunaga, T.; et al. IGF1 suppresses cholesterol accumulation in the liver of growth hormone-deficient mice via the activation of ABCA1. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1232–E1241. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Kobayashi, T.; Dong, T.; Saheki, T.; Matsumoto, M.; Iwama, H.; Zhang, H.; et al. Role of ATP-binding cassette transporter A1 in suppressing lipid accumulation by glucagon-like peptide-1 agonist in hepatocytes. Mol. Metab. 2020, 34, 16–26. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Liu, M.; Chung, S.; Shelness, G.S.; Parks, J.S. Hepatic ABCA1 and VLDL triglyceride production. Biochim. Biophys. Acta 2012, 1821, 770–777. [Google Scholar] [CrossRef]
- Koganti, S.; Snyder, R.; Gumaste, U.; Karamyan, V.T.; Thekkumkara, T. 2-methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Eur. J. Pharmacol. 2014, 723, 131–140. [Google Scholar] [CrossRef]
- Meyer, M.R.; Fredette, N.C.; Howard, T.A.; Hu, C.; Ramesh, C.; Daniel, C.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. G protein-coupled estrogen receptor protects from atherosclerosis. Sci. Rep. 2014, 4, 7564. [Google Scholar] [CrossRef]
- Wei, T.; Chen, W.; Wen, L.; Zhang, J.; Zhang, Q.; Yang, J.; Liu, H.; Chen, B.W.; Zhou, Y.; Feng, X.; et al. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett. 2016, 382, 195–202. [Google Scholar] [CrossRef]
- Vijayanathan, V.; Venkiteswaran, S.; Nair, S.K.; Verma, A.; Thomas, T.J.; Zhu, B.T.; Thomas, T. Physiologic levels of 2-methoxyestradiol interfere with nongenomic signaling of 17beta-estradiol in human breast cancer cells. Clin. Cancer Res. 2006, 12, 2038–2048. [Google Scholar] [CrossRef]
- Koganti, S.; Snyder, R.; Thekkumkara, T. Pharmacologic effects of 2-methoxyestradiol on angiotensin type 1 receptor down-regulation in rat liver epithelial and aortic smooth muscle cells. Gend. Med. 2012, 9, 76–93. [Google Scholar] [CrossRef][Green Version]
- Verenich, S.; Gerk, P.M. Therapeutic promises of 2-methoxyestradiol and its drug disposition challenges. Mol. Pharm. 2010, 7, 2030–2039. [Google Scholar] [CrossRef]
- Kanasaki, M.; Srivastava, S.P.; Yang, F.; Xu, L.; Kudoh, S.; Kitada, M.; Ueki, N.; Kim, H.; Li, J.; Takeda, S.; et al. Deficiency in catechol-o-methyltransferase is linked to a disruption of glucose homeostasis in mice. Sci. Rep. 2017, 7, 7927. [Google Scholar] [CrossRef]
- Htun, N.C.; Miyaki, K.; Song, Y.; Ikeda, S.; Shimbo, T.; Muramatsu, M. Association of the catechol-O-methyl transferase gene Val158Met polymorphism with blood pressure and prevalence of hypertension: Interaction with dietary energy intake. Am. J. Hypertens 2011, 24, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Xiu, L.; Lin, M.; Liu, W.; Kong, D.; Liu, Z.; Zhang, Y.; Ouyang, P.; Liang, Y.; Zhong, S.; Chen, C.; et al. Association of DRD3, COMT, and SLC6A4 Gene Polymorphisms with Type 2 Diabetes in Southern Chinese: A Hospital-Based Case-Control Study. Diabetes Technol. Ther. 2015, 17, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Najib, J. Entacapone: A catechol-O-methyltransferase inhibitor for the adjunctive treatment of Parkinson’s disease. Clin. Ther. 2001, 23, 802–832. [Google Scholar] [CrossRef]
- Borges, N. Tolcapone in Parkinson’s disease: Liver toxicity and clinical efficacy. Expert. Opin. Drug Saf. 2005, 4, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, E.; Cosentino, M.; Ferrari, M.; Porta, G.; Mattarucchi, E.; Marino, F.; Lecchini, S.; Nappi, G. Two patients with COMT inhibitor-induced hepatic dysfunction and UGT1A9 genetic polymorphism. Neurology 2005, 65, 1820–1822. [Google Scholar] [CrossRef]
- Chen, W.; Cui, Y.; Zheng, S.; Huang, J.; Li, P. Simoncini, T.; Zhang, Y.; Fu, X. 2-methoxyestradiol induces vasodilation by stimulating NO release via PPARgamma/PI3K/Akt pathway. PLoS ONE 2015, 10, e0118902. [Google Scholar]
- Dong, X.C. FOXO transcription factors in non-alcoholic fatty liver disease. Liver Res. 2017, 1, 168–173. [Google Scholar] [CrossRef]
- Prasad, S.B.; Yadav, S.S.; Das, M.; Govardhan, H.B.; Pandey, L.K.; Singh, S.; Pradhan, S.; Narayan, G. Down Regulation of FOXO1 Promotes Cell Proliferation in Cervical Cancer. J. Cancer 2014, 5, 655–662. [Google Scholar] [CrossRef]
- Nozhat, Z.; Mohammadi-Yeganeh, S.; Azizi, F.; Zarkesh, M.; Hedayati, M. Effects of metformin on the PI3K/AKT/FOXO1 pathway in anaplastic thyroid Cancer cell lines. Daru 2018, 26, 93–103. [Google Scholar] [CrossRef]
- Kamagate, A.; Qu, S.; Perdomo, G.; Su, D.; Kim, D.H.; Slusher, S.; Meseck, M.; Dong, H.H. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J. Clin. Investig. 2008, 118, 2347–2364. [Google Scholar] [CrossRef]
- Cheng, Z.; White, M.F. Targeting Forkhead box O1 from the concept to metabolic diseases: Lessons from mouse models. Antioxid. Redox Signal. 2011, 14, 649–661. [Google Scholar] [CrossRef]
- Valenti, L.; Rametta, R.; Dongiovanni, P.; Maggioni, M.; Fracanzani, A.L.; Zappa, M.; Lattuada, E.; Roviaro, G.; Fargion, S. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes 2008, 57, 1355–1362. [Google Scholar] [CrossRef]
- Banerjee, A.; Meyer, K.; Mazumdar, B.; Ray, R.B.; Ray, R. Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin-induced metabolic gene expression. J. Virol. 2010, 84, 5936–5946. [Google Scholar] [CrossRef]
- Liu, X.; Qiao, A.; Ke, Y.; Kong, X.; Liang, J.; Wang, R.; Ouyang, X.; Zuo, J.; Chang, Y.; Fang, F. FoxO1 represses LXRalpha-mediated transcriptional activity of SREBP-1c promoter in HepG2 cells. FEBS Lett. 2010, 584, 4330–4334. [Google Scholar] [CrossRef]
- Zhu, Y.; Liao, H.; Xie, X.; Yuan, Y.; Lee, T.; Wang, N.; Wang, X.; Shyy, J.Y.-J.; Stemerman, M.B. Oxidized LDL downregulates ATP-binding cassette transporter-1 in human vascular endothelial cells via inhibiting liver X receptor (LXR). Cardiovasc. Res. 2005, 68, 425–432. [Google Scholar] [CrossRef]
- Lake, N.J.; Taylor, R.L.; Trahair, H.; Harikrishnan, K.N.; Curran, J.E.; Almeida, M.; Kulkarni, H.; Mukhamedova, N.; Hoang, A.; Low, H.; et al. TRAK2, a novel regulator of ABCA1 expression, cholesterol efflux and HDL biogenesis. Eur. Heart J. 2017, 38, 3579–3587. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibata, T.; Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Kobayashi, T.; Saheki, T.; Yoshimura, T.; Murao, K. Effects of 2-Methoxyestradiol, a Main Metabolite of Estradiol on Hepatic ABCA1 Expression in HepG2 Cells. Nutrients 2022, 14, 288. https://doi.org/10.3390/nu14020288
Ibata T, Lyu J, Imachi H, Fukunaga K, Sato S, Kobayashi T, Saheki T, Yoshimura T, Murao K. Effects of 2-Methoxyestradiol, a Main Metabolite of Estradiol on Hepatic ABCA1 Expression in HepG2 Cells. Nutrients. 2022; 14(2):288. https://doi.org/10.3390/nu14020288
Chicago/Turabian StyleIbata, Tomohiro, Jingya Lyu, Hitomi Imachi, Kensaku Fukunaga, Seisuke Sato, Toshihiro Kobayashi, Takanobu Saheki, Takafumi Yoshimura, and Koji Murao. 2022. "Effects of 2-Methoxyestradiol, a Main Metabolite of Estradiol on Hepatic ABCA1 Expression in HepG2 Cells" Nutrients 14, no. 2: 288. https://doi.org/10.3390/nu14020288
APA StyleIbata, T., Lyu, J., Imachi, H., Fukunaga, K., Sato, S., Kobayashi, T., Saheki, T., Yoshimura, T., & Murao, K. (2022). Effects of 2-Methoxyestradiol, a Main Metabolite of Estradiol on Hepatic ABCA1 Expression in HepG2 Cells. Nutrients, 14(2), 288. https://doi.org/10.3390/nu14020288