Abstract
ATP-binding cassette transporter A1 (ABCA1) is a key regulator of lipid efflux, and the absence of ABCA1 induces hepatic lipid accumulation, which is one of the major causes of fatty liver. 2-Methoxyestradiol (2-ME2) has been demonstrated to protect against fatty liver. In this study, we investigated the effects of 2-ME2 on the hepatic lipid content and ABCA1 expression. We found that 2-ME2 dose-dependently increased ABCA1 expression, and therefore, the lipid content was significantly decreased in HepG2 cells. 2-ME2 enhanced the ABCA1 promoter activity; however, this effect was reduced after the inhibition of the PI3K pathway. The overexpression of Akt or p110 induced ABCA1 promoter activity, while dominant-negative Akt diminished the ability of 2-ME2 on ABCA1 promoter activity. Further, 2-ME2 stimulated the rapid phosphorylation of Akt and FoxO1 and reduced the nuclear accumulation of FoxO1. Chromatin immunoprecipitation confirmed that FoxO1 bonded to the ABCA1 promoter region. The binding was reduced by 2-ME2, which facilitated ABCA1 gene transcription. Furthermore, mutating FoxO1-binding sites in the ABCA1 promoter region or treatment with FoxO1-specific siRNA disrupted the effect of 2-ME2 on ABCA1 expression. All of our results demonstrated that 2-ME2 might upregulate ABCA1 expression via the PI3K/Akt/FoxO1 pathway, which thus reduces the lipid content in hepatocytes.
1. Introduction
2-Methoxyestradiol (2-ME2) is a main metabolite of estradiol and is generated by a sequential process: 17β-estradiol (E2) produces 2-hydroxyestradiol (2HE) via the enzyme cytochrome P450 isoform 1A1, and then, 2HE generates 2-ME2 by a conjugation reaction catalyzed via the enzyme catechol-O-methyltransferase (COMT) [1]. Estrogen protects women from NAFLD before menopause by reducing the TG accumulation in the liver via the activation of the estrogen receptor (ER), which is the main receptor of E2, or the G protein-coupled estrogen receptor (GPER or Gpr30), a receptor of 2-ME2 [2]. Differently from E2, 2-ME2 has a very low ability of binding to classical ER, indicating a lack of cytotoxicity [3,4].
As an important lipid transporter, ATP-binding cassette transporter A1 (ABCA1) is a 254-kD membrane protein to transport cholesterol from the cytoplasm to apolipoproteins, and it plays a critical role in the process of reverse cholesterol transport [5]. ABCA1 is firstly detected as a mutated molecule in patients with Tangier disease, and the reduction of ABCA1 results in a severe deficiency of high-density lipoprotein (HDL) and cholesterol accumulation in the liver [6]. Several reports have indicated that the secretion of TG and accumulation of lipids in hepatocytes is significantly increased after blocking the ABCA1 function, pointing out the close relationship between ABCA1 expression and fatty liver disease [7,8].
The liver is an important organ that performs varying metabolic functions, including the metabolism of lipids. As part of multiple lipid-processing functions performed in hepatocytes, overproduced lipids in the cytoplasm are normally stored in triglyceride (TG)-enriched lipoproteins, such as low-density lipoprotein cholesterol (LDL-C) and very low-density lipoprotein cholesterol (VLDL-C) in the cytoplasm. Abnormal lipid accumulation in hepatocytes leads to several metabolic diseases, including non-alcoholic fatty liver disease (NAFLD) [9]. Recently, 2-ME2 has been proven to protect the liver from ischemia/reperfusion-induced damage in alcoholic fatty livers [10]. However, little is known about the role of 2-ME2 in regulating the hepatic lipid content. In the present study, we studied the detailed mechanisms of 2-ME2-stimulating ABCA1 expression in HepG2 cells.
2. Materials and Methods
2.1. Cell Culture
HepG2 cells (Riken Cell Bank, Japan) were cultured in Dulbecco’s modified Eagle’s minimal essential medium with glucose of 1000 mg/L (L-DMEM; Gibco) supplemented with 10% heat-inactivated certified fetal bovine serum (Thermo Scientific; Scoresby VIC, Australia), 0.1-mg/mL streptomycin, and 100-U/mL penicillin, as described previously [11]. For treatment, HepG2 cells were starved in phenol-free L-DMEM supplemented with 0.5% charcoal/dextran-treated FBS for 6 h and then were treated with 2-ME2 at varying doses (0.01, 0.1, 1, and 10 μM) for 24 h or 2-ME2 at 1 μM for varying times (5, 10,15, and 30 min). For treatment with inhibitors, after 6 h of starvation, HepG2 cells were firstly treated with PI3K inhibitor LY294002 at 10 μM, MEK/ERK inhibitor PD98059 at 10 μM, or PKC inhibitor bisindolylmaleimide I at 1 μM for 30 min and then were incubated with 2-ME2 at 1 μM for 24 h.
2.2. Western Blot Analysis
The total proteins and nuclear proteins were extracted from HepG2 cells, as described previously [12]. Concentrations of the proteins were determined with GeneQuant 1300.
As described previously, after separation by 7.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel, the proteins were transferred to polyvinylidene difluoride membranes for immunoblotting [13]. The membranes with proteins were blocked with skim milk overnight and then incubated with 3% BSA diluted in PBS with 0.1% Tween 20 (PBS-T) containing rabbit polyclonal antibody, the first antibody against ABCA1 (1:500; Santa Cruz, sc-20794), Akt (Upstate, 28740; 1:1000), FoxO1 (1:1000; CST, 2880s), p-Akt Ser 473 (CST, 9271s; 1:1000), p-FoxO1 Ser256 (1:1000; CST, 9461s), GAPDH (1:5000; TREVIGEN, 2275-PC-100), or nuclear TFIID (1:200; Santa Cruz, sc-273) overnight and incubated with horseradish peroxidase (HRP)-linked antirabbit IgG secondary antibody (1:2000; DakoCytomation, P0448) for 1 h at 4 °C. Then, the targeted proteins were detected by ECL (GE Healthcare). The analysis of the protein bands by Western blot was carried out under a Luminescent image analyzer LAS-1000 Plus (Fuji Film, Japan).
2.3. Reverse Transcription-Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from HepG2 cells treated with 2-ME2 at varying doses for 24 h in RNA-Bee reagent, according to the manufacturer’s protocols. Each sample was repeated 3 times. We synthesized cDNA from 1 μg of total RNA with SuperScript II reverse transcriptase (Invitrogen), according to the manufacturer’s protocols. PCR was performed with a final volume of a 10-µL mixture of cDNA and LightCycler 480 SYBR Green Master (Roche) by using CFX96 Touch Real-Time PCR Detection Systems (Bio-Rad), as described previously [14]. The sense and antisense primers were shown as the following: human ABCA1 5′-AACAGTTTGTGGCCCTTTTG-3′ and 5′-AGTTCCAGGCTGGGGTACTT-3′ [15]; human GAPDH 5′-ATGGGGAAGGTGAAGGTCG-3′ and 5′-GGGGTCATTGATGGCAACAATA-3′. All primers used were amplified in the same PCR conditions using 60 °C as the annealing temperature. In each set of PCR reactions, we used water as the negative control and 5 dilutions of the control cDNA as the standards. Each sample was repeated 3 times with human GAPDH as the reference gene, as described previously [11].
2.4. Transfection and Luciferase Reporter Gene Assay
The human ABCA1 promoter region was constructed with luciferase reporter plasmid (pABCA1-LUC), and FoxO1 response sequence (FRS)-mutated plasmid (pABCA1-mt-LUC) was generated by a site-directed mutagenesis kit, as described previously [16,17]. Purified promoter plasmid was transfected into HepG2 cells using Lipofectamine (Life Technologies, Gaithersburg, MD, USA). For co-transfection, a vector expressing the p110 catalytic subunit of PI3K (p110), Akt, or the dominant-negative mutant of Akt (Akt with a K197M mutation, Akt-DN) was co-transfected with pABCA1-LUC using Lipofectamine in HepG2 cells. Cells transfected with pABCA1-LUC were treated with or without 2-ME2 at 1 µM for 24 h before harvest. HepG2 cells without plasmid-transfection were used as a normalization of each experiment. The luciferase activity was checked in an aliquot of the cytoplasmic preparation, as described in the manufacturer’s instructions (ToykoInk, Tokyo, Japan) by a microplate reader (SH-9000Lab; CORONA), as previously described [17]. One microgram of Rous sarcoma virus-β-galactosidase (RSV-β Gal) plasmid was added to all transfections to monitor the efficiency of the DNA uptake by HepG2 cells, as described in a previous study [18]. All assays were corrected for β-galactosidase activity, and the total amount of protein in each reaction was identical.
2.5. Cholesterol and Triglyceride Content Assay
To measure the total cholesterol and triglyceride concentration, the cells were washed by PBS and lysed by HEPES buffer with 1% triton X-100. A colorimetric assay was used to measure the cholesterol content, which was carried out by random-access chemistry analyzer ARCHITECT c8000 with reagents widely combined with cholesterol [17].
2.6. Oil Red O Staining
HepG2 cells were seeded on coverslips and then were treated with 2-ME2 for 24 h. Cells were fixed by 4% paraformaldehyde (PFA) for 30 min at room temperature and washed by PBS, pH 7.2 three times. Fixed cells were incubated with Oil Red O working solution for 15 min. After washing three times with PBS, the nucleus was stained with hematoxylin solution for 30 s. After washing three times with PBS again, the cells were mounted, and lipid droplets in the cells were monitored by an Olympus upright microscope (BX-51/DP-72).
2.7. Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation (ChIP) assay was performed by the ChIP-ITTM kit (Active Motif). Chromatin was immunoprecipitated with 2 μg of antibody of either FoxO1, TFIID, or control IgG. The harvested DNA was detected by real-time PCR and PCR to harvest the FRS fragment from −641 to −472 in the human ABCA1 promoter sequence by using sense and antisense primers 5′-AATCTCCAAGGCAGTAGGTCG-3′ and 5′-GAATCTCCCTCAGGACGCCAA-3′ [17]. PCR was performed with the TAKARA PCR thermal cycler MP in the amplification program: 95 °C for 5 min, followed by 36 cycles of 95 °C for 20 s, 61 °C for 30 s, and 72 °C for 30 s. The products of the PCR (169 bp) were detected by 3% agarose gel electrophoresis.
2.8. Transfection of Small Interfering RNA (siRNA)
siRNAs were designed to target the following cDNA sequences: scrambled, 5′-GGCTTATTGTTCTTAGTAAGA-3′; FoxO1-siRNA, 5′-AATGGCGTGCACTTTCTGCAG-3′ [17]. The transfection of siRNA was performed by using siPORT Lipid (Ambion), as described in a previous study [16].
2.9. Statistical Analysis
Data were represented as the mean ± SEM (n = 3) of separate experiments for each group. Statistical analyses were performed using Excel software and SPSS software. Statistical comparisons were made by one-way ANOVA or Student’s t-test; p < 0.05 was considered significant.
3. Results
3.1. 2-Methoxyestradiol (2-ME2) Upregulates the Expression of ABCA1 in HepG2 Cells
To check the effect of 2-ME2 on hepatic ABCA1 expression, we treated HepG2 cells with 2-ME2 at varying concentrations (0–10 μM) for 24 h. The results showed that both the protein level and mRNA levels of hepatic ABCA1 increased with an increase in the dose of 2-ME2 (Figure 1A,B).
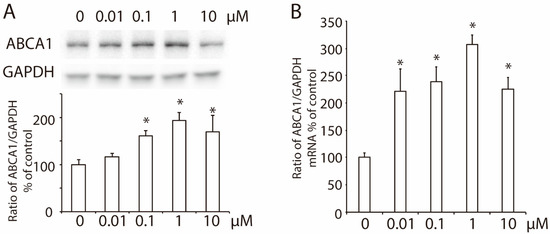
Figure 1.
Effects of 2-ME2 on the expression of ABCA1 in HepG2 cells. (A) ABCA1 protein expression in HepG2 cells treated with 2-ME2 at varying doses for 24 h. (B) Effect of 2-ME2 on the mRNA level of ABCA1 expression. An abundance of GAPDH served as the control, and the relative expression of ABCA1 to GAPDH is shown as a percent of the control in the histogram. The histogram shows the mean ± SEM (n = 3) of separate experiments for each group. * p < 0.05 compared to 0.
3.2. 2-ME2 Enhances the Promoter Activity of ABCA1 via the PI3K/Akt Pathway in HepG2 Cells
A luciferase reporter system constructed with an ABCA1 promoter (pABCA1-LUC) was employed to investigate the role of 2-ME2 in ABCA1 transcription. Similar to that in ABCA1 expression, 2-ME2 significantly enhanced ABCA1 promoter activity, and the strongest promoter activity was induced by 1 μM of 2-ME2 (Figure 2A). To detect the pathways in 2-ME2-mediated ABCA1 promoter activity, we treated HepG2 cells with inhibitors that separately block PI3K (10-μM LY294002), MEK/ERK (10-μM PD98059), or PKC (1-μM bisindolylmaleimide I) before the 2-ME2 treatment. As shown in Figure 2B, the inhibitors of MEK/ERK or PKC did not alter the action of 2-ME2, while LY, the inhibitor of PI3K, blocked the enhancement of ABCA1 promoter activity by 2-ME2. This result was confirmed by Western blotting that the inhibition of PI3K with LY blocked 2-ME2-upregulated ABCA1 expression, while the other two inhibitors did not significantly affect ABCA1 expression, suggesting that 2-ME2 stimulates ABCA1 promoter activity via the PI3K pathway. Next, we overexpressed the p110 catalytic subunit of PI3K or Akt, which are downstream of the PI3K cascade pathway, in HepG2 cells, and the ABCA1 promoter activity was enhanced significantly (Figure 2D). However, domain-negative Akt (Akt-DN) reduced the ABCA1 promoter activity enhanced by 2-ME2 (Figure 2D). These results demonstrated that 2-ME2-induced ABCA1 transcription required activation of the PI3K/Akt signaling pathway.
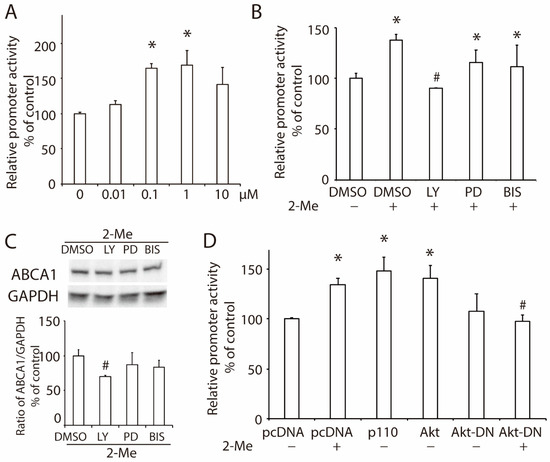
Figure 2.
2-ME2 enhances the promoter activity of ABCA1 via the PI3K/Akt signaling pathway. (A) ABCA1 promoter activity in HepG2 cells treated with 2-ME2 at varying doses for 24 h. (B) Effects of a PI3K inhibitor LY294002 (LY), an MEK/ERK inhibitor PD98059 (PD), or a PKC inhibitor bisindolylmaleimide I (BIS) on 2-ME2-induced ABCA1 promoter activity in HepG2 cells. (C) Protein expression of ABCA1 in HepG2 cells treated with 2-ME2 with or without LY, PD, or BIS. (D) Role of the PI3K/Akt pathway on ABCA1 promoter activity induced by 2-ME2. pcDNA, control vector; p110, overexpression of the p110 catalytic subunit of PI3K; Akt, overexpression of Akt; Akt-DN, domain-negative Akt. Each data shows the mean ± SEM (n = 3) of separate experiments for each group. * p < 0.05 compared to control or pcDNA; # p < 0.05 compared to 2-ME2.
3.3. 2-ME2 Reduces Lipid Content via the PI3K Pathway in HepG2 Cells
As a cholesterol exporter, ABCA1 exports intracellular cholesterol to HDL in the presence of ApoA-I. Our previous study showed that the upregulation of hepatic ABCA1 induced by insulin-like growth factor-1 (IGF-1) or glucagon-like peptide-1 (GLP-1) decreased the lipid content in HepG2 cells [19,20]. In this study, we demonstrated that 2-ME2 significantly reduced the cholesterol content to 78 ± 6% of that in the control group. However, blocking the PI3K pathway by its specific inhibitor, LY, canceled this reduction (Figure 3A). Further, the triglyceride content was significantly decreased by 2-ME2, which was blocked by LY (Figure 3B). Consistently, Oil Red O staining also showed that HepG2 cells with 2-ME2 treatment had fewer and smaller lipid droplets compared to the control group (Figure 3C). These data indicated that 2-ME2 could reduce the hepatic lipid content via the PI3K pathway.
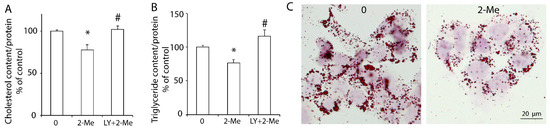
Figure 3.
2-ME2 decreases the lipid content in HepG2 cells via the PI3K pathway. Cholesterol content (A), triglyceride content (B), or Oil Red O staining (C) in HepG2 cells with varying treatments. The cholesterol content was normalized by the protein concentration. 2-ME2, 2-ME2 at 1 μM; LY, PI3K inhibitor LY294002 at 10 μM. The results are represented as the mean ± SEM (n = 3) of separate experiments for each group. * p < 0.05 compared to control; # p < 0.05 compared to 2-ME2.
3.4. 2-ME2 Stimulates the Activation of Akt and Transcription Factor FoxO1 in HepG2 Cells
To further study the activation of Akt, HepG2 cells were treated with 1 μM of 2-ME2 for different time periods. As shown in Figure 4A, the phosphorylation of Akt was detected at 5 min and 15 min after the 2-ME2 treatment. Downstream of the PI3K/Akt pathway, FoxO1 belongs to the FoxO transcription factor family responding to the activation of Akt [21]. Next, we examined the role of FoxO1 in the effect of 2-ME2 on ABCA1 expression. The results showed that the phosphorylation of FoxO1 was stimulated by 2-ME2 from 5 min to 15 min (Figure 4A). Moreover, treatment with 2-ME2 for 24 h decreased the expression of FoxO1 in HepG2 cells (Figure 4B). Once FoxO1 was phosphorylated, it was translocated from the nucleus to the cytoplasm, which allowed stimulating ABCA1 gene transcription. As shown in Figure 4C, the nuclear abundance of FoxO1 was reduced by 2-ME2. These results demonstrated that 2-ME2 activated the phosphorylation of Akt and FoxO1, which may contribute to the regulation of ABCA1 transcription.
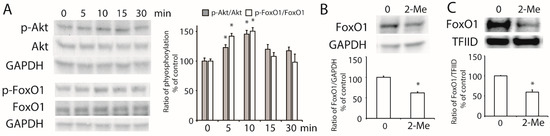
Figure 4.
Activation of Akt and FoxO1 induced by 2-ME2. (A) 2-ME2 stimulated the phosphorylation of Akt at the Ser473 site and FoxO1 at the Ser256 site. A histogram shows the mean ± SEM (n = 3) of phosphorylation for each group. (B) Effect of 2-ME2 on the expression of FoxO1. (C) Nuclear abundance of FoxO1 in HepG2 cells treated with 2-ME2. Abundance of GAPDH or TFIID served as the control. A histogram shows the mean ± SEM (n = 3) of separate experiments for each group. * p < 0.05 compared to 0.
3.5. 2-ME2 Increases ABCA1 Transcription via FoxO1
To study the effect of FoxO1 on the transcription of ABCA1, we co-transfected the plasmid of FoxO1 and pABCA1-LUC into HepG2 cells. As shown in Figure 5A, the overexpression of FoxO1 reduced the ABCA1 promoter activity. Previously, we reported that there is a FoxO1 response sequence (FRS) motif in the ABCA1 promoter region [16]. Based on the results of the ChIP assay, we confirmed that FoxO1 directly bonded to the ABCA1 promoter region (Figure 5B), and ChIP real-time PCR demonstrated that this binding was significantly decreased by the treatment with 2-ME2 (Figure 5C). Next, we generated a luciferase-reporter plasmid with a mutation of the FRS motif in the ABCA1 promoter (FoxO1-binding-mutated ABCA1 promoter) by site-directed mutagenesis and found that 2-ME2 did not alter the FoxO1-mutated promoter activity (Figure 5D). Moreover, the overexpression of FoxO1, p110, or Akt could not affect the FoxO1-binding-mutated ABCA1 promoter activity (Figure 5E), suggesting that the FRS motif is required in 2-ME2-regulated ABCA1 transcription.
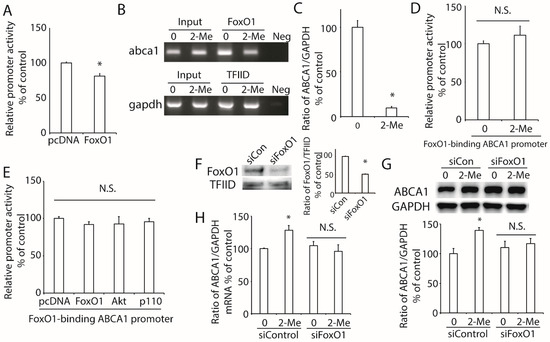
Figure 5.
Role of FoxO1 in 2-ME2-mediated ABCA1 transcription in HepG2 cells. (A) Effect of FoxO1 on ABCA1 promoter activity. (B) Binding of FoxO1 to the ABCA1 promoter region. FoxO1 specifically immunoprecipitates ABCA1 chromatin by the ChIP assay. No ChIP was detected when the chromatin was immunoprecipitated with an unspecific negative control IgG (IgG). (C) ChIP-real time PCR showed that 2-ME2 reduced the binding of FoxO1 to ABCA1 promoter. (D), Effect of 2-ME2 on FoxO1-binding sites-mutated promoter activity (5′-AACA-3′ to 5′-GGAG-3′). (E) Effect of FoxO1, Akt, or p110 on FoxO1-binding sites-mutated promoter activity. (F) Expression of nuclear FoxO1 after silencing FoxO1 with specific siRNA. (G,H) Effect of 2-ME2 on the protein (G) and mRNA (H) expression of ABCA1 after silencing FoxO1. siCon, scramble siRNA; siFoxO1, FoxO1-specific siRNA. Each data point shows the mean ± SEM (n = 3) of separate experiments for each group. * p < 0.05 compared to 0; N.S., no significant difference.
To further examine the effect of FoxO1 on the regulation of ABCA1 expression, we tried to silence FoxO1 expression by its specific siRNA in HepG2 cells. The results showed that the expression of FoxO1 was remarkably decreased by FoxO1-specific siRNA but not by a scrambled siRNA (Figure 5F). Then, we treated these HepG2 cells with 2-ME2 and found that the protein and mRNA levels of ABCA1 were not changed by 2-ME2 after the silencing of FoxO1 (Figure 5G,H).
These findings support the idea that FoxO1 is critical for 2-ME2-stimulated ABCA1 expression.
4. Discussion
In this study, we found that 2-ME2 upregulated the expression of hepatic ABCA1 through the PI3K/Akt/FoxO1 signaling pathway (shown in Figure 6). ABCA1 is a membrane protein that plays an important role in HDL formation and in cholesterol efflux. Active transport via transporters, including the best characterized ABCA1, ABCG1, and SR-BI, is mainly responsible for the bulk efflux of cholesterol from the cells onto extracellular acceptors. Here, we reported that 2-ME2 induces the expression of ABCA1 and lowers the intracellular cholesterol content, but how the cholesterol efflux is involved in this mechanism is for further study. The dysfunction of ABCA1 in Tangier disease results in cholesterol accumulation in the peripheral tissue and causes severe HDL deficiency [5]. Typically, a specific hepatic ABCA1 knockout decreases the generation of heterogeneous-sized nascent HDL particles in vivo, leading to large TG-rich very low-density lipoprotein (VLDL) accumulation in the liver [22]. Thus, ABCA1 has a beneficial effect to protect the liver from cholesterol accumulation. This is also confirmed by our recent study that the upregulation of hepatic ABCA1 expression by insulin-like growth factor-1 (IGF-1) or glucagon-like peptide-1 (GLP-1) reduced hepatic cholesterol accumulation in mice [19,20]. In this study, hepatic ABCA1 expression was elevated by 2-ME2, which contributed to the reduction of the lipid content in hepatocytes. As reported, GPER or GPR30 is a high-affinity membrane receptor of 2-ME2 and shows its protective effect on the cardiovascular system [23]. The global knockout of GPER increased the total and low-density lipoprotein-cholesterol (LDL-C) levels in mice, while the activation of GPER by its small molecule agonist rescued this effect [24]. Moreover, the deficiency of GPER accelerates liver inflammation and fibrosis [25]. However, the specific receptor of 2-ME2 responsible for these activities remains unknown. Thus, further studying needs to be done to investigate the receptor of 2-ME2 and its role in regulating hepatic lipid metabolism.
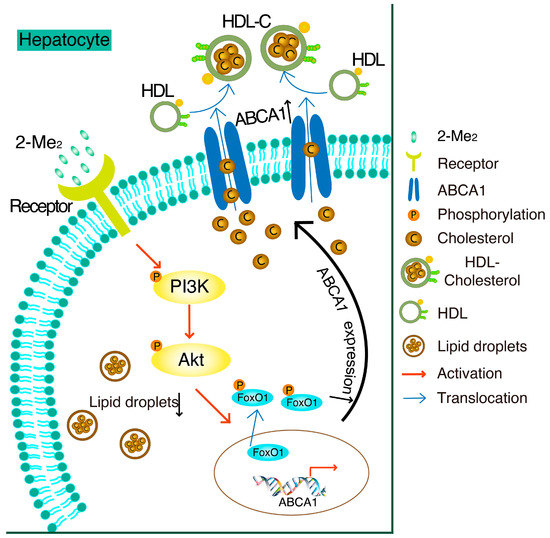
Figure 6.
Schematic diagram of the mechanisms in 2-ME2-suppressed cholesterol accumulation by the upregulation of ABCA1 in HepG2 cells.
In HepG2 cells, treatment with 2-ME2 induces ABCA1 expression by activation of the PI3K/Akt pathway and phosphorylation of FoxO1. As a result, cholesterol accumulation is suppressed by 2-ME2.
Physiologically, the range of the plasma 2-ME2 level in women is 10–55 pg/mL (0.03–0.18 nM). During pregnancy, the level of 2-ME2 increases to 1000 times [26]. A previous study suggested that 2-ME2 at a physiological concentration may regulate some biological processes, and its higher dose may induce pathophysiological processes in tissues [1]. However, the concentration of 2-ME2 varies a lot in different tissues, and it is even higher in certain tissues than in the plasma [27]. Thus, we treated HepG2 cells with varying concentrations of 2-ME2, starting from the lower physiological dose (0.01 μM), and analyzed the protein and mRNA expression, as well as the transcription activity of ABCA1. Our results demonstrated that even a lower physiological concentration of 2-ME2 (0.1 μM) could stimulate the expression and transcription of ABCA1, and 1 μM of this metabolite exhibited the strongest effect. This corroborates the findings of a previous study that 1 μM of 2-ME2 significantly reduced the angiotensin type 1 receptor in rat liver epithelial and aortic smooth muscle cells [27]. Since the bioavailability of 2-ME2 is only 1 to 2% [28], the dosage needs to be carefully determined before the administration of 2-ME2 in vivo.
2-ME2 as the main metabolite of estradiol is generated by a sequential process of 17β-estradiol (E2) via the enzyme cytochrome P450 isoform 1A1 (CYP1A1) to produce 2-hydroxyestradiol (2HE) and then a conjugation reaction catalyzed by the enzyme catechol-O-methyltransferase (COMT) [29]. In humans, the COMT single-nucleotide polymorphism rs4680 (COMT158Val-Met) reduces the enzymatic activity and stability in Met allele carriers. COMT158Val-Met is associated with many diseases, including diabetes, obesity, and fatty liver [30,31]. Genetic variations in COMT with lower enzymatic activity could be a trigger for the onset of metabolic syndromes and liver damage. Although entacapone, the inhibitor of COMT, is commercially available for the treatment of Parkinson’s disease and a few patients exhibit the onset of clinical apparent liver damage [32], this may be due to the short duration of this medication. The long-acting COMT inhibitor tolcapone has been associated with severe liver injury [33,34]. Further studying is required to understand the role of 2-ME2 on liver functions clinically.
One of the aims of this study was to investigate the signal transduction pathways activated by 2-ME2 in hepatocytes. Our results showed that the phosphorylation of Akt was significantly activated by 2-ME2, which is consistent with a previous report that 2-ME2 stimulates the PI3K/Akt pathway to induce the release of nitric oxide [35]. Our previous study proved that the activation of the PI3K/Akt pathway increases the transcription activity of ABCA1 in hepatocytes and pancreatic beta cells [16,19]. In this study, the results showed that the activation of the PI3K/Akt pathway by overexpression of Akt or the p110 catalytic subunit of PI3K remarkably enhanced the ABCA1 promoter activity. However, the inhibition of the PI3K/Akt pathway by its inhibitor, LY294002, or domain-negative Akt canceled the 2-ME2-induced ABCA1 promoter activity, confirming that activation of the PI3K/Akt pathway is required for the stimulation of ABCA1 transcription. However, in order to conclude that the activation of PI3K affects the expression of ABCA1, it will be necessary to conduct experiments with stable activated cells of PI3K in the future. Further, owing to the reduction of ABCA1 after inhibition of the PI3K/Akt pathway, the cholesterol content was increased compared to the treatment of 2-ME2.
The transcription factor forkhead box O (FoxO) family functions downstream of the PI3K/Akt pathway and plays an important role in maintaining the lipid homeostasis and metabolism in the liver [36]. Previous studies demonstrated that activation of the PI3K pathway suppressed the expression of FoxO1 in regulating the cell proliferation of several cancer cells [37,38]. Particularly in hepatocytes, the overexpression of FoxO1 induces the accumulation of triglycerides [39], and targeting the FoxO1 expression in mouse livers may contribute to reversed metabolic syndrome [40]. In our study, 2-ME2 decreased the expression of FoxO1, which was consistent with the reduction of the triglyceride content and total cholesterol content in HepG2 cells. Further, the phosphorylation of FoxO isoforms by the PI3K/Akt pathway led to their nuclear exclusion and inhibited the transcription of target genes. Previously, we reported that FoxO1 was phosphorylated by the activation of the PI3K/Akt pathway to regulate the IGF-1-mediated ABCA1 expression and hepatic cholesterol accumulation in growth hormone-deficient mice [19]. In the present study, FoxO1 was consistently phosphorylated by 2-ME2 via the PI3K/Akt pathway to stimulate the expression of hepatic ABCA1. In addition, 2-ME2 reduced the binding of FoxO1 to the ABCA1 promoter region to enhance the transcription of the ABCA1 gene. Further, the mutation of FRS in the ABCA1 promoter region or silencing of FoxO1 blocked the effect of 2-ME2 on ABCA1 expression. In clinical settings, the expression, nuclear accumulation, and dephosphorylation at Ser256 of FoxO1 were reported in patients with NASH, consistent with the reduction of Akt activation [41]. In addition, FoxO1 expression was stimulated by hepatitis C virus (HCV) infection to suppress the activation of Akt, which resulted in HCV-induced insulin resistance [42]. These reports suggested that the inhibition of hepatic FoxO1 may have a protective effect in the liver. However, FoxO1 was reported to repress LXR-mediated gene transcription via LXR responsive elements (LXREs) in HepG2 cells [43], and LXR is a well-known transcription factor that positively regulates ABCA1 gene transcription [44,45]. This evidence may explain the slight upregulation of the ABCA1 protein (about 10% upregulation compared to the siControl; p = 0.51) induced by the silencing of FoxO1 in Figure 5G. Thus, the detailed mechanism of FoxO1-regulated hepatic ABCA1 expression needs to be further investigated.
5. Conclusions
In summary, our findings showed that 2-ME2 reduces the lipid accumulation in hepatocytes through the elevation of hepatic ABCA1, which might be mediated by the PI3K/Akt/FoxO1 signaling pathway. This study may contribute to the understanding of how 2-ME2 protects the liver functions.
Author Contributions
Conceptualization, T.I., J.L., H.I. and K.M.; methodology, T.I., J.L., H.I., K.F., S.S., T.K., T.S. and T.Y.; validation, H.I., K.F., S.S., T.K., T.S. and T.Y.; investigation, T.I. and J.L.; resources, T.I., J.L., H.I., K.F., S.S., T.K., T.S. and T.Y.; data curation, T.I. and J.L.; writing-original draft preparation, T.I., J.L. and K.M.; writing-review and editing, J.L. and K.M.; supervision, H.I. and K.M.; project administration, K.M.; funding acquisition, K.M., J.L. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded, in part, by Grant-in-Aid for the Ministry of Education, Culture, Sports, Science and Technology, Japan (18K08518) to Koji Murao, JSPS KAKENHI Grant-in-Aid for Research activity start-up (19K23970) to Seisuke Sato, and JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (20K17513) to Jingya Lyu. This work was funded, in part, by the Medical Scientific Research Foundation of Guangdong Province of China (A2021249) to Jingya Lyu.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, K.J., upon reasonable request.
Acknowledgments
We thank Azusa Sugimoto and Miyo Ozaki for their technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Parada-Bustamante, A.; Valencia, C.; Reuquen, P.; Diaz, P.; Rincion-Rodriguez, R.; Orihuela, P.A. Role of 2-methoxyestradiol, an Endogenous Estrogen Metabolite, in Health and Disease. Mini. Rev. Med. Chem. 2015, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [PubMed]
- Dubey, R.K.; Jackson, E.K. Potential vascular actions of 2-methoxyestradiol. Trends Endocrinol. Metab. 2009, 20, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Mueck, A.O.; Seeger, H. 2-Methoxyestradiol—Biology and mechanism of action. Steroids 2010, 75, 625–631. [Google Scholar] [CrossRef]
- Fielding, C.J.; Fielding, P.E. Molecular physiology of reverse cholesterol transport. J. Lipid Res. 1995, 36, 211–228. [Google Scholar] [CrossRef]
- Oram, J.F. Tangier disease and ABCA1. Biochim. Biophys. Acta 2000, 1529, 321–330. [Google Scholar] [CrossRef]
- Liu, W.; Qin, L.; Yu, H.; Lv, F.; Wang, Y. Apolipoprotein A-I and adenosine triphosphate-binding cassette transporter A1 expression alleviates lipid accumulation in hepatocytes. J. Gastroenterol. Hepatol. 2014, 29, 614–622. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, Y.; Wang, Y.; An, W. Suppression of ABCA1 by unsaturated fatty acids leads to lipid accumulation in HepG2 cells. Biochimie 2010, 92, 958–963. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Cho, H.I.; Seo, M.J.; Lee, S.M. 2-Methoxyestradiol protects against ischemia/reperfusion injury in alcoholic fatty liver by enhancing sirtuin 1-mediated autophagy. Biochem. Pharmacol. 2017, 131, 40–51. [Google Scholar] [CrossRef]
- Murao, K.; Imachi, H.; Yu, X.; Cao, W.M.; Nishiuchi, T.; Chen, K.; Li, J.; Ahmed, R.A.M.; Wong, N.C.W.; Ishida, T. Interferon alpha decreases expression of human scavenger receptor class BI, a possible HCV receptor in hepatocytes. Gut 2008, 57, 664–671. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Ibata, T.; Kobayashi, T.; Dong, T.; Yoshimoto, T.; Yonezaki, K.; Nagata, H.; et al. Angiotensin II induces cholesterol accumulation and impairs insulin secretion by regulating ABCA1 in beta cells. J. Lipid Res. 2018, 59, 1906–1915. [Google Scholar] [CrossRef]
- Murao, K.; Li, J.; Imachi, H.; Muraoka, T.; Masugata, H.; Zhang, G.X.; Kobayashi, R.; Ishida, T.; Tokumitsu, H. Exendin-4 regulates glucokinase expression by CaMKK/CaMKIV pathway in pancreatic beta-cell line. Diabetes Obes. Metab. 2009, 11, 939–946. [Google Scholar] [CrossRef]
- Yu, X.; Murao, K.; Imachi, H.; Li, J.; Nishiuchi, T.; Hosomi, N.; Masugata, H.; Zhang, G.X.; Iwama, H.; Ishida, T. Hyperglycemia suppresses ABCA1 expression in vascular smooth muscle cells. Horm. Metab. Res. 2010, 42, 241–246. [Google Scholar] [CrossRef]
- Sporstol, M.; Tapia, G.; Malerod, L.; Mousavi, S.A.; Berg, T. Pregnane X receptor-agonists down-regulate hepatic ATP-binding cassette transporter A1 and scavenger receptor class B type I. Biochem. Biophys. Res. Commun. 2005, 331, 1533–1541. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Iwama, H.; Zhang, H.; Murao, K. Insulin-like Growth Factor 1 Regulates the Expression of ATP-Binding Cassette Transporter A1 in Pancreatic Beta Cells. Horm. Metab. Res. 2016, 48, 338–344. [Google Scholar] [CrossRef]
- Nishiuchi, Y.; Murao, K.; Imachi, H.; Nishiuchi, T.; Iwama, H.; Ishida, T. Transcriptional factor prolactin regulatory element-binding protein-mediated gene transcription of ABCA1 via 3′,5′-cyclic adenosine-5′-monophosphate. Atherosclerosis 2010, 212, 418–425. [Google Scholar] [CrossRef]
- Murao, K.; Wada, Y.; Nakamura, T.; Taylor, A.H.; Mooradian, A.D.; Wong, N.C. Effects of glucose and insulin on rat apolipoprotein A-I gene expression. J. Biol. Chem. 1998, 273, 18959–18965. [Google Scholar] [CrossRef]
- Fukunaga, K.; Imachi, H.; Lyu, J.; Dong, T.; Sato, S.; Ibata, T.; Kobayashi, T.; Yoshimoto, T.; Yonezaki, K.; Matsunaga, T.; et al. IGF1 suppresses cholesterol accumulation in the liver of growth hormone-deficient mice via the activation of ABCA1. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1232–E1241. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Kobayashi, T.; Dong, T.; Saheki, T.; Matsumoto, M.; Iwama, H.; Zhang, H.; et al. Role of ATP-binding cassette transporter A1 in suppressing lipid accumulation by glucagon-like peptide-1 agonist in hepatocytes. Mol. Metab. 2020, 34, 16–26. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Liu, M.; Chung, S.; Shelness, G.S.; Parks, J.S. Hepatic ABCA1 and VLDL triglyceride production. Biochim. Biophys. Acta 2012, 1821, 770–777. [Google Scholar] [CrossRef]
- Koganti, S.; Snyder, R.; Gumaste, U.; Karamyan, V.T.; Thekkumkara, T. 2-methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Eur. J. Pharmacol. 2014, 723, 131–140. [Google Scholar] [CrossRef]
- Meyer, M.R.; Fredette, N.C.; Howard, T.A.; Hu, C.; Ramesh, C.; Daniel, C.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. G protein-coupled estrogen receptor protects from atherosclerosis. Sci. Rep. 2014, 4, 7564. [Google Scholar] [CrossRef]
- Wei, T.; Chen, W.; Wen, L.; Zhang, J.; Zhang, Q.; Yang, J.; Liu, H.; Chen, B.W.; Zhou, Y.; Feng, X.; et al. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett. 2016, 382, 195–202. [Google Scholar] [CrossRef]
- Vijayanathan, V.; Venkiteswaran, S.; Nair, S.K.; Verma, A.; Thomas, T.J.; Zhu, B.T.; Thomas, T. Physiologic levels of 2-methoxyestradiol interfere with nongenomic signaling of 17beta-estradiol in human breast cancer cells. Clin. Cancer Res. 2006, 12, 2038–2048. [Google Scholar] [CrossRef]
- Koganti, S.; Snyder, R.; Thekkumkara, T. Pharmacologic effects of 2-methoxyestradiol on angiotensin type 1 receptor down-regulation in rat liver epithelial and aortic smooth muscle cells. Gend. Med. 2012, 9, 76–93. [Google Scholar] [CrossRef][Green Version]
- Verenich, S.; Gerk, P.M. Therapeutic promises of 2-methoxyestradiol and its drug disposition challenges. Mol. Pharm. 2010, 7, 2030–2039. [Google Scholar] [CrossRef]
- Kanasaki, M.; Srivastava, S.P.; Yang, F.; Xu, L.; Kudoh, S.; Kitada, M.; Ueki, N.; Kim, H.; Li, J.; Takeda, S.; et al. Deficiency in catechol-o-methyltransferase is linked to a disruption of glucose homeostasis in mice. Sci. Rep. 2017, 7, 7927. [Google Scholar] [CrossRef]
- Htun, N.C.; Miyaki, K.; Song, Y.; Ikeda, S.; Shimbo, T.; Muramatsu, M. Association of the catechol-O-methyl transferase gene Val158Met polymorphism with blood pressure and prevalence of hypertension: Interaction with dietary energy intake. Am. J. Hypertens 2011, 24, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Xiu, L.; Lin, M.; Liu, W.; Kong, D.; Liu, Z.; Zhang, Y.; Ouyang, P.; Liang, Y.; Zhong, S.; Chen, C.; et al. Association of DRD3, COMT, and SLC6A4 Gene Polymorphisms with Type 2 Diabetes in Southern Chinese: A Hospital-Based Case-Control Study. Diabetes Technol. Ther. 2015, 17, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Najib, J. Entacapone: A catechol-O-methyltransferase inhibitor for the adjunctive treatment of Parkinson’s disease. Clin. Ther. 2001, 23, 802–832. [Google Scholar] [CrossRef]
- Borges, N. Tolcapone in Parkinson’s disease: Liver toxicity and clinical efficacy. Expert. Opin. Drug Saf. 2005, 4, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, E.; Cosentino, M.; Ferrari, M.; Porta, G.; Mattarucchi, E.; Marino, F.; Lecchini, S.; Nappi, G. Two patients with COMT inhibitor-induced hepatic dysfunction and UGT1A9 genetic polymorphism. Neurology 2005, 65, 1820–1822. [Google Scholar] [CrossRef]
- Chen, W.; Cui, Y.; Zheng, S.; Huang, J.; Li, P. Simoncini, T.; Zhang, Y.; Fu, X. 2-methoxyestradiol induces vasodilation by stimulating NO release via PPARgamma/PI3K/Akt pathway. PLoS ONE 2015, 10, e0118902. [Google Scholar]
- Dong, X.C. FOXO transcription factors in non-alcoholic fatty liver disease. Liver Res. 2017, 1, 168–173. [Google Scholar] [CrossRef]
- Prasad, S.B.; Yadav, S.S.; Das, M.; Govardhan, H.B.; Pandey, L.K.; Singh, S.; Pradhan, S.; Narayan, G. Down Regulation of FOXO1 Promotes Cell Proliferation in Cervical Cancer. J. Cancer 2014, 5, 655–662. [Google Scholar] [CrossRef]
- Nozhat, Z.; Mohammadi-Yeganeh, S.; Azizi, F.; Zarkesh, M.; Hedayati, M. Effects of metformin on the PI3K/AKT/FOXO1 pathway in anaplastic thyroid Cancer cell lines. Daru 2018, 26, 93–103. [Google Scholar] [CrossRef]
- Kamagate, A.; Qu, S.; Perdomo, G.; Su, D.; Kim, D.H.; Slusher, S.; Meseck, M.; Dong, H.H. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J. Clin. Investig. 2008, 118, 2347–2364. [Google Scholar] [CrossRef]
- Cheng, Z.; White, M.F. Targeting Forkhead box O1 from the concept to metabolic diseases: Lessons from mouse models. Antioxid. Redox Signal. 2011, 14, 649–661. [Google Scholar] [CrossRef]
- Valenti, L.; Rametta, R.; Dongiovanni, P.; Maggioni, M.; Fracanzani, A.L.; Zappa, M.; Lattuada, E.; Roviaro, G.; Fargion, S. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes 2008, 57, 1355–1362. [Google Scholar] [CrossRef]
- Banerjee, A.; Meyer, K.; Mazumdar, B.; Ray, R.B.; Ray, R. Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin-induced metabolic gene expression. J. Virol. 2010, 84, 5936–5946. [Google Scholar] [CrossRef]
- Liu, X.; Qiao, A.; Ke, Y.; Kong, X.; Liang, J.; Wang, R.; Ouyang, X.; Zuo, J.; Chang, Y.; Fang, F. FoxO1 represses LXRalpha-mediated transcriptional activity of SREBP-1c promoter in HepG2 cells. FEBS Lett. 2010, 584, 4330–4334. [Google Scholar] [CrossRef]
- Zhu, Y.; Liao, H.; Xie, X.; Yuan, Y.; Lee, T.; Wang, N.; Wang, X.; Shyy, J.Y.-J.; Stemerman, M.B. Oxidized LDL downregulates ATP-binding cassette transporter-1 in human vascular endothelial cells via inhibiting liver X receptor (LXR). Cardiovasc. Res. 2005, 68, 425–432. [Google Scholar] [CrossRef]
- Lake, N.J.; Taylor, R.L.; Trahair, H.; Harikrishnan, K.N.; Curran, J.E.; Almeida, M.; Kulkarni, H.; Mukhamedova, N.; Hoang, A.; Low, H.; et al. TRAK2, a novel regulator of ABCA1 expression, cholesterol efflux and HDL biogenesis. Eur. Heart J. 2017, 38, 3579–3587. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).