
Gut-Flora-Dependent Metabolite Trimethylamine-N-Oxide Promotes Atherosclerosis-Associated Inflammation Responses by Indirect ROS Stimulation and Signaling Involving AMPK and SIRT1



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cell Viability Assay
2.3. Measurement of Reactive Oxygen Species
2.4. Western Blotting Analysis
2.5. Quantitative Real-Time PCR
2.6. Animals and Group Allocation
2.7. Plaque Lesion Analysis
2.8. Lipid Profile Analysis
2.9. 16 S rRNA Gene Sequencing
2.10. Statistical Analysis
3. Results
3.1. TMAO-Induced Inflammation by Stimulating ROS in VSMCs and HUVEC
3.2. TMAO-Induced ROS Generation and Upregulation of Inflammatory Cytokines Were Mediated by SIRT1 in VSMCs and HUVECs
3.3. TMAO-Modulated SIRT1 Expression That Is Dependent on AMKP Activity in VSMCs and HUVECs
3.4. TMAO-Induced ROS Accumulation and Inflammatory Cytokine Release Via AMPK-SIRT1 Pathway in VSMCs and HUVECs
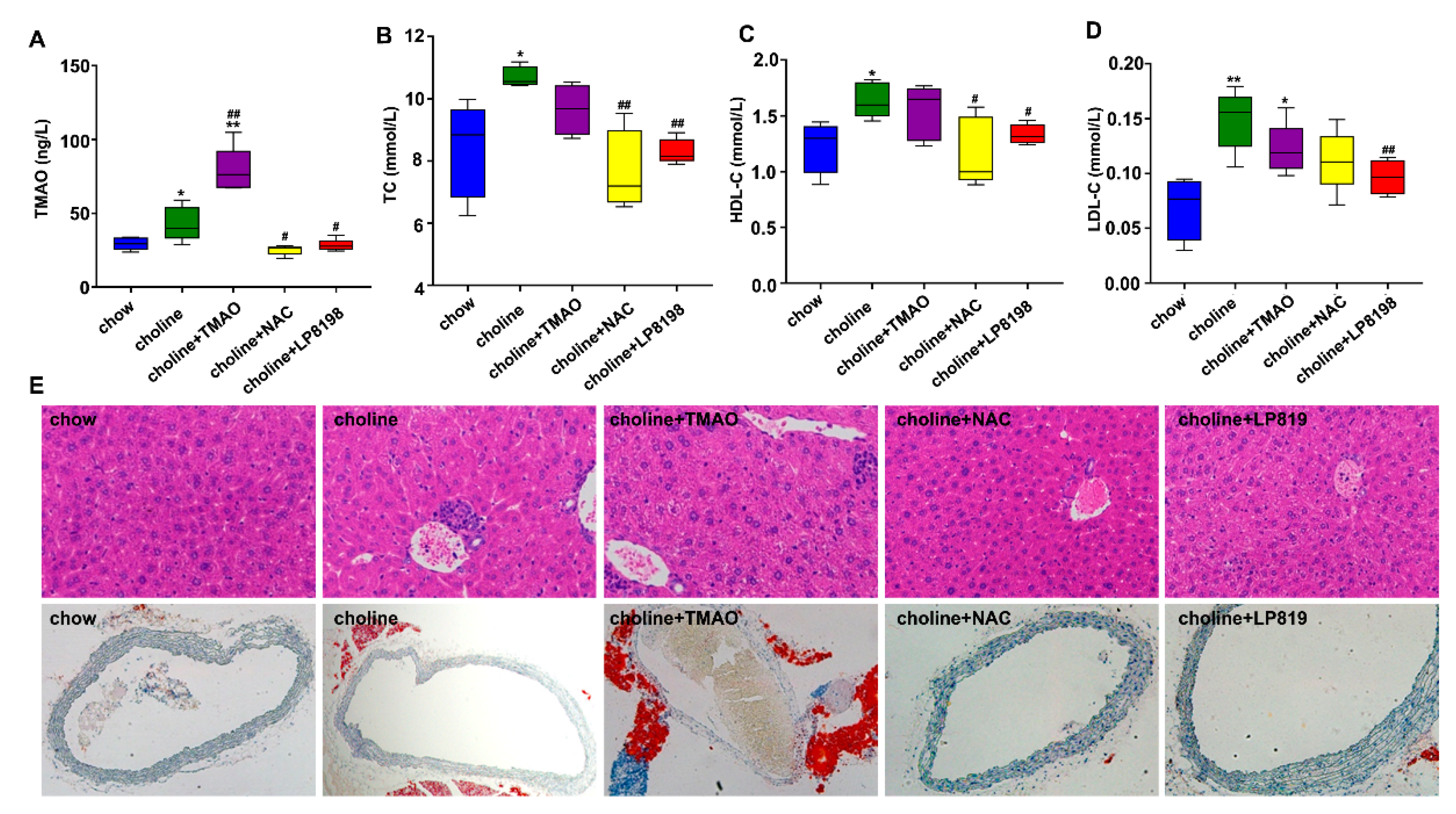
3.5. TMAO and Choline Feeding Promotes Atherosclerosis Development in C57BL/6 Mice
3.6. Effects of TMAO, NAC and LP8198 on Community Diversity and Richness of the Gut Microbiota in Choline-Fed Mice
3.7. Principal Component Analysis (PCA) and Species Composition Analysis of Intestinal Flora in Choline, Choline+TMAO, Choline+NAC and Choline+LP 8198 Mice
3.8. Effects of Choline Diet, NAC and LP8198 Exposure on Composition of the Intestinal Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–82. [Google Scholar] [CrossRef] [PubMed]
- Micha, R.; Wallace, S.K.; Mozaffarian, D. Red and processed meat consumption and risk of incident coronary heart disease, stroke, and diabetes mellitus: A systematic review and meta-analysis. Circulation 2010, 121, 2271–2283. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Skagen, K.; Troseid, M.; Ueland, T.; Holm, S.; Abbas, A.; Gregersen, I.; Kummen, M.; Bjerkeli, V.; Reier-Nilsen, F.; Russell, D.; et al. The Carnitine-butyrobetaine-trimethylamine-N-oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis 2016, 247, 64–69. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Wang, Z.; Wu, Y.; Fan, Y.; Koeth, R.A.; Hazen, S. Prognostic value of elevated levels of intestinal microflora-generated metabolite trimethylamine n-oxide in patients with heart failure: The gut hypothesis revisited. J. Am. Coll. Cardiol. 2013, 61, E750. [Google Scholar] [CrossRef]
- Troseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjorndal, B.; Halvorsen, B.; et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 2015, 277, 717–726. [Google Scholar] [CrossRef]
- Yin, J.; Liao, S.; He, Y.; Wang, S.; Xia, G.; Liu, F.; Zhu, J.; You, C.; Chen, Q.; Zhou, L.; et al. Dysbiosis of Gut Microbiota With Reduced Trimethylamine-N-Oxide Level in Patients With Large-Artery Atherosclerotic Stroke or Transient Ischemic Attack. J. Am. Heart Assoc. 2015, 4, e002699. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Jazwiec, R.; Dadlez, M.; Drapala, A.; Sikora, M.; Skrzypecki, J. Trimethylamine-N-Oxide: A Carnitine-Derived Metabolite That Prolongs the Hypertensive Effect of Angiotensin II in Rats. Can. J. Cardiol. 2014, 30, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappa B. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease—Reply. N. Engl. J. Med. 2005, 353, 429–430. [Google Scholar]
- Libby, P.; Ridker, P.M. Inflammation and atherothrombosis—From population biology and bench research to clinical practice. J. Am. Coll. Cardiol. 2006, 48, A33–A46. [Google Scholar] [CrossRef]
- Maskrey, B.H.; Megson, I.L.; Whitfield, P.D.; Rossi, A.G. Mechanisms of Resolution of Inflammation A Focus on Cardiovascular Disease. Arterioscl. Throm. Vas. 2011, 31, 1001–1006. [Google Scholar] [CrossRef]
- Wong, B.W.; Meredith, A.; Lin, D.; McManus, B.M. The Biological Role of Inflammation in Atherosclerosis. Can. J. Cardiol. 2012, 28, 631–641. [Google Scholar] [CrossRef]
- Inoue, T.; Node, K. Vascular failure: A new clinical entity for vascular disease. J. Hypertens. 2006, 24, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Roustazadeh, A.; Alipoor, B. Ox-LDL Particles: Modified Components, Cellular Uptake, Biological Roles and Clinical Assessments. Cardiovasc. Hematol. Disord. Drug Targets 2011, 11, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.E., Jr.; Pruitt, W.M.; Pruitt, K. Diverse Roles of SIRT1 in Cancer Biology and Lipid Metabolism. Int. J. Mol. Sci. 2015, 16, 950–965. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Huang, G.; Fan, W.; Chen, Y.; Ward, J.M.; Xu, X.; Xu, Q.; Kang, A.; McBurney, M.W.; Fargo, D.C.; et al. SIRT1-Mediated Deacetylation of CRABPII Regulates Cellular Retinoic Acid Signaling and Modulates Embryonic Stem Cell Differentiation. Mol. Cell. 2014, 55, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Simic, P.; Zainabadi, K.; Bell, E.; Sykes, D.B.; Saez, B.; Lotinun, S.; Baron, R.; Scadden, D.; Schipani, E.; Guarente, L. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating beta-catenin. EMBO Mol. Med. 2013, 5, 430–440. [Google Scholar] [CrossRef]
- Rajendran, R.; Garva, R.; Krstic-Demonacos, M.; Demonacos, C. Sirtuins: Molecular Traffic Lights in the Crossroad of Oxidative Stress, Chromatin Remodeling, and Transcription. J. Biomed. Biotechnol. 2011, 2011, 368276. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol Ameliorates Muscular Pathology in the Dystrophic mdx Mouse, a Model for Duchenne Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784794. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Zeng, Z.; Wu, J.; Zhang, Y.; Chen, Z. Sirt1 Inhibits Oxidative Stress in Vascular Endothelial Cells. Oxid. Med. Cell. Longev. 2017, 2017, 7543973. [Google Scholar] [CrossRef]
- Gu, X.; Luo, X.; Wang, C.; Ma, D.; Wang, Y.; He, Y.; Li, W.; Zhou, H.; Zhang, T. Cloning and analysis of bile salt hydrolase genes from Lactobacillus plantarum CGMCC No. 8198. Biotechnol. Lett. 2014, 36, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.Q.; Hu, H.J.; Luo, X.G.; Wang, Q.T.; Gu, X.C.; Zhou, H.; Zhou, W.J.; Ni, X.M.; Zhang, T.C. Complete Genome Sequence of Lactobacillus plantarum CGMCC 8198. Genome Announc. 2017, 5, e01559-16. [Google Scholar] [CrossRef]
- Khosravi, M.; Poursaleh, A.; Ghasempour, G.; Farhad, S.; Najafi, M. The effects of oxidative stress on the development of atherosclerosis. Biol. Chem. 2019, 400, 711. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef]
- Chen, H.; Lu, Y.; Cao, Z.; Ma, Q.; Pi, H.; Fang, Y.; Yu, Z.; Hu, H.; Zhou, Z. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol. Lett. 2016, 246, 7–16. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Cobessi, D.; Tung, E.Y.; Berry, E.A. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: A new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J. Mol. Biol. 2005, 351, 573–597. [Google Scholar] [CrossRef]
- Won, J.; Park, S.; Hong, S.; Son, S.; Yu, J. Rotenone-induced Impairment of Mitochondrial Electron Transport Chain Confers a Selective Priming Signal for NLRP3 Inflammasome Activation. J. Biol. Chem. 2015, 290, 27425–27437. [Google Scholar] [CrossRef]
- Park, S.; Shin, J.; Bae, J.; Han, D.; Park, S.; Shin, J.; Lee, S.K.; Park, H. SIRT1 Alleviates LPS-Induced IL-1β Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts. Cells 2020, 9, 728. [Google Scholar] [CrossRef]
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 2016, 155, 10–21. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Fernández-Friera, L.; Fuster, V.; López-Melgar, B.; Oliva, B.; García-Ruiz, J.M.; Mendiguren, J.; Bueno, H.; Pocock, S.; Ibáñez, B.; Fernández-Ortiz, A.; et al. Normal LDL-Cholesterol Levels Are Associated With Subclinical Atherosclerosis in the Absence of Risk Factors. J. Am. Coll. Cardiol. 2017, 70, 2979–2991. [Google Scholar] [CrossRef]
- Quispe, R.; Elshazly, M.B.; Zhao, D.; Toth, P.P.; Puri, R.; Virani, S.S.; Blumenthal, R.S.; Martin, S.S.; Jones, S.R.; Michos, E.D. Total cholesterol/HDL-cholesterol ratio discordance with LDL-cholesterol and non-HDL-cholesterol and incidence of atherosclerotic cardiovascular disease in primary prevention: The ARIC study. Eur. J. Prev. Cardiol. 2020, 27, 1597–1605. [Google Scholar] [CrossRef]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Dobrowsky, R.T.; Fernyhough, P. Nutrient excess and altered mitochondrial proteome and function contribute to neurodegeneration in diabetes. Mitochondrion 2011, 11, 845–854. [Google Scholar] [CrossRef]
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6, e006347. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Witkowski, M.; Friebel, J.; Buffa, J.A.; Li, X.S.; Wang, Z.; Sangwan, N.; Li, L.; DiDonato, J.A.; Tizian, C.; et al. Vascular endothelial tissue factor contributes to trimethylamine N-oxide-enhanced arterial thrombosis. Cardiovasc. Res. 2022, 118, 2367–2384. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Bioph. Res. Co. 2016, 481, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Chen, J.; Chen, J.; Tao, J.; Wu, S.; Xu, G.; Wang, Z.; Wei, D.; Yin, W. Trimethylamine N-oxide promotes apoE−/− mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J. Cell. Physiol. 2020, 235, 6582–6591. [Google Scholar] [CrossRef]
- Chang, Q.X.; Chen, X.; Ming-Xin, Y.; Zang, N.L.; Li, L.Q.; Zhong, N.; Xia, L.X.; Huang, Q.T.; Zhong, M. Trimethylamine N-Oxide increases soluble fms-like tyrosine Kinase-1 in human placenta via NADPH oxidase dependent ROS accumulation. Placenta 2021, 103, 134–140. [Google Scholar] [CrossRef]
- Habas, K.; Shang, L. Alterations in intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in human endothelial cells. Tissue Cell 2018, 54, 139–143. [Google Scholar] [CrossRef]
- Marui, N.; Offermann, M.K.; Swerlick, R.; Kunsch, C.; Rosen, C.A.; Ahmad, M.; Alexander, R.W.; Medford, R.M. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J. Clin. Investig. 1993, 92, 1866–1874. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschop, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef]
- Owczarczyk, A.B.; Schaller, M.A.; Reed, M.; Rasky, A.J.; Lombard, D.B.; Lukacs, N.W. Sirtuin 1 Regulates Dendritic Cell Activation and Autophagy during Respiratory Syncytial Virus-Induced Immune Responses. J. Immunol. 2015, 195, 1637–1646. [Google Scholar] [CrossRef]
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 2012, 287, 25758–25769. [Google Scholar] [CrossRef]
- Liu, T.F.; Yoza, B.K.; El, G.M.; Vachharajani, V.T.; McCall, C.E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef]
- Karbasforooshan, H.; Karimi, G. The role of SIRT1 in diabetic retinopathy. Biomed. Pharmacother. 2018, 97, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Yuen, D.A.; Connelly, K.A.; Zhang, Y.; Advani, S.L.; Thai, K.; Kabir, G.; Kepecs, D.; Spring, C.; Smith, C.; Batruch, I.; et al. Early outgrowth cells release soluble endocrine antifibrotic factors that reduce progressive organ fibrosis. Stem Cells 2013, 31, 2408–2419. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD+ elevation. Aging Cell. 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Ni, M.; De Vries, G.W. Implication of S-adenosylhomocysteine hydrolase in inhibition of TNF-alpha- and IL-1beta-induced expression of inflammatory mediators by AICAR in RPE cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1274–1281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feng, K.; Chen, Z.; Pengcheng, L.; Zhang, S.; Wang, X. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J. Cell. Physiol. 2019, 234, 18192–18205. [Google Scholar] [CrossRef]
- Pal, P.B.; Sonowal, H.; Shukla, K.; Srivastava, S.K.; Ramana, K.V. Aldose reductase regulates hyperglycemia-induced HUVEC death via SIRT1/AMPK-α1/mTOR pathway. J. Mol. Endocrinol. 2019, 63, 11–25. [Google Scholar] [CrossRef]
- Nagappan, A.; Kim, J.; Jung, D.Y.; Jung, M.H. Cryptotanshinone from the Salvia miltiorrhiza Bunge Attenuates Ethanol-Induced Liver Injury by Activation of AMPK/SIRT1 and Nrf2 Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 265. [Google Scholar] [CrossRef]
- Long, J.; Dai, W.; Zheng, Y.; Zhao, S. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol. Med. 2019, 25, 26. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, X.; Zhu, Y.; Li, Z.; Zhu, Y.T.; Wu, J.C.; Qin, Z.H.; Xiang, M.; Lin, F. Exercise activates lysosomal function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neurosci. Ther. 2019, 25, 796–807. [Google Scholar] [CrossRef]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef]
- Gao, B.; Chi, L.; Mahbub, R.; Bian, X.; Tu, P.; Ru, H.; Lu, K. Multi-Omics Reveals that Lead Exposure Disturbs Gut Microbiome Development, Key Metabolites, and Metabolic Pathways. Chem. Res. Toxicol. 2017, 30, 996–1005. [Google Scholar] [CrossRef]
- Qu, W.; Nie, C.; Zhao, J.; Ou, X.; Zhang, Y.; Yang, S.; Bai, X.; Wang, Y.; Wang, J.; Li, J. Microbiome-Metabolomics Analysis of the Impacts of Long-Term Dietary Advanced-Glycation-End-Product Consumption on C57BL/6 Mouse Fecal Microbiota and Metabolites. J. Agric. Food Chem. 2018, 66, 8864–8875. [Google Scholar] [CrossRef]
- Li, X.; Su, C.; Jiang, Z.; Yang, Y.; Zhang, Y.; Yang, M.; Zhang, X.; Du, Y.; Zhang, J.; Wang, L.; et al. Berberine attenuates choline-induced atherosclerosis by inhibiting trimethylamine and trimethylamine-N-oxide production via manipulating the gut microbiome. NPJ Biofilms Microbiomes 2021, 7, 36. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GenBank Accession Number | Gene | Sequences (5′-3′) |
|---|---|---|
| NR_146119.1 | 18S | Forward CAGCCACCCGAGATTGAGCA |
| Reverse TAGTAGCGACGGGCGGTGTG | ||
| NM_000576.3 | IL-1β | Forward GCTCTCCACCTCCAGGGACA |
| Reverse AGGCCCAAGGCCACAGGTAT | ||
| NM_000600.5 | IL-6 | Forward CACACAGACAGCCACTCACC |
| Reverse CCAGTGCCTCTTTGCTGCTT | ||
| NM_000594.4 | TNF-α | Forward TCTGGCCCAGGCAGTCAGAT |
| Reverse ATTGGCCAGGAGGGCATTGG | ||
| NM_001079821.3 | NLRP3 | Forward GACCCAAACCCACCCATCCA |
| Reverse AGCGGTCCTATGTGCTCGTC | ||
| NM_004994.3 | MMP9 | Forward ACAGCCGGGACGCAGACATC |
| Reverse GGCCAGGAGGAAAGGCGTGT | ||
| NM_001165412.2 | NF-κB | Forward CAGCAGATGGCCCATACCTT |
| Reverse TTTTCACTAGAGGCACCAGG | ||
| NM_001142498.2 | SIRT1 | Forward GCTGGAACAGGTTGCGGGAA |
| Reverse GCTGGGCACCTAGGACATCG | ||
| NM_001355028.2 | AMPK | Forward ACAGCCGAGAAGCAGAAACA |
| Reverse CTTCACTTTGCCGAAGGTGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Xue, J.; Shan, J.; Hong, Y.; Zhu, W.; Nie, Z.; Zhang, Y.; Ji, N.; Luo, X.; Zhang, T.; et al. Gut-Flora-Dependent Metabolite Trimethylamine-N-Oxide Promotes Atherosclerosis-Associated Inflammation Responses by Indirect ROS Stimulation and Signaling Involving AMPK and SIRT1. Nutrients 2022, 14, 3338. https://doi.org/10.3390/nu14163338
Zhou S, Xue J, Shan J, Hong Y, Zhu W, Nie Z, Zhang Y, Ji N, Luo X, Zhang T, et al. Gut-Flora-Dependent Metabolite Trimethylamine-N-Oxide Promotes Atherosclerosis-Associated Inflammation Responses by Indirect ROS Stimulation and Signaling Involving AMPK and SIRT1. Nutrients. 2022; 14(16):3338. https://doi.org/10.3390/nu14163338
Chicago/Turabian StyleZhou, Sa, Jiamin Xue, Jingbo Shan, Yingxiang Hong, Wenkang Zhu, Zhiyan Nie, Yujie Zhang, Nanxi Ji, Xuegang Luo, Tongcun Zhang, and et al. 2022. "Gut-Flora-Dependent Metabolite Trimethylamine-N-Oxide Promotes Atherosclerosis-Associated Inflammation Responses by Indirect ROS Stimulation and Signaling Involving AMPK and SIRT1" Nutrients 14, no. 16: 3338. https://doi.org/10.3390/nu14163338
APA StyleZhou, S., Xue, J., Shan, J., Hong, Y., Zhu, W., Nie, Z., Zhang, Y., Ji, N., Luo, X., Zhang, T., & Ma, W. (2022). Gut-Flora-Dependent Metabolite Trimethylamine-N-Oxide Promotes Atherosclerosis-Associated Inflammation Responses by Indirect ROS Stimulation and Signaling Involving AMPK and SIRT1. Nutrients, 14(16), 3338. https://doi.org/10.3390/nu14163338