
Gut Microbiota Markers and Dietary Habits Associated with Extreme Longevity in Healthy Sardinian Centenarians

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Characteristics of Subjects
2.2. Sampling
2.3. Total DNA Extraction from Fecal Sample and Quantification of Bacterial DNA
2.4. 16S Libraries Preparation and Sequencing
2.5. Data and Statistical Analysis
3. Results
3.1. Clinical and Lifestyle Data of Subjects
3.2. Gut Microbiota Analysis
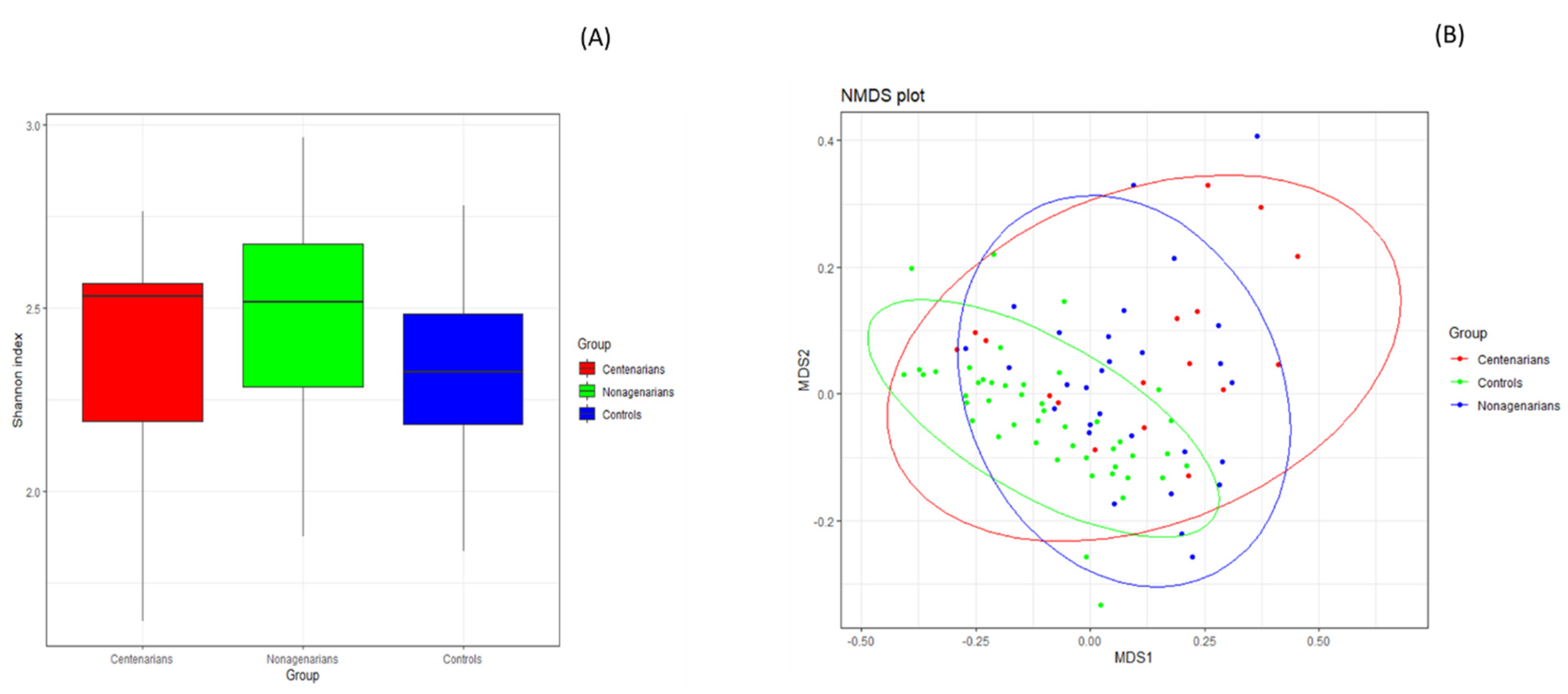
3.2.1. Alpha and Beta Diversity Analysis
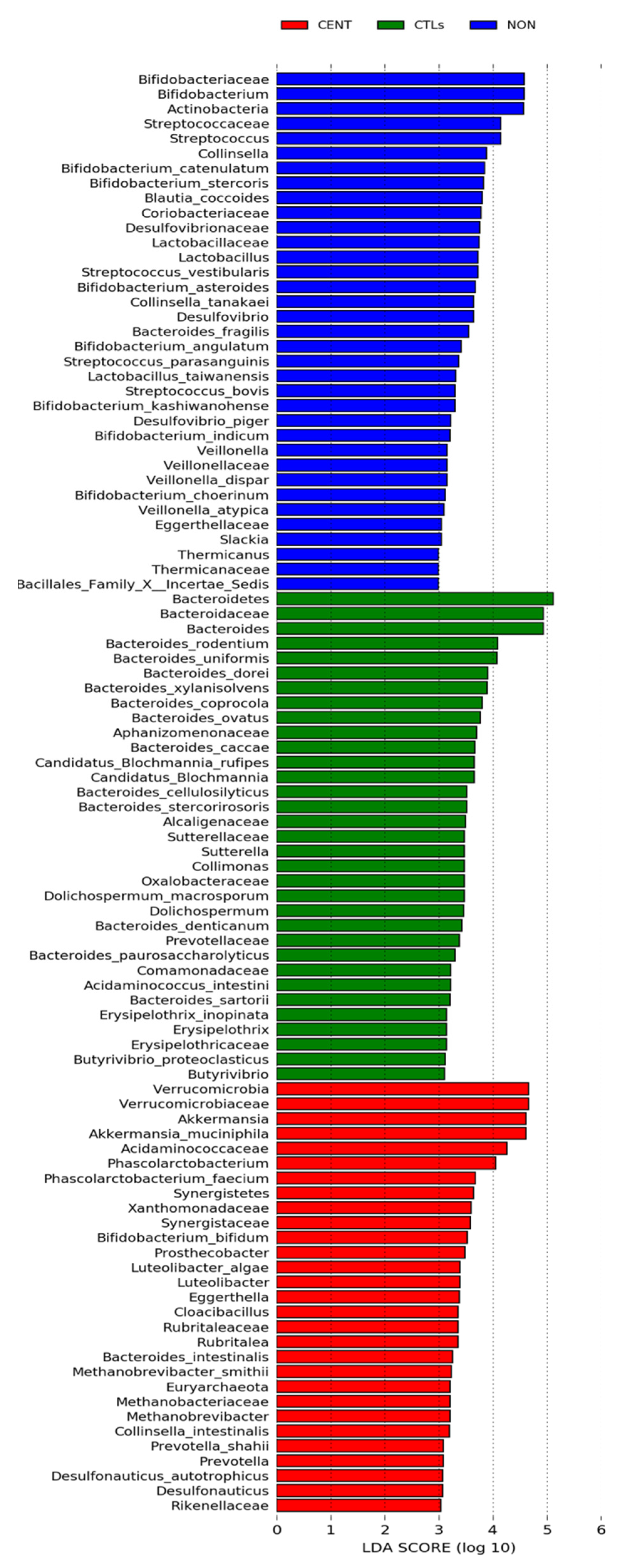
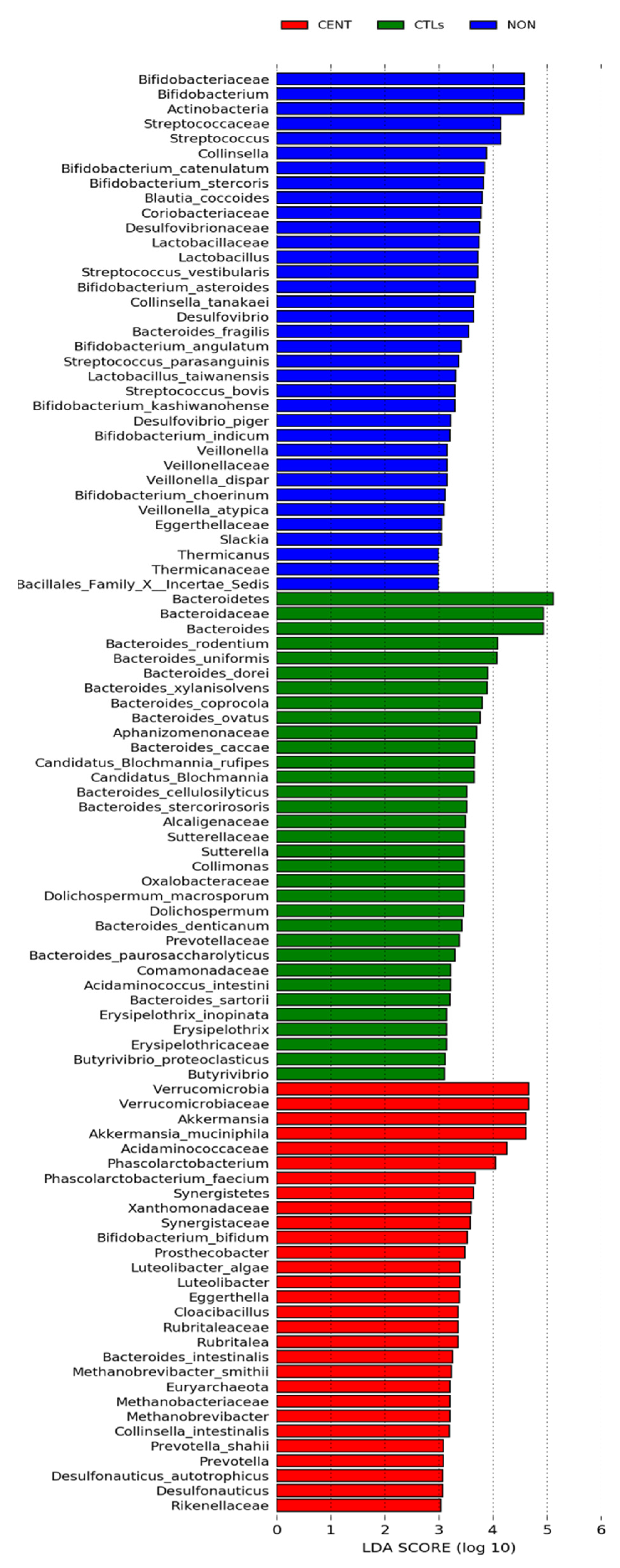
3.2.2. Compositional Analysis of the Gut Microbiota
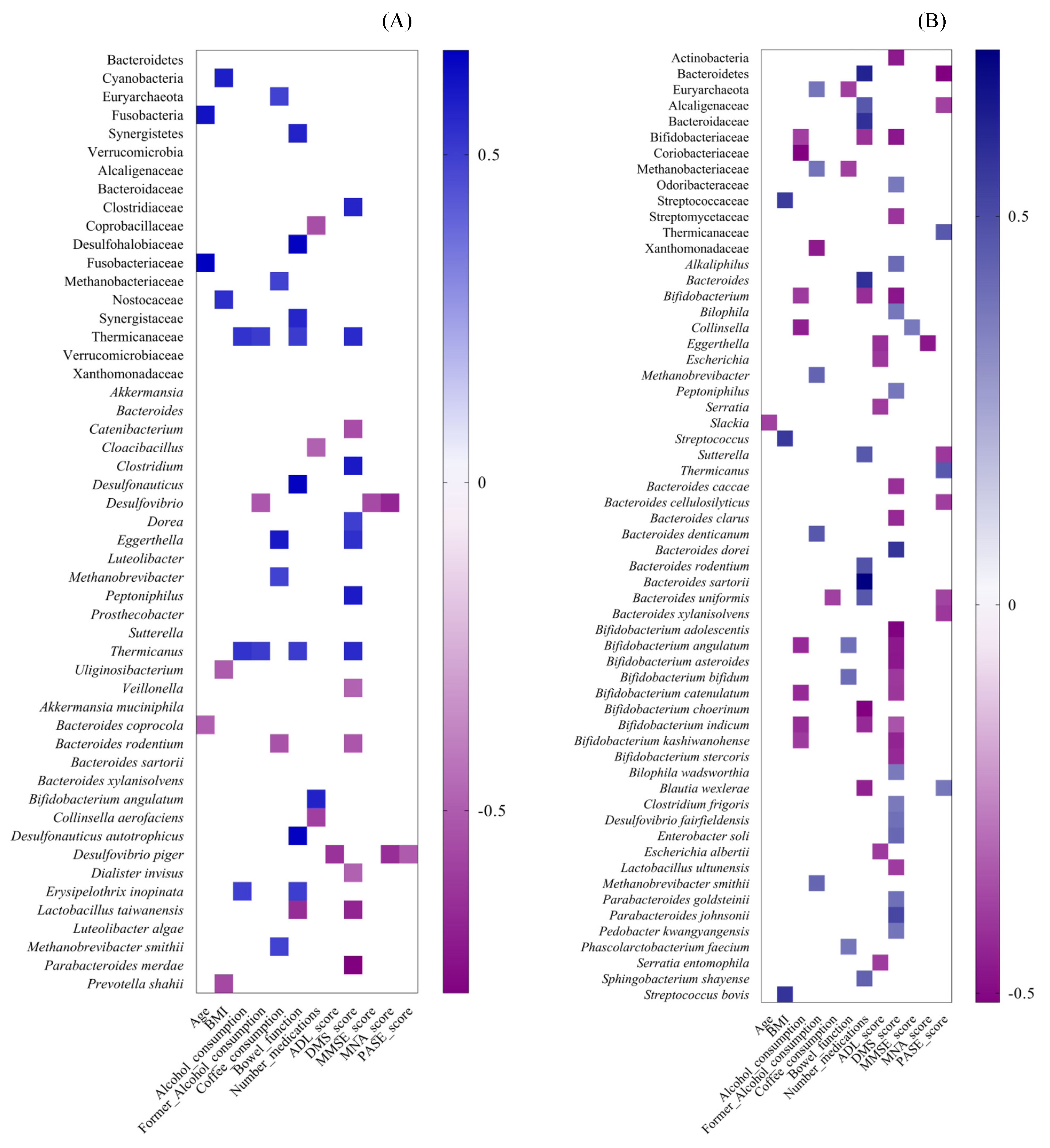
3.2.3. Spearman Correlation between Gut Microbiota Alterations and Dietary, Lifestyle and Clinical Variables in CENT and Non
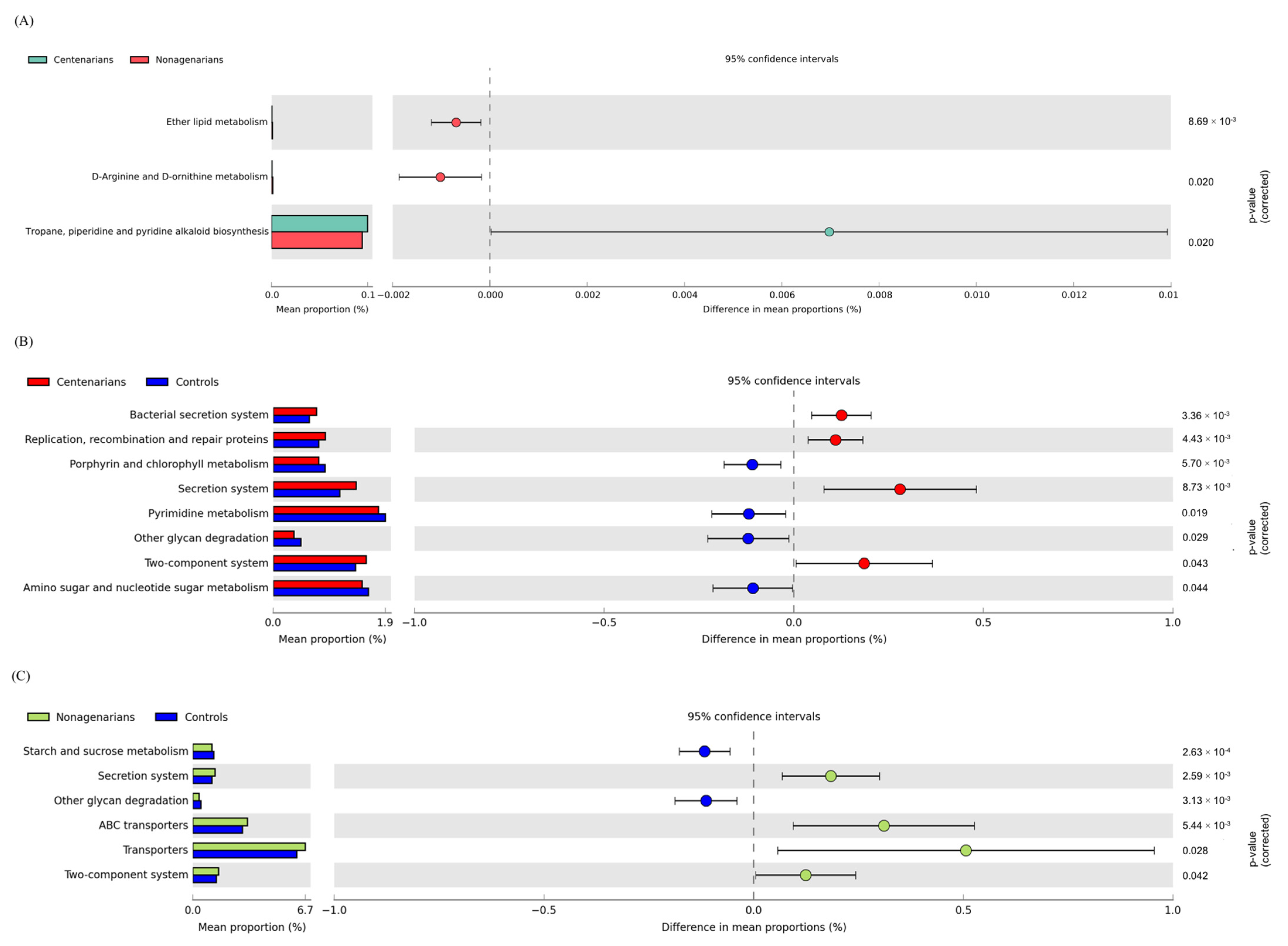
3.2.4. Functional Metagenome Prediction Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rogina, B.; Reenan, R.A.; Nilsen, S.P.; Helfand, S.L. Extended Life-Span Conferred by Cotransporter Gene Mutations in Drosophila. Science 2000, 290, 2137–2140. [Google Scholar] [CrossRef] [PubMed]
- Cevenini, E.; Invidia, L.; Lescai, F.; Salvioli, S.; Tieri, P.; Castellani, G.; Franceschi, C. Human models of aging and longevity. Expert Opin. Biol. Ther. 2008, 8, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.B.; Pettersson, S.; Melby, M.; Bosch, T.C.G. The Microbiome Mediates Environmental Effects on Aging. BioEssays News Rev. Mol. Cell. Dev. Biol. 2019, 41, e1800257. [Google Scholar] [CrossRef]
- Terry, D.F.; Wilcox, M.A.; McCormick, M.A.; Pennington, J.Y.; Schoenhofen, E.A.; Andersen, S.; Perls, T. Lower All-Cause, Cardiovascular, and Cancer Mortality in Centenarians’ Offspring. J. Am. Geriatr. Soc. 2004, 52, 2074–2076. [Google Scholar] [CrossRef]
- Bucci, L.; Ostan, R.; Cevenini, E.; Pini, E.; Scurti, M.; Vitale, G.; Mari, D.; Caruso, C.; Sansoni, P.; Fanelli, F.; et al. Centenarians’ offspring as a model of healthy aging: A reappraisal of the data on Italian subjects and a comprehensive overview. Aging 2016, 8, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Badal, V.D.; Vaccariello, E.D.; Murray, E.R.; Yu, K.E.; Knight, R.; Jeste, D.V.; Nguyen, T.T. The Gut Microbiome, Aging, and Longevity: A Systematic Review. Nutrients 2020, 12, 3759. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.G.; Cossu, G.; Pintus, E.; Zaccheddu, R.; Callia, O.; Conti, G.; Pintus, M.; Gonzalez, C.I.A.; Massidda, M.V.; Mura, G.; et al. Moderate Exercise Improves Cognitive Function in Healthy Elderly People: Results of a Randomized Controlled Trial. Clin. Pract. Epidemiol. Ment. Health 2021, 17, 75–80. [Google Scholar] [CrossRef]
- Carta, M.G.; Cossu, G.; Pintus, E.; Zoccheddu, R.; Callia, O.; Conti, G.; Pintus, M.; Gonzalez, C.I.A.; Massidda, M.V.; Mura, G.; et al. Active elderly and health—can moderate exercise improve health and wellbeing in older adults? Protocol for a randomized controlled trial. Trials 2021, 22, 331. [Google Scholar] [CrossRef]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; De Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.G.; Fitzgerald, G.F.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108, 4586–4591. [Google Scholar] [CrossRef] [Green Version]
- Mangiola, F.; Nicoletti, A.; Gasbarrini, A.; Ponziani, F.R. Gut microbiota and aging. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7404–7413. [Google Scholar] [CrossRef]
- Soenen, S.; Rayner, C.K.; Jones, K.L.; Horowitz, M. The ageing gastrointestinal tract. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.; Biagi, E.; Brigidi, P.; O’Toole, P.; De Vos, W.M. Maintenance of a healthy trajectory of the intestinal microbiome during aging: A dietary approach. Mech. Ageing Dev. 2014, 136–137, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Rajoka, M.S.R.; Zhao, H.; Li, N.; Lu, Y.; Lian, Z.; Shao, D.; Jin, M.; Li, Q.; Zhao, L.; Shi, J. Origination, change, and modulation of geriatric disease-related gut microbiota during life. Appl. Microbiol. Biotechnol. 2018, 102, 8275–8289. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Rampelli, S.; Turroni, S.; Quercia, S.; Candela, M.; Brigidi, P. The gut microbiota of centenarians: Signatures of longevity in the gut microbiota profile. Mech. Ageing Dev. 2017, 165, 180–184. [Google Scholar] [CrossRef]
- Lynch, D.; Jeffery, I.; Cusack, S.; O’Connor, E.; O’Toole, P. Diet-Microbiota-Health Interactions in Older Subjects: Implications for Healthy Aging. Interdiscip. Top. Gerontol. 2014, 40, 141–154. [Google Scholar] [CrossRef]
- Inzitari, M.; For the International Association of Gerontology and Geriatrics (IAGG) Task Force for Nutrition in the Elderly; Doets, E.; Bartali, B.; Benetou, V.; Di Bari, M.; Visser, M.; Volpato, S.; Gambassi, G.; Topinkova, E.; et al. Nutrition in the age-related disablement process. J. Nutr. Health Aging 2011, 15, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, T.S.; Shanahan, F.; O’Toole, P.W. The gut microbiome as a modulator of healthy ageing. Nat. Rev. Gastroenterol. Hepatol. 2022, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, M.; Knight, R.; Leibel, R.L. The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol. Metab. 2015, 26, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Littman, D.R. The Microbiome in Infectious Disease and Inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Wang, X.; Feng, W.; Liu, Q.; Zhou, S.; Liu, Q.; Cai, L. The gut microbiota and its interactions with cardiovascular disease. Microb. Biotechnol. 2020, 13, 637–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vascellari, S.; Palmas, V.; Melis, M.; Pisanu, S.; Cusano, R.; Uva, P.; Perra, D.; Madau, V.; Sarchioto, M.; Oppo, V.; et al. Gut Microbiota and Metabolome Alterations Associated with Parkinson’s Disease. mSystems 2020, 5, e00561-20. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.; Vascellari, S.; Santoru, M.L.; Oppo, V.; Fabbri, M.; Sarchioto, M.; Murgia, D.; Zibetti, M.; Lopiano, L.; Serra, A.; et al. Gut microbiota and metabolome distinctive features in Parkinson disease: Focus on levodopa and levodopa-carbidopa intrajejunal gel. Eur. J. Neurol. 2020, 28, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Melis, M.; Palmas, V.; Pisanu, S.; Serra, A.; Perra, D.; Santoru, M.; Oppo, V.; Cusano, R.; Uva, P.; et al. Clinical Phenotypes of Parkinson’s Disease Associate with Distinct Gut Microbiota and Metabolome Enterotypes. Biomolecules 2021, 11, 144. [Google Scholar] [CrossRef]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The gut microbiome in human neurological disease: A review. Ann. Neurol. 2017, 81, 369–382. [Google Scholar] [CrossRef]
- Raja, G.; Gupta, H.; Gebru, Y.A.; Youn, G.S.; Choi, Y.R.; Kim, H.S.; Yoon, S.J.; Kim, D.J.; Kim, T.-J.; Suk, K.T. Recent Advances of Microbiome-Associated Metabolomics Profiling in Liver Disease: Principles, Mechanisms, and Applications. Int. J. Mol. Sci. 2021, 22, 1160. [Google Scholar] [CrossRef]
- Knip, M.; Siljander, H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Palmas, V.; Pisanu, S.; Madau, V.; Casula, E.; Deledda, A.; Cusano, R.; Uva, P.; Vascellari, S.; Loviselli, A.; Manzin, A.; et al. Gut microbiota markers associated with obesity and overweight in Italian adults. Sci. Rep. 2021, 11, 5532. [Google Scholar] [CrossRef]
- Gérard, P. Gut microbiota and obesity. Exp. 2015, 73, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orrù, S.; Blois, S.; et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Kostic, A.; Xavier, R.J. An Integrative View of Microbiome-Host Interactions in Inflammatory Bowel Diseases. Cell Host Microbe 2015, 17, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisanu, S.; Palmas, V.; Madau, V.; Casula, E.; Deledda, A.; Cusano, R.; Uva, P.; Vascellari, S.; Boi, F.; Loviselli, A.; et al. Impact of a Moderately Hypocaloric Mediterranean Diet on the Gut Microbiota Composition of Italian Obese Patients. Nutrients 2020, 12, 2707. [Google Scholar] [CrossRef]
- Deledda, A.; Annunziata, G.; Tenore, G.; Palmas, V.; Manzin, A.; Velluzzi, F. Diet-Derived Antioxidants and Their Role in Inflammation, Obesity and Gut Microbiota Modulation. Antioxidants 2021, 10, 708. [Google Scholar] [CrossRef]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C.; et al. Through Ageing, and Beyond: Gut Microbiota and Inflammatory Status in Seniors and Centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in Fecal Microbiota in Different European Study Populations in Relation to Age, Gender, and Country: A Cross-Sectional Study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Cătoi, A.F.; Corina, A.; Katsiki, N.; Vodnar, D.C.; Andreicuț, A.D.; Stoian, A.P.; Rizzo, M.; Pérez-Martínez, P. Gut microbiota and aging-A focus on centenarians. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165765. [Google Scholar] [CrossRef]
- Santoro, A.; Ostan, R.; Candela, M.; Biagi, E.; Brigidi, P.; Capri, M.; Franceschi, C. Gut microbiota changes in the extreme decades of human life: A focus on centenarians. Exp. 2017, 75, 129–148. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Poulain, M.; Pes, G.M.; Grasland, C.; Carru, C.; Ferrucci, L.; Baggio, G.; Franceschi, C.; Deiana, L. Identification of a geographic area characterized by extreme longevity in the Sardinia island: The AKEA study. Exp. Gerontol. 2004, 39, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Calò, C.M.; Vona, G.; Robledo, R.; Francalacci, P. From old markers to next generation: Reconstructing the history of the peopling of Sardinia. Ann. Hum. Biol. 2021, 48, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zeng, T.; Zinellu, A.; Rubino, S.; Kelvin, D.J.; Carru, C. A Cross-Sectional Study of Compositional and Functional Profiles of Gut Microbiota in Sardinian Centenarians. mSystems 2019, 4, e00325-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zeng, T.; Deligios, M.; Milanesi, L.; Langille, M.G.I.; Zinellu, A.; Rubino, S.; Carru, C.; Kelvin, D.J. Age-Related Variation of Bacterial and Fungal Communities in Different Body Habitats across the Young, Elderly, and Centenarians in Sardinia. mSphere 2020, 5, e00558-19. [Google Scholar] [CrossRef] [Green Version]
- Crum, R.M.; Anthony, J.C.; Bassett, S.S.; Folstein, M.F. Population-based norms for the Mini-Mental State Exam-ination by age and educational level. JAMA 1993, 269, 2386–2391. [Google Scholar] [CrossRef]
- Katz, S.; Downs, T.D.; Cash, H.R.; Grotz, R.C. Progress in Development of the Index of ADL. Gerontologist 1970, 10, 20–30. [Google Scholar] [CrossRef]
- Guigoz, Y.; Lauque, S.; Vellas, B.J. Identifying the elderly at risk for malnutrition: The Mini Nutritional Assessment. Clin. Geriatr. Med. 2002, 18, 737–757. [Google Scholar] [CrossRef]
- Valentini, A.; Federici, M.; Cianfarani, M.A.; Tarantino, U.; Bertoli, A. Frailty and nutritional status in older people: The Mini Nutritional Assessment as a screening tool for the identification of frail subjects. Clin. Interv. Aging 2018, 13, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Washburn, R.A.; Smith, K.W.; Jette, A.M.; Janney, C.A. The physical activity scale for the elderly (PASE): Development and evaluation. J. Clin. Epidemiol. 1993, 46, 153–162. [Google Scholar] [CrossRef]
- Panagiotakos, D.B.; Pitsavos, C.; Arvaniti, F.; Stefanadis, C. Adherence to the Mediterranean food pattern predicts the prevalence of hypertension, hypercholesterolemia, diabetes and obesity, among healthy adults; the accuracy of the MedDietScore. Prev. Med. 2007, 44, 335–340. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peñña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.-Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.; Hua, Y.; Zeng, B.; Ning, R.; Li, Y.; Zhao, J. Gut microbiota signatures of longevity. Curr. Biol. 2016, 26, R832–R833. [Google Scholar] [CrossRef] [Green Version]
- Rampelli, S.; Candela, M.; Turroni, S.; Biagi, E.; Collino, S.; Franceschi, C.; O’Toole, P.W.; Brigidi, P. Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging 2013, 5, 902–912. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Wu, X.; Qiu, L.; Wang, D.; Gan, M.; Chen, X.; Wei, H.; Xu, F. Analysis of the intestinal microbial community structure of healthy and long-living elderly residents in Gaotian Village of Liuyang City. Appl. Microbiol. Biotechnol. 2015, 99, 9085–9095. [Google Scholar] [CrossRef]
- Allesina, S.; Tang, S. Stability criteria for complex ecosystems. Nature 2012, 483, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Verdi, S.; Jackson, M.; Beaumont, M.; Bowyer, R.C.E.; Bell, J.; Spector, T.D.; Steves, C.J. An Investigation into Physical Frailty as a Link Between the Gut Microbiome and Cognitive Health. Front. Aging Neurosci. 2018, 10, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashtanova, D.A.; Tkacheva, O.N.; Doudinskaya, E.N.; Strazhesko, I.D.; Kotovskaya, Y.V.; Popenko, A.S.; Tyakht, A.V.; Alexeev, D.G. Gut Microbiota in Patients with Different Metabolic Statuses: Moscow Study. Microorganisms 2018, 6, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, J.C.; Manasson, J.; Scher, J.U. The role of the gut microbiome in systemic inflammatory disease. BMJ 2018, 360, j5145. [Google Scholar] [CrossRef] [PubMed]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimaraes, V.D.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.-P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Tuikhar, N.; Keisam, S.; Labala, R.K.; Imrat; Ramakrishnan, P.; Arunkumar, M.C.; Ahmed, G.; Biagi, E.; Jeyaram, K. Comparative analysis of the gut microbiota in centenarians and young adults shows a common signature across genotypically non-related populations. Mech. Ageing Dev. 2019, 179, 23–35. [Google Scholar] [CrossRef]
- Kim, B.-S.; Choi, C.W.; Shin, H.; Jin, S.-P.; Bae, J.-S.; Han, M.; Seo, E.Y.; Chun, J.; Chung, J.H. Comparison of the Gut Microbiota of Centenarians in Longevity Villages of South Korea with Those of Other Age Groups. J. Microbiol. Biotechnol. 2019, 29, 429–440. [Google Scholar] [CrossRef]
- Belzer, C.; De Vos, W.M. Microbes inside—from diversity to function: The case of Akkermansia. ISME J. 2012, 6, 1449–1458. [Google Scholar] [CrossRef]
- Ganesh, B.; Klopfleisch, R.; Loh, G.; Blaut, M. Commensal Akkermansia muciniphila Exacerbates Gut Inflammation in Salmonella Typhimurium-Infected Gnotobiotic Mice. PLoS ONE 2013, 8, e74963. [Google Scholar] [CrossRef]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Routy, B.; le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, R.L.; Dong, X.; de Moraes, A.; Zielke, R.A.; Fernandes, G.R.; Peremyslova, E.; Vasquez-Perez, S.; Schoenborn, A.A.; Gomes, E.P.; Pereira, A.C.; et al. Akkermansia muciniphila mediates negative effects of IFNγ on glucose metabolism. Nat. Commun. 2016, 7, 13329. [Google Scholar] [CrossRef] [PubMed]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M.; Koliada, A.; Marotta, F. Gut microbiota: A player in aging and a target for anti-aging intervention. Ageing Res. Rev. 2017, 35, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Duranti, S.; Milani, C.; Lugli, G.A.; van Sinderen, D.; Ventura, M. Bifidobacterium bifidum: A Key Member of the Early Human Gut Microbiota. Microorganisms 2019, 7, 544. [Google Scholar] [CrossRef] [Green Version]
- Drago, L.; Toscano, M.; Rodighiero, V.; De Vecchi, E.; Mogna, G. Cultivable and Pyrosequenced Fecal Microflora in Centenarians and Young Subjects. J. Clin. Gastroenterol. 2012, 46, S81–S84. [Google Scholar] [CrossRef]
- Yasuma, T.; Toda, M.; Abdel-Hamid, A.; D’Alessandro-Gabazza, C.; Kobayashi, T.; Nishihama, K.; D’Alessandro, V.; Pereira, G.; Mackie, R.; Gabazza, E.; et al. Degradation Products of Complex Arabinoxylans by Bacteroides intestinalis Enhance the Host Immune Response. Microorganisms 2021, 9, 1126. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Y.; Xu, J.; Xue, Z.; Zhang, M.; Pang, X.; Zhang, X.; Zhao, L. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci. Rep. 2015, 5, 14405. [Google Scholar] [CrossRef] [Green Version]
- Panasevich, M.R.; Morris, E.M.; Chintapalli, S.V.; Wankhade, U.D.; Shankar, K.; Britton, S.L.; Koch, L.G.; Thyfault, J.P.; Rector, R.S. Gut microbiota are linked to increased susceptibility to hepatic steatosis in low-aerobic-capacity rats fed an acute high-fat diet. Am. J. Physiol. Liver Physiol. 2016, 311, G166–G179. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Hullar, M.A.J.; Schwarz, Y.; Lampe, J.W. Human Gut Bacterial Communities Are Altered by Addition of Cruciferous Vegetables to a Controlled Fruit- and Vegetable-Free Diet. J. Nutr. 2009, 139, 1685–1691. [Google Scholar] [CrossRef]
- Wang, F.; Yu, T.; Huang, G.; Cai, D.; Liang, X.; Su, H.; Zhu, Z.; Li, D.; Yang, Y.; Shen, P.; et al. Gut Microbiota Community and Its Assembly Associated with Age and Diet in Chinese Centenarians. J. Microbiol. Biotechnol. 2015, 25, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Gaci, N.; Borrel, G.; Tottey, W.; O’Toole, P.; Brugère, J.-F. Archaea and the human gut: New beginning of an old story. World J. Gastroenterol. 2014, 20, 16062–16078. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Gordon, J.I. A humanized gnotobiotic mouse model of host–archaeal–bacterial mutualism. Proc. Natl. Acad. Sci. USA 2006, 103, 10011–10016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut Bifidobacteria Populations in Human Health and Aging. Front. Microbiol. 2016, 7, 1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Xu, W.; Ibrahim, S.A.; Jin, J.; Feng, J.; Jiang, J.; Meng, J.; Ren, F. Effects of Age and Region on Fecal Microflora in Elderly Subjects Living in Bama, Guangxi, China. Curr. Microbiol. 2010, 62, 64–70. [Google Scholar] [CrossRef]
- Wang, F.; Huang, G.; Cai, D.; Li, D.; Liang, X.; Yu, T.; Shen, P.; Su, H.; Liu, J.; Gu, H.; et al. Qualitative and Semiquantitative Analysis of Fecal Bifidobacterium Species in Centenarians Living in Bama, Guangxi, China. Curr. Microbiol. 2015, 71, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Tiihonen, K.; Ouwehand, A.; Rautonen, N. Human intestinal microbiota and healthy ageing. Ageing Res. Rev. 2010, 9, 107–116. [Google Scholar] [CrossRef]
- Nowak, A.; Paliwoda, A.; Błasiak, J. Anti-proliferative, pro-apoptotic and anti-oxidative activity of Lactobacillus and Bifidobacterium strains: A review of mechanisms and therapeutic perspectives. Crit. Rev. Food Sci. Nutr. 2018, 59, 3456–3467. [Google Scholar] [CrossRef]
- Ni, Y.; Yang, X.; Zheng, L.; Wang, Z.; Wu, L.; Jiang, J.; Yang, T.; Ma, L.; Fu, Z. Lactobacillus and Bifidobacterium Improves Physiological Function and Cognitive Ability in Aged Mice by the Regulation of Gut Microbiota. Mol. Nutr. Food Res. 2019, 63, e1900603. [Google Scholar] [CrossRef]
- Finamore, A.; Roselli, M.; Donini, L.M.; Brasili, E.; Rami, R.; Carnevali, P.; Mistura, L.; Pinto, A.; Giusti, A.; Mengheri, E. Supplementation with Bifidobacterium longum Bar33 and Lactobacillus helveticus Bar13 mixture improves immunity in elderly humans (over 75 years) and aged mice. Nutrition 2019, 63–64, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Bogert, B.V.D.; Boekhorst, J.; Smid, E.J.; Zoetendal, E.G.; Kleerebezem, M. Draft Genome Sequence of Veillonella parvula HSIVP1, Isolated from the Human Small Intestine. Genome Announc. 2013, 1, e00861-13. [Google Scholar] [CrossRef] [Green Version]
- Rigsbee, L.; Agans, R.; Shankar, V.; Kenche, H.; Khamis, H.J.; Michail, S.; Paliy, O. Quantitative Profiling of Gut Microbiota of Children with Diarrhea-Predominant Irritable Bowel Syndrome. Am. J. Gastroenterol. 2012, 107, 1740–1751. [Google Scholar] [CrossRef]
- Tana, C.; Umesaki, Y.; Imaoka, A.; Handa, T.; Kanazawa, M.; Fukudo, S. Altered profiles of intestinal microbiota and organic acids may be the origin of symptoms in irritable bowel syndrome. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2009, 22, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Nankova, B.B.; Agarwal, R.; Macfabe, D.F.; La Gamma, E.F. Enteric Bacterial Metabolites Propionic and Butyric Acid Modulate Gene Expression, Including CREB-Dependent Catecholaminergic Neurotransmission, in PC12 Cells—Possible Relevance to Autism Spectrum Disorders. PLoS ONE 2014, 9, e103740. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.K.; Ben Bacha, A.; Kotb, M. Etiology of autistic features: The persisting neurotoxic effects of propionic acid. J. Neuroinflammation 2012, 9, 74. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef]
- Rowan, F.; Docherty, N.G.; Murphy, M.; Murphy, B.; Coffey, J.C.; O’Connell, P.R. Desulfovibrio Bacterial Species Are Increased in Ulcerative Colitis. Dis. Colon Rectum 2010, 53, 1530–1536. [Google Scholar] [CrossRef]
- Kushugulova, A.; Kozhakhmetov, S.; Baiskhanova, D.; Tynybayeva, I.K.; Shapekova, N.; Kakimova, A.; Saduakhasova, S.; Shakhabayeva, G.; Khassenbekova, Z.; Nurgozhin, T.; et al. Gut microbiome diversity in Kazakhstani women of different age groups. Biotechnol. Theory Pract. 2014, 10, 4–9. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Porras, D.; García-Mediavilla, M.V.; Martínez-Flórez, S.; Juarez-Fernández, M.; Cuevas, M.J.; Mauriz, J.L.; González-Gallego, J.; Nistal, E.; Sánchez-Campos, S. Beneficial effects of exercise on gut microbiota functionality and barrier integrity, and gut-liver crosstalk in an in vivo model of early obesity and non-alcoholic fatty liver disease. Dis. Model. Mech. 2019, 12, dmm039206. [Google Scholar] [CrossRef] [Green Version]
- Nagy-Szakal, D.; Hollister, E.B.; Luna, R.A.; Szigeti, R.; Tatevian, N.; Smith, C.W.; Versalovic, J.; Kellermayer, R. Cellulose Supplementation Early in Life Ameliorates Colitis in Adult Mice. PLoS ONE 2013, 8, e56685. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Zhang, K.; Ma, X.; He, P. Clostridium species as probiotics: Potentials and challenges. J. Anim. Sci. Biotechnol. 2020, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, I.; Wallace, G.; Zhang, C.; Legge, R.; Benson, A.K.; Carr, T.P.; Moriyama, E.N.; Walter, J. Diet-Induced Metabolic Improvements in a Hamster Model of Hypercholesterolemia Are Strongly Linked to Alterations of the Gut Microbiota. Appl. Environ. Microbiol. 2009, 75, 4175–4184. [Google Scholar] [CrossRef] [Green Version]
- Fleissner, C.K.; Huebel, N.; El-Bary, M.M.A.; Loh, G.; Klaus, S.; Blaut, M. Absence of intestinal microbiota does not protect mice from diet-induced obesity. Br. J. Nutr. 2010, 104, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Kaakoush, N.O. Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef] [Green Version]
- Shankar, V.; Gouda, M.; Moncivaiz, J.; Gordon, A.; Reo, N.V.; Hussein, L.; Paliy, O. Differences in Gut Metabolites and Microbial Composition and Functions between Egyptian and U.S. Children Are Consistent with Their Diets. mSystems 2017, 2, e00169-16. [Google Scholar] [CrossRef] [Green Version]
- Koropatkin, N.M.; Cameron, E.A.; Martens, E.C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Genet. 2012, 10, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M.; Ambudkar, S.V. Overview: ABC Transporters and Human Disease. J. Bioenerg. Biomembr. 2001, 33, 453–458. [Google Scholar] [CrossRef]
- Wolanin, P.M.; Thomason, P.A.; Stock, J.B. Histidine protein kinases: Key signal transducers outside the animal kingdom. Genome Biol. 2002, 3, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Capra, E.J.; Laub, M.T. Evolution of Two-Component Signal Transduction Systems. Annu. Rev. Microbiol. 2012, 66, 325–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CENT | NON | CTLs | p CENT vs NON | p CENT vs CTLs | p NON vs CTLs | ||
|---|---|---|---|---|---|---|---|
| n | 17 | 29 | 46 | ||||
| Demographic data | |||||||
| Age (M ± SD) | 102.2 ± 2.3 | 93.1 ± 2.7 | 50.9 ± 8.3 | 1.62 × 10−14 | 1.11 × 10−33 | 3.46 × 10−39 | |
| Female (n, %) | 14, 82.3 | 23, 79.3 | 37, 80.4 | 0.802 | 0.863 | 0.906 | |
| Male (n, %) | 3, 17.6 | 6, 20.7 | 9, 19.6 | 0.802 | 0.863 | 0.906 | |
| Anthropometric data | |||||||
| BMI (M ± SD) | 26.12 ± 4.5 | 27.14 ± 4.2 | 22.75, 2.8 | 0.274 | 0.001 | 0 | |
| Lifestyle factors | |||||||
| Current smoking status (n, %) | 0, 0.0 | 0, 0.0 | 7, 15.2 | - | 0.088 | 0.027 | |
| Former smoking status (n, %) | 2, 11.8 | 3, 10.3 | 2, 4.3 | 0.881 | 0.785 | 0.881 | |
| Current alcohol consumption (n, %) | 8, 47.1 | 11, 37.9 | 9, 19.6 | 0.544 | 0.701 | 0.829 | |
| Former alcohol consumption (n, %) | 13, 76.5 | 17, 58.6 | n.d. | 0.22 | - | - | |
| Coffee consumption (n, %) | 13, 76.5 | 21, 72.4 | 33, 71.7 | 0.762 | 0.982 | 0.677 | |
| Bowel function | |||||||
| Movements/week (M ± SD) | 4.4 ± 2.2 | 4.9 ± 2.1 | n.d. | 0.193 | - | - | |
| From 1 to 3 (movements/week, %) | 8, 47.1 | 10, 30.5 | n.d. | n.d. | - | - | |
| From 4 to 5 (movements/week, %) | 3, 17.6 | 2, 6.9 | n.d. | n.d. | - | - | |
| From 6 to 7 (movements/week, %) | 6, 35.3 | 17, 58.6 | n.d. | n.d. | - | - | |
| Medications, n/day (M ± SD) | 3.4 ± 2.9 | 4.5 ±2.8 | n.d. | 0.209 | 4.43 × 10−09 | 1.12 × 10−14 | |
| MMSE score (M ± SD) | 19.3 ± 4.3 | 25.3 ± 4.3 | n.d. | 0.001 | - | - | |
| MDS (M ± SD) | 31.0 ± 5.3 | 30.7 ± 4.5 | 32.9 ± 3.7 | 0.426 | 0.067 | 0.036 | |
| ADL score (M ± SD) | 2.7 ± 2.3 | 4.2 ± 2.3 | n.d. | 0.03 | - | - | |
| PASE score (M ± SD) | 11.2 ± 12.3 | 31.7 ± 24.1 | n.d. | 0.024 | - | - | |
| MNA score (M ± SD) | 24.1 ± 3.4 | 24.0 ± 6.9 | n.d. | 0.782 | - | - | |
| Comorbidities (n, %) | 16, 94.1 | 27, 93.1 | 6, 13.0 | 0.893 | 0 | 0 | |
| CPAR | COFF | p CPAR vs COFF | |
|---|---|---|---|
| n | 7 | 7 | |
| Demographic data | |||
| Age (M ± SD) | 102 ± 1.9 | 65.4 ± 6.6 | 0.000 |
| Female (n, %) | 5, 71.4 | 4, 57.1 | 1.000 |
| Anthropometric data | |||
| BMI (M ± SD) | 27.76 ± 5.3 | 25.93 ± 1.9 | 0.425 |
| Lifestyle factors | |||
| Current smoking status (n, %) | 0, 0.0 | 1, 14.3 | 1.000 |
| Former smoking status (n, %) | 5, 71.4 | 6, 85.7 | 0.500 |
| Current alcohol consumption (n, %) | 3, 42.9 | 6, 85.7 | 0.375 |
| Former alcohol consumption (n, %) | 5, 71.4 | 6, 85.7 | 1.000 |
| Coffee consumption (n, %) | 6, 85.7 | 7, 100 | 1.000 |
| Bowel function | |||
| Movements/week (M ± SD) | 3.7 ± 1.9 | 6.4 ± 1.1 | 0.037 |
| From 1 to 3 (movements/week, %) | 4, 57.1 | 0, 0.0 | n.d. |
| From 4 to 5 (movements/week, %) | 2, 28.6 | 1, 14.3 | n.d.. |
| From 6 to 7 (movements/week, %) | 1, 14.3 | 6, 85.7 | n.d. |
| Medications, n/day (M ± SD) | 5.0 ± 3.6 | 2.1 ± 1.7 | 0.041 |
| MMSE score (M ± SD) | 20.5 ± 4.3 | 27.2 ± 2.7 | 0.014 |
| MDS (M ± SD) | 28.6 ± 7.9 | 27.0 ± 7.0 | 0.323 |
| ADL score (M ± SD) | 2.4 ± 2.6 | 6.0 ± 0.0 | 0.010 |
| PASE score (M ± SD) | 10.5 ± 12.8 | 133.5 ± 22.3 | 0.000 |
| MNA score (M ± SD) | 24.0 ± 4.2 | 27.0 ± 2.5 | 0.023 |
| Comorbidities (n, %) | 7, 100 | 7, 100 | 1.000 |
| MEAN ± SD | OVERALL P | BONFERRONI P (CENT VS. CTLS) | BONFERRONI P (NON VS. CTLS) | |
|---|---|---|---|---|
| 0.003 | 0.015 | 0.022 | ||
| CENT | 4.88 ± 4.79 | |||
| NON | 3.83 ± 4.28 | |||
| CTLS | 1.73 ± 1.85 | |||
| 0.499 | ||||
| CPAR | 4.88 ± 4.79 | |||
| COFF | 3.83 ± 4.28 |
| Post hoc Analysis, Bonferroni Method (Only for Significant Bacteria) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Phylum | Family | Genus | Species | Kruskal-Wallis p-Value | Pairwise Group | Pairwise p-Value | Chi Square (χ2) | ↓/↑ | Mean ± SD CENT | Mean ± SD NON | Mean ± SD CTLs |
| Actinobacteria | 0.0018 | CENT- CTLs | 0.0393 | 12.66 | ↑ | 8.04 ± 10.23 | 9.12 ± 9.91 | 3.09 ± 3.61 | |||
| NON- CTLs | 0.0039 | ↑ | |||||||||
| Actinobacteria | Bifidobacteriaceae | 0.0037 | NON- CTLs | 0.0058 | 11.2 | ↑ | 6.95 ± 10.25 | 8.01 ± 9.71 | 2.08 ± 3.03 | ||
| Actinobacteria | Coriobacteriaceae | 0.0003 | CENT- CTLs | 0.0445 | 16.16 | ↑ | 1.10 ± 1.13 | 1.17 ± 1.41 | 0.56 ± 0.87 | ||
| NON- CTLs | 0.0004 | ↑ | |||||||||
| Actinobacteria | Bifidobacteriaceae | Bifidobacterium | 0.0039 | NON- CTLs | 0.0061 | 11.1 | ↑ | 6.91 ± 10.18 | 7.98 ± 9.67 | 2.07 ± 3.02 | |
| Actinobacteria | Coriobacteriaceae | Collinsella | 0.0022 | NON- CTLs | 0 | 12.26 | ↑ | 0.66 ± 0.73 | 0.77 ± 1.28 | 0.00 ± 0.00 | |
| CENT- CTLs | 0 | ↑ | |||||||||
| Actinobacteria | Coriobacteriaceae | Eggerthella | 0.0075 | CENT- CTLs | 0.0275 | 9.79 | ↑ | 0.10 ± 0.22 | 0.05 ± 0.05 | 0.03 ± 0.05 | |
| NON- CTLs | 0.0394 | ↑ | |||||||||
| Actinobacteria | Eggerthellaceae | Slackia | 0.0024 | NON- CTLs | 0.0028 | 12.07 | ↑ | 0.27 ± 0.29 | 0.29 ± 0.26 | 0.17 ± 0.26 | |
| Actinobacteria | Bifidobacteriaceae | Bifodobacterium | B. angulatum | 0 | NON- CTLs | 0 | 24.71 | ↑ | 0.01 ± 0.03 | 0.18 ± 0.60 | 0.00 ± 0.01 |
| CENT- NON | 0.0035 | 24.71 | ↓ | ||||||||
| Actinobacteria | Bifidobacteriaceae | Bifodobacterium | B. asteroides | 0.0006 | CENT- CTLs | 0.0294 | 14.95 | ↑ | 0.11 ± 0.18 | 0.11 ± 0.14 | 0.02 ± 0.03 |
| NON- CTLs | 0.0011 | ↑ | |||||||||
| Actinobacteria | Bifidobacteriaceae | Bifodobacterium | B. bifidum | 0.0175 | NON- CTLs | 0.0418 | 8.09 | ↑ | 0.29 ± 0.48 | 0.11 ± 0.17 | 0.04 ± 0.17 |
| Actinobacteria | Bifidobacteriaceae | Bifodobacterium | B. catenulatum | 0.0025 | CENT- NON | 0.0347 | 11.95 | ↓ | 0.92 ± 3.03 | 1.03 ± 2.04 | 0.12 ± 0.33 |
| NON- CTLs | 0.0031 | 11.95 | ↑ | ||||||||
| Actinobacteria | Bifidobacteriaceae | Bifodobacterium | B. choerinum | 0.0118 | NON- CTLs | 0.033 | 8.88 | ↑ | 0.14 ± 0.19 | 0.24 ± 0.33 | 0.09 ± 0.19 |
| Actinobacteria | Bifidobacteriaceae | Bifodobacterium | B. indicum | 0.0018 | NON- CTLs | 0.0022 | 12.68 | ↑ | 0.14 ± 0.25 | 0.23 ± 0.25 | 0.06 ± 0.10 |
| Actinobacteria | Bifidobacteriaceae | Bifodobacterium | B. kashiwanohense | 0.0011 | NON- CTLs | 0.0008 | 13.55 | ↑ | 0.12 ± 0.34 | 0.16 ± 0.29 | 0.02 ± 0.05 |
| Actinobacteria | Bifidobacteriaceae | Bifodobacterium | B. stercoris | 0.04 | NS | NS | 6.44 | 1.30 ± 2.75 | 1.26 ± 2.20 | 0.27 ± 0.46 | |
| Actinobacteria | Coriobacteriaceae | Collinsella | C. aerofaciens | 0.0122 | NON- CTLs | 0.0126 | 8.82 | ↑ | 0.36 ± 0.42 | 0.40 ± 0.48 | 0.21 ± 0.41 |
| Actinobacteria | Coriobacteriaceae | Collinsella | C. intestinalis | 0.0048 | CENT- CTLs | 0.0295 | 10.66 | ↑ | 0.13 ± 0.22 | 0.06 ± 0.06 | 0.09 ± 0.33 |
| NON- CTLs | 0.0192 | ↓ | |||||||||
| Actinobacteria | Coriobacteriaceae | Collinsella | C. tanakaei | 0.001 | NON- CTLs | 0.0007 | 13.77 | ↑ | 0.09 ± 0.34 | 0.29 ± 1.26 | 0.01 ± 0.02 |
| Bacteroidetes | 0.0002 | CENT- CTLs | 0.0032 | 16.95 | ↓ | 24.74 ± 24.26 | 25.28 ± 17.60 | 43.59 ± 21.64 | |||
| NON- CTLs | 0.0019 | ↓ | |||||||||
| Bacteroidetes | Bacteroidaceae | 0.0007 | CENT- CTLs | 0.0085 | 14.54 | ↓ | 15.77 ± 17.53 | 15.03 ± 12.52 | 28.05 ± 18.05 | ||
| NON- CTLs | 0.004 | ↓ | |||||||||
| Bacteroidetes | Rikenellaceae | 0.0023 | CENT- CTLs | 0.0034 | 12.17 | ↑ | 0.12 ± 0.18 | 0.06 ± 0.11 | 0.07 ± 0.14 | ||
| Bacteroidetes | Bacteroidaceae | Bacteroides | 0.0007 | CENT- CTLs | 0.0085 | 14.54 | ↓ | 15.77 ± 17.53 | 15.03 ± 12.52 | 28.05 ± 18.05 | |
| NON- CTLs | 0.004 | ↓ | |||||||||
| Bacteroidetes | Prevotellaceae | Paraprevotella | 0.0495 | NS | NS | NS | 0.11 ± 0.20 | 0.18 ± 0.36 | 0.34 ± 0.51 | ||
| Bacteroidetes | Porphyromonadaceae | Porphyromonas | 0.1131 | NS | NS | 4.36 | 0.02 ± 0.02 | 0.19 ± 0.48 | 0.06 ± 0.09 | ||
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. caccae | 0.0005 | NON- CTLs | 0.0003 | 15.31 | ↓ | 0.56 ± 0.76 | 0.27 ± 0.65 | 0.90 ± 1.09 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. cellulosilyticus | 0.011 | NON- CTLs | 0.0091 | 9.02 | ↓ | 0.37 ± 0.74 | 0.19 ± 0.56 | 0.61 ± 1.22 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. coprocola | 0.0209 | CENT- CTLs | 0.0333 | 7.73 | ↓ | 0.64 ± 2.45 | 0.28 ± 0.83 | 0.98 ± 3.70 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. denticanum | 0.0154 | NON- CTLs | 0.0363 | 8.35 | ↓ | 0.13 ± 0.31 | 0.12 ± 0.23 | 0.52 ± 1.20 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. dorei | 0.0338 | NS | NS | 6.78 | 1.05 ± 2.14 | 1.47 ± 3.18 | 2.21 ± 3.23 | |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. fragilis | 0.0172 | NON- CTLs | 0.0316 | 8.13 | ↑ | 0.22 ± 0.30 | 0.68 ± 1.54 | 0.24 ± 0.71 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. intestinalis | 0.0184 | CENT- NON | 0.0202 | 7.99 | ↑ | 0.22 ± 0.53 | 0.00 ± 0.01 | 0.11 ± 0.54 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. ovatus | 0.0085 | CENT- CTLs | 0.0179 | 9.54 | ↓ | 0.27 ± 0.51 | 0.44 ± 0.63 | 1.27 ± 2.57 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. paurosaccharolyticus | 0.0167 | CENT- CTLs | 0.0176 | 8.18 | ↓ | 0.10 ± 0.13 | 0.15 ± 0.18 | 0.18 ± 0.17 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. rodentium | 0.0002 | CENT- CTLs | 0.0446 | 17.37 | ↓ | 1.82 ± 2.47 | 0.95 ± 1.11 | 2.65 ± 2.36 |
| NON- CTLs | 0.0002 | ↓ | |||||||||
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. sartorii | 0.0002 | CENT- CTLs | 0.0006 | 17.3 | ↓ | 0.22 ± 0.60 | 0.12 ± 0.10 | 0.25 ± 0.20 |
| NON- CTLs | 0.0102 | ↓ | |||||||||
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. stercorirosoris | 0 | NON- CTLs | 0 | 23.17 | ↓ | 0.40 ± 0.41 | 0.22 ± 0.24 | 0.62 ± 0.52 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. uniformis | 0.0043 | NON- CTLs | 0.0042 | 10.89 | ↓ | 3.05 ± 5.92 | 1.43 ± 2.30 | 3.09 ± 3.36 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. xylanisolvens | 0.0005 | CENT- CTLs | 0.0017 | 15.19 | ↓ | 0.50 ± 0.47 | 0.74 ± 0.82 | 1.74 ± 2.57 |
| NON- CTLs | 0.014 | ↓ | |||||||||
| Bacteroidetes | Prevotellaceae | Paraprevotella | P. clara | 0.0378 | CENT- CTLs | 0.0453 | 6.55 | ↓ | 0.04 ± 0.08 | 0.06 ± 0.11 | 0.16 ± 0.28 |
| Bacteroidetes | Prevotellaceae | Prevotella | P. shahii | 0.0276 | NS | NS | 7.18 | 0.14 ± 0.59 | 0.02 ± 0.08 | 0.07 ± 0.21 | |
| Bacteroidetes | Sphingobacteriaceae | Sphingobacterium | S. shayense | 0.0389 | NON- CTLs | 0.0421 | 4.49 | ↓ | 0.09 ± 0.10 | 0.08 ± 0.10 | 0.18 ± 0.27 |
| Chloroflexi | Caldilineaceae | 0.0288 | NON- CTLs | 0.0267 | 7.09 | ↓ | 0.06 ± 0.04 | 0.05 ± 0.06 | 0.12 ± 0.13 | ||
| Cyanobacteria | 0.0093 | NON- CTLs | 0.0071 | 9.36 | ↓ | 0.62 ± 0.81 | 0.31 ± 0.45 | 0.95 ± 1.63 | |||
| Cyanobacteria | Aphanizomenonaceae | Dolichospermum | 0.0019 | CENT- CTLs | 0.0045 | 12.57 | ↓ | 0.00 ± 0.00 | 0.01 ± 0.01 | 0.34 ± 1.30 | |
| NON- CTLs | 0.0348 | ↓ | |||||||||
| Cyanobacteria | Aphanizomenonaceae | Dolichospermum | D. macrosporum | 0 | CENT- CTLs | 0.0005 | 26.32 | ↓ | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.34 ± 1.30 |
| NON- CTLs | 0 | ↓ | |||||||||
| Euryarchaeota | 0 | CENT- CTLs | 0 | 19.43 | ↑ | 0.29 ± 0.65 | 0.12 ± 0.36 | 0.00 ± 0.00 | |||
| NON- CTLs | 0.0131 | ↑ | |||||||||
| Euryarchaeota | Methanobacteriaceae | 0 | CENT- CTLs | 0 | 40.68 | ↑ | 0.29 ± 0.65 | 0.12 ± 0.36 | 0.03 ± 0.20 | ||
| NON- CTLs | 0.013 | ↑ | |||||||||
| Euryarchaeota | Methanobacteriaceae | Methanobrevibacter | M. smithii | 0 | CENT- CTLs | 0 | 25.27 | ↑ | 0.28 ± 0.61 | 0.11 ± 0.34 | 0.04 ± 0.19 |
| NON- CTLs | 0.0116 | ↑ | |||||||||
| Euryarchaeota | Methanobacteriaceae | Methanobrevibacter | 0 | CENT- CTLs | 0 | 24.45 | ↑ | 0.29 ± 0.65 | 0.12 ± 0.36 | 0.04 ± 0.20 | |
| NON- CTLs | 0.0189 | ↑ | |||||||||
| Firmicutes | Eubacteriaceae | 0.0179 | NON- CTLs | 0.0445 | 8.05 | ↑ | 0.13 ± 0.10 | 0.14 ± 0.12 | 0.09 ± 0.08 | ||
| Firmicutes | Lactobacillaceae | 0.009 | CENT- NON | 0.0267 | 9.43 | ↓ | 0.23 ± 0.47 | 1.04 ± 3.22 | 0.13 ± 0.17 | ||
| NON- CTLs | 0.0235 | 9.43 | ↑ | ||||||||
| Firmicutes | Streptococcaceae | 0 | NON- CTLs | 0 | 23.55 | ↑ | 0.72 ± 0.87 | 1.80 ± 2.18 | 0.19 ± 0.26 | ||
| Firmicutes | Synergistaceae | 0 | CENT- CTLs | 0.0067 | 30.49 | ↑ | 0.48 ± 0.88 | 0.17 ± 0.63 | 0.03 ± 0.13 | ||
| Firmicutes | Thermicanaceae | 0.0051 | CENT- CTLs | 0.0244 | 10.55 | ↑ | 0.11 ± 0.21 | 0.14 ± 0.23 | 0.03 ± 0.06 | ||
| NON- CTLs | 0.0251 | ↑ | |||||||||
| Firmicutes | Acidaminococcaceae | Acidaminococcus | 0.0084 | NON- CTLs | 0.006 | 9.56 | ↓ | 0.36 ± 1.22 | 0.04 ± 0.13 | 0.54 ± 1.64 | |
| Firmicutes | Lachnospiraceae | Blautia | 0.0376 | CENT- CTLs | 0.0335 | 6.56 | ↓ | 3.63 ± 2.36 | 5.48 ± 3.94 | 6.50 ± 4.86 | |
| Firmicutes | Lachnospiraceae | Butyrivibrio | 0.0016 | CENT- CTLs | 0.0038 | 12.93 | ↓ | 0.02 ± 0.02 | 0.02 ± 0.03 | 0.12 ± 0.34 | |
| NON- CTLs | 0.0318 | ↓ | |||||||||
| Firmicutes | Syntrophomonadaceae | Caldicellulosiruptor | 0.0102 | CENT- CTLs | 0.0148 | 9.17 | ↑ | 0.11 ± 0.11 | 0.08 ± 0.10 | 0.06 ± 0.07 | |
| Firmicutes | Eubacteriaceae | Eubacterium | 0.0296 | CENT- NON | 0.0283 | 7.04 | ↓ | 0.13 ± 0.21 | 0.30 ± 0.82 | 0.05 ± 0.10 | |
| Firmicutes | Lactobacillaceae | Lactobacillus | 0.0064 | NON- CTLs | 0.0174 | 10.12 | ↑ | 0.22 ± 0.46 | 1.00 ± 3.09 | 0.12 ± 0.16 | |
| CENT- NON | 0.0204 | 10.12 | ↓ | ||||||||
| Firmicutes | Acidaminococcaceae | Phascolarctobacterium | 0.0197 | NON- CTLs | 0.0393 | 7.85 | ↓ | 1.84 ± 2.79 | 0.29 ± 0.44 | 1.35 ± 2.00 | |
| Firmicutes | Streptococcaceae | Streptococcus | 0 | NON- CTLs | 0 | 23.67 | ↑ | 0.70 ± 0.87 | 1.78 ± 2.16 | 0.19 ± 0.25 | |
| Firmicutes | Bacillales_Family X_Incertae Sedis | Thermicanus | 0.0051 | CENT- CTLs | 0.0244 | 10.55 | ↑ | 0.11 ± 0.21 | 0.14 ± 0.23 | 0.03 ± 0.06 | |
| NON- CTLs | 0.0251 | ↑ | |||||||||
| Firmicutes | Acidaminococcaceae | Acidaminococcus | A. intestini | 0.001 | NON- CTLs | 0.0009 | 13.81 | ↓ | 0.02 ± 0.06 | 0.00 ± 0.00 | 0.14 ± 0.42 |
| Firmicutes | Lachnospiraceae | Blautia | B. coccoides | 0.0072 | CENT- CTLs | 0.0059 | 9.87 | ↓ | 0.63 ± 0.46 | 1.40 ± 1.31 | 1.40 ± 1.05 |
| CENT- NON | 0.0375 | 9.87 | ↓ | ||||||||
| Firmicutes | Lachnospiraceae | Blautia | B. wexlerae | 0.0526 | NS | NS | 5.89 | 0.29 ± 0.42 | 0.59 ± 0.84 | 0.88 ± 1.84 | |
| Firmicutes | Lachnospiraceae | Butyrivibrio | B. proteoclasticus | 0.0016 | CENT- CTLs | 0.0039 | 12.89 | ↓ | 0.02 ± 0.02 | 0.02 ± 0.03 | 0.12 ± 0.34 |
| NON- CTLs | 0.0327 | ↓ | |||||||||
| Firmicutes | Erysipelothricaceae | Erysipelothrix | E. inopinata | 0.0317 | CENT- CTLs | 0.0258 | 6.9 | ↓ | 0.06 ± 0.13 | 0.13 ± 0.29 | 0.16 ± 0.49 |
| Firmicutes | Lactobacillaceae | Lactobacillus | L. taiwanensis | 0.0001 | CENT- CTLs | 0.0313 | 18.86 | ↑ | 0.02 ± 0.05 | 0.16 ± 0.84 | 0.00 ± 0.00 |
| NON- CTLs | 0.0001 | ↑ | |||||||||
| Firmicutes | Acidaminococcaceae | Phascolarctobacterium | P. faecium | 0.0048 | NON- CTLs | 0.0252 | 10.67 | ↓ | 0.68±1.10 | 0.04±0.12 | 0.45±1.03 |
| CENT- NON | 0.0095 | 10.67 | ↑ | ||||||||
| Firmicutes | Ruminococcaceae | Ruminococcus | R. torques | 0.0437 | NS | NS | NS | 0.27 ± 0.58 | 0.13 ± 0.26 | 0.14 ± 0.30 | |
| Firmicutes | Streptococcaceae | Streptococcus | S. bovis | 0 | NON- CTLs | 0 | 20.22 | ↑ | 0.03 ± 0.03 | 0.24 ± 0.55 | 0.03 ± 0.13 |
| Firmicutes | Streptococcaceae | Streptococcus | S. parasanguinis | 0 | NON- CTLs | 0 | 24.27 | ↑ | 0.05 ± 0.08 | 0.19 ± 0.30 | 0.01 ± 0.01 |
| Firmicutes | Streptococcaceae | Streptococcus | S. vestibularis | 0.0066 | NON- CTLs | 0.0046 | 10.05 | ↑ | 0.18 ± 0.31 | 0.57 ± 0.89 | 0.05 ± 0.10 |
| Firmicutes | Veillonellaceae | Veillonella | V. atypica | 0.0122 | NON- CTLs | 0.0091 | 8.82 | ↑ | 0.05 ± 0.14 | 0.12 ± 0.22 | 0.04 ± 0.11 |
| Firmicutes | Veillonellaceae | Veillonella | V. dispar | 0.0195 | NON- CTLs | 0.0155 | 7.87 | ↑ | 0.05 ± 0.18 | 0.11 ± 0.28 | 0.02 ± 0.04 |
| Fusobacteria | Fusobacteriaceae | 0.0266 | NS | NS | 7.26 | 0.03 ± 0.10 | 0.10 ± 0.35 | 0.21 ± 1.39 | |||
| Proteobacteria | Alcaligenaceae | 0.0003 | CENT- CTLs | 0.0065 | 16.38 | ↓ | 0.46 ± 1.31 | 0.46 ± 1.43 | 0.83 ± 0.86 | ||
| NON- CTLs | 0.0014 | ↓ | |||||||||
| Proteobacteria | Comamonadaceae | 0.0357 | NS | NS | NS | 0.07 ± 0.11 | 0.11 ± 0.33 | 0.13 ± 0.18 | |||
| Proteobacteria | Desulfohalobiaceae | 0.014 | CENT- CTLs | 0.0109 | 8.53 | ↑ | 0.23 ± 0.27 | 0.11 ± 0.09 | 0.15 ± 0.27 | ||
| Proteobacteria | Xanthomonadaceae | 0 | CENT- CTLs | 0.0015 | 45.43 | ↑ | 0.13 ± 0.15 | 0.09 ± 0.11 | 0.01 ± 0.03 | ||
| NON- CTLs | 0.0021 | ↑ | |||||||||
| Proteobacteria | Oxalobacteraceae | Collimonas | 0 | CENT- CTLs | 0 | 61.68 | ↓ | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.32 ± 0.54 | |
| NON- CTLs | 0 | ↓ | |||||||||
| Proteobacteria | Desulfohalobiaceae | Desulfonauticus | 0.0142 | CENT- CTLs | 0.011 | 8.51 | ↑ | 0.23 ± 0.27 | 0.11 ± 0.09 | 0.15 ± 0.27 | |
| Proteobacteria | Desulfovibrionaceae | Desulfovibrio | 0.0079 | CENT- CTLs | 0.0177 | 9.68 | ↑ | 0.44 ± 0.48 | 0.70 ± 1.66 | 0.19 ± 0.34 | |
| Proteobacteria | Enterobacteriaceae | Enterobacter | 0.0246 | NON- CTLs | 0.0201 | 7.41 | ↑ | 0.34 ± 0.67 | 0.76 ± 2.35 | 0.11 ± 0.35 | |
| Proteobacteria | Enterobacteriaceae | Escherichia | 0.0022 | NON- CTLs | 0.0019 | 12.28 | ↑ | 7.00 ± 12.18 | 3.14 ± 6.70 | 0.22 ± 0.65 | |
| Proteobacteria | Yersiniaceae | Serratia | 0.0007 | NON- CTLs | 0.0005 | 14.56 | ↑ | 1.15 ± 1.92 | 0.67 ± 1.10 | 0.06 ± 0.13 | |
| Proteobacteria | Sutterellaceae | Sutterella | 0.0003 | CENT- CTLs | 0.0068 | 16.32 | ↓ | 0.43 ± 1.17 | 0.46 ± 1.43 | 0.80 ± 0.84 | |
| NON- CTLs | 0.0015 | ↓ | |||||||||
| Proteobacteria | Zoogloeaceae | Uliginosibacterium | 0.0327 | NON- CTLs | 0.0356 | 6.84 | ↓ | 0.02 ± 0.07 | 0.00 ± 0.00 | 0.20 ± 1.25 | |
| Proteobacteria | Enterobacteriaceae | Candidatus Blochmannia | C. B. rufipes | 0.0118 | NON- CTLs | 0.0123 | 8.89 | ↓ | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.65 ± 0.76 |
| Proteobacteria | Desulfohalobiaceae | Desulfonauticus | D. autotrophicus | 0.0142 | CENT- CTLs | 0.011 | 8.51 | ↑ | 0.23 ± 0.27 | 0.11 ± 0.09 | 0.15 ± 0.27 |
| Proteobacteria | Desulfovibrionaceae | Desulfovibrio | D. piger | 0.0004 | CENT- CTLs | 0.0093 | 15.42 | ↑ | 0.14 ± 0.36 | 0.20 ± 0.72 | 0.03 ± 0.09 |
| NON- CTLs | 0.002 | ↑ | |||||||||
| Proteobacteria | Enterobacteriaceae | Escherichia | E. albertii | 0.004 | NON- CTLs | 0.0037 | 11.06 | ↑ | 5.67 ± 9.94 | 2.54 ± 5.38 | 0.20 ± 0.60 |
| Proteobacteria | Yersiniaceae | Serratia | S. entomophila | 0.0009 | NON- CTLs | 0.0007 | 14.12 | ↑ | 1.13 ± 1.90 | 0.66 ± 1.09 | 0.06 ± 0.13 |
| Synergistetes | 0.0007 | CENT- CTLs | 0.0024 | 14.54 | ↑ | 0.56 ± 0.95 | 0.28 ± 0.70 | 0.04 ± 0.07 | |||
| NON- CTLs | 0.0154 | ↑ | |||||||||
| Synergistetes | Synergistaceae | Cloacibacillus | 0.0028 | CENT- CTLs | 0.0018 | 11.77 | ↑ | 0.27 ± 0.71 | 0.11 ± 0.57 | 0.02 ± 0.11 | |
| Synergistetes | Synergistaceae | Synergistes | 0.0191 | CENT- CTLs | 0.0148 | 7.92 | ↑ | 0.15 ± 0.45 | 0.03 ± 0.09 | 0.01 ± 0.06 | |
| Verrucomicrobia | 0.0032 | CENT- CTLs | 0.0036 | 11.47 | ↑ | 10.26 ± 14.88 | 6.46 ± 10.39 | 2.20 ± 4.71 | |||
| Verrucomicrobia | Verrucomicrobiaceae | 0.0047 | CENT- CTLs | 0.0054 | 10.72 | ↑ | 10.19 ± 14.84 | 6.43 ± 10.36 | 2.19 ± 4.70 | ||
| Verrucomicrobia | Verrucomicrobiaceae | Akkermansia | 0.0054 | CENT- CTLs | 0.0058 | 10.45 | ↑ | 9.02 ± 13.17 | 5.67 ± 9.17 | 1.91 ± 4.12 | |
| Verrucomicrobia | Verrucomicrobiaceae | Luteolibacter | 0.001 | CENT- CTLs | 0.0012 | 13.78 | ↑ | 0.49 ± 0.70 | 0.31 ± 0.50 | 0.11 ± 0.24 | |
| Verrucomicrobia | Verrucomicrobiaceae | Prosthecobacter | 0.0072 | CENT- CTLs | 0.0098 | 9.86 | ↑ | 0.16 ± 0.22 | 0.10 ± 0.15 | 0.04 ± 0.08 | |
| Verrucomicrobia | Rubritaleaceae | Rubritalea | 0.0035 | CENT- CTLs | 0.0045 | 11.29 | ↑ | 0.38 ± 0.53 | 0.24 ± 0.39 | 0.09 ± 0.18 | |
| Verrucomicrobia | Verrucomicrobiaceae | Akkermansia | A. muciniphila | 0.0054 | CENT- CTLs | 0.0058 | 10.45 | ↑ | 9.02 ± 13.16 | 5.67 ± 9.17 | 1.91 ± 4.12 |
| Verrucomicrobia | Verrucomicrobiaceae | Luteolibacter | L. algae | 0.001 | CENT- CTLs | 0.0012 | 13.78 | ↑ | 0.49 ± 0.70 | 0.31 ± 0.50 | 0.11 ± 0.24 |
| Post-Hoc Analysis, Bonferroni Method (only for Significant Bacteria) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Phylum | Family | Genus | Species | Kruskal-Wallis p-Value | Bonferroni p | ↓/↑ | Mean ± SD CPAR | Mean ± SD COFF |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. denticanum | 0.028 | 1.37 | ↓ | 0.05 ± 0.03 | 0.97 ± 0.84 |
| Bacteroidetes | Bacteroidaceae | Bacteroides | B. plebeius | 0.043 | 1.98 | ↓ | 0.06 ± 0.14 | 2.03 ± 2.10 |
| Firmicutes | Ruminococcaceae | Faecalibacterium | 0.018 | 0.90 | ↓ | 7.52 ± 4.48 | 12.83 ± 8.04 | |
| Firmicutes | Ruminococcaceae | Faecalibacterium | F. prausnitzii | 0.028 | 1.37 | ↓ | 1.81 ± 1.34 | 3.34 ± 2.63 |
| Firmicutes | Lachnospiraceae | Roseburia | R. faecis | 0.028 | 1.37 | ↓ | 0.32 ± 0.23 | 1.26 ± 0.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmas, V.; Pisanu, S.; Madau, V.; Casula, E.; Deledda, A.; Cusano, R.; Uva, P.; Loviselli, A.; Velluzzi, F.; Manzin, A. Gut Microbiota Markers and Dietary Habits Associated with Extreme Longevity in Healthy Sardinian Centenarians. Nutrients 2022, 14, 2436. https://doi.org/10.3390/nu14122436
Palmas V, Pisanu S, Madau V, Casula E, Deledda A, Cusano R, Uva P, Loviselli A, Velluzzi F, Manzin A. Gut Microbiota Markers and Dietary Habits Associated with Extreme Longevity in Healthy Sardinian Centenarians. Nutrients. 2022; 14(12):2436. https://doi.org/10.3390/nu14122436
Chicago/Turabian StylePalmas, Vanessa, Silvia Pisanu, Veronica Madau, Emanuela Casula, Andrea Deledda, Roberto Cusano, Paolo Uva, Andrea Loviselli, Fernanda Velluzzi, and Aldo Manzin. 2022. "Gut Microbiota Markers and Dietary Habits Associated with Extreme Longevity in Healthy Sardinian Centenarians" Nutrients 14, no. 12: 2436. https://doi.org/10.3390/nu14122436
APA StylePalmas, V., Pisanu, S., Madau, V., Casula, E., Deledda, A., Cusano, R., Uva, P., Loviselli, A., Velluzzi, F., & Manzin, A. (2022). Gut Microbiota Markers and Dietary Habits Associated with Extreme Longevity in Healthy Sardinian Centenarians. Nutrients, 14(12), 2436. https://doi.org/10.3390/nu14122436