
Alkaline Phosphatase: An Old Friend as Treatment Target for Cardiovascular and Mineral Bone Disorders in Chronic Kidney Disease

 ,
,  ,
, 
 , and
, and
Abstract
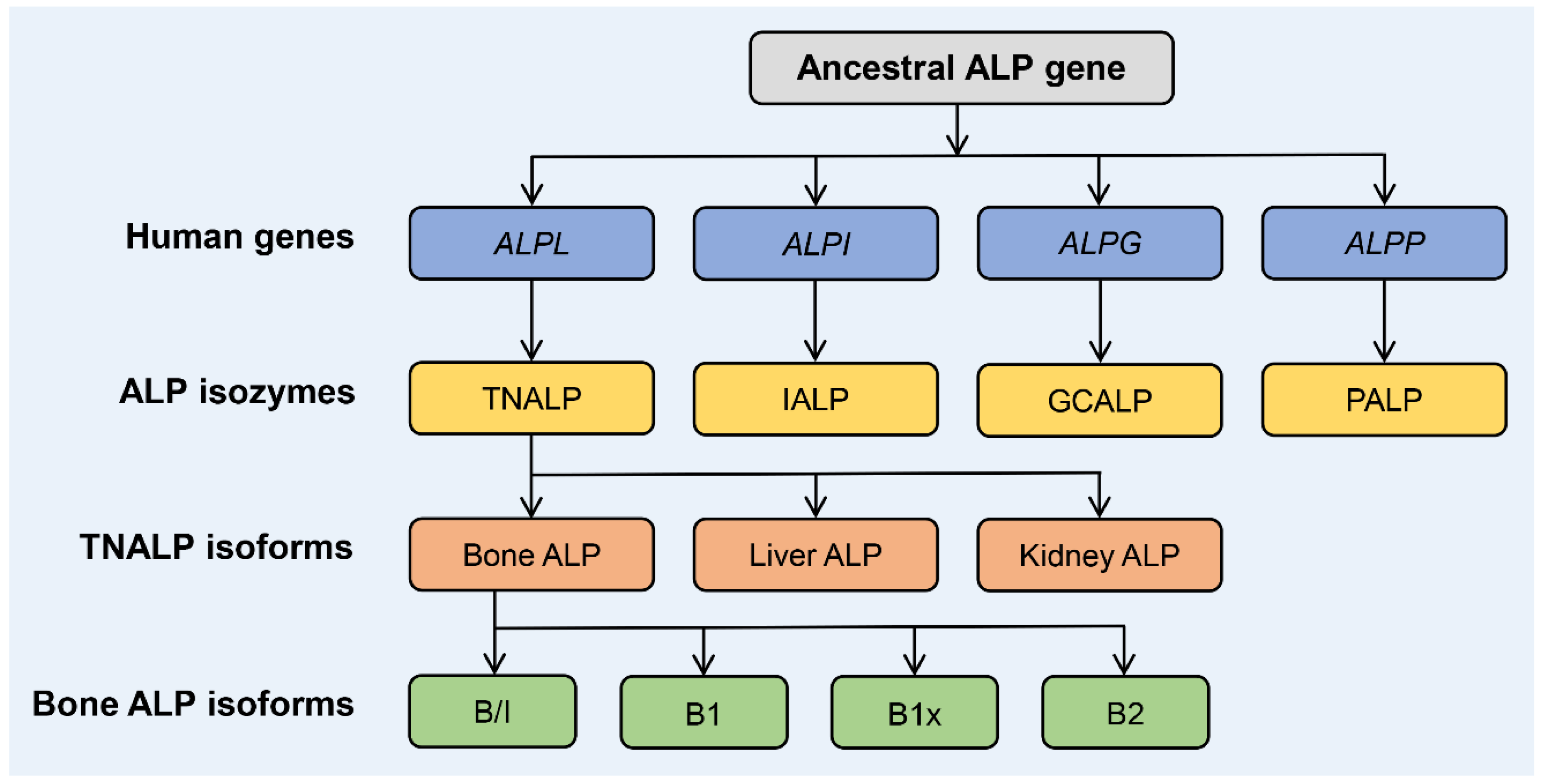
1. Biology and Structure of ALP Isozymes and Isoforms
2. Function of BALP
3. The Role of ALP/BALP in Inflammation, Metabolic Syndrome, and Proteinuria
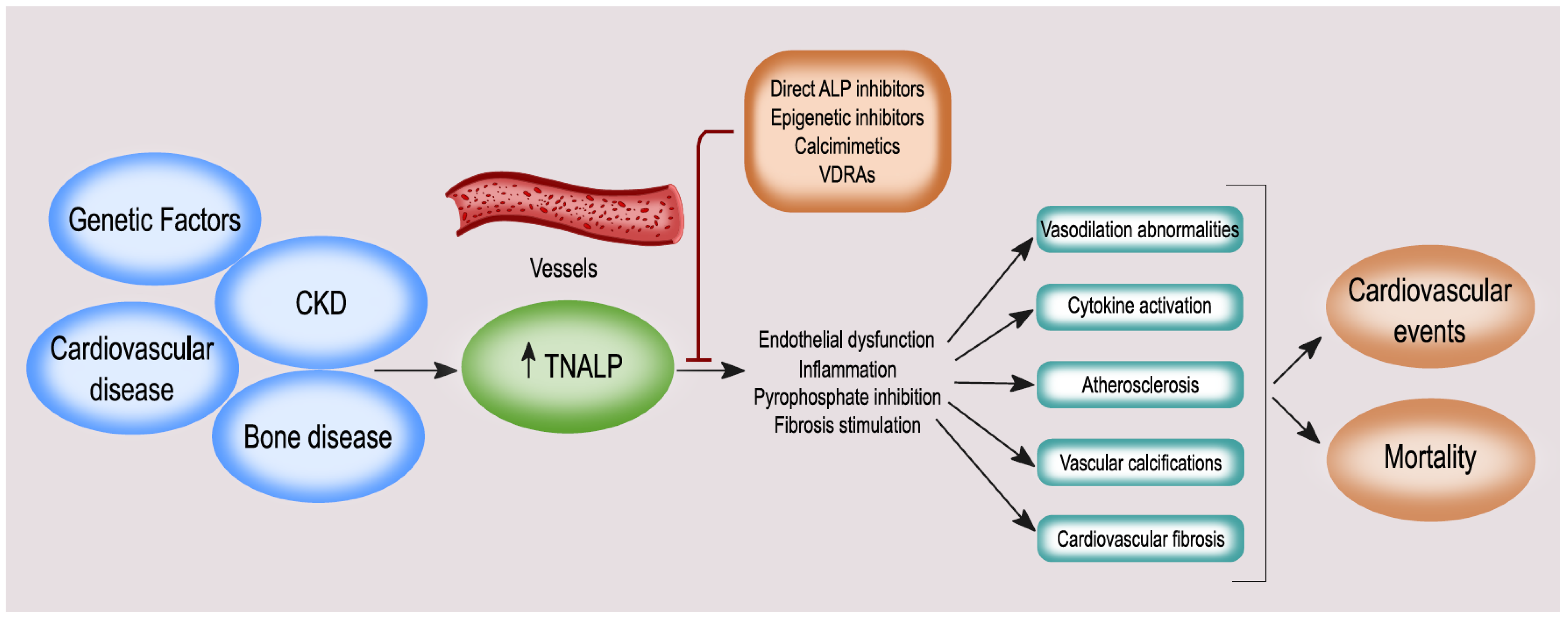
4. ALP and BALP: Vascular Calcification, Endothelial Dysfunction, Cardiovascular Disease (CVD), and Mortality
5. The Association between ALP, Bone Turnover, and Fracture Risk
6. Vitamin K and BALP
7. Treatment Strategies Targeting ALP
7.1. Modulation of ALP Expression: Serum ALP as Interventional Treatment Target
7.2. Effect of ALP Modulation on Clinical Outcomes: From CKD-MBD Treatment to Anti-Fracture Treatment
7.2.1. Apabetalone
7.2.2. Vitamin D
7.2.3. Phosphate Binders
7.2.4. Calcimimetics
7.2.5. Denosumab
7.2.6. Bisphosphonates
7.2.7. Teriparatide
7.2.8. Romosozumab
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALP | alkaline phosphatase |
| BALP | bone alkaline phosphatase |
| BET | bromodomain and extraterminal |
| BMD | bone mineral density |
| CKD | chronic kidney disease |
| CKD-MBD | chronic kidney disease-mineral bone disorder |
| CRP | C-reactive protein |
| CVD | cardiovascular disease |
| ESRD | end-stage renal disease |
| GCALP | germ cell alkaline phosphatase |
| GPI | glycosylphosphatidylinositol |
| HAoSMC | human aortic smooth muscle cells |
| HPP | hypophosphatasia |
| IALP | intestinal alkaline phosphatase |
| IL | interleukin |
| iNOS | inducible NO synthase |
| MGP | matrix Gla protein |
| MKn | menaquinone |
| NF-κB | nuclear factor κB |
| PALP | placental alkaline phosphatase |
| PD | peritoneal dialysis |
| PK | phylloquinone |
| PPi | inorganic pyrophosphate |
| PTH | parathyroid hormone |
| SXR | steroid xenobiotic receptor |
| TNALP | tissue-nonspecific alkaline phosphatase |
| TNF | tumor necrosis factor |
| TRACP5b | tartrate-resistant acid phosphatase isoform 5b |
| VSMC | vascular smooth muscle cell |
References
- Millán, J.L. Alkaline Phosphatases. Purinergic Signal. 2006, 2, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Swallow, D.M.; Povey, S.; Parkar, M.; Andrews, P.W.; Harris, H.; Pym, B.; Goodfellow, P. Mapping of the Gene Coding for the Human Liver/Bone/Kidney Isozyme of Alkaline Phosphatase to Chromosome 1. Ann. Hum. Genet. 1986, 50, 229–235. [Google Scholar] [CrossRef]
- Smith, M.; Weiss, M.J.; Griffin, C.A.; Murray, J.C.; Buetow, K.H.; Emanuel, B.S.; Henthorn, P.S.; Harris, H. Regional Assignment of the Gene for Human Liver/Bone/Kidney Alkaline Phosphatase to Chromosome 1p36.1-P34. Genomics 1988, 2, 139–143. [Google Scholar] [CrossRef]
- Weiss, M.J.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the Human Liver/Bone/Kidney Alkaline Phosphatase Gene. J. Biol. Chem. 1988, 263, 12002–12010. [Google Scholar] [CrossRef]
- Gainer, A.L.; Stinson, R.A. Evidence That Alkaline Phosphatase from Human Neutrophils Is the Same Gene Product as the Liver/Kidney/Bone Isoenzyme. Clin. Chim. Acta 1982, 123, 11–17. [Google Scholar] [CrossRef]
- Nwafor, D.C.; Brichacek, A.L.; Ali, A.; Brown, C.M. Tissue-Nonspecific Alkaline Phosphatase in Central Nervous System Health and Disease: A Focus on Brain Microvascular Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 5257. [Google Scholar] [CrossRef]
- Haarhaus, M.; Arnqvist, H.J.; Magnusson, P. Calcifying Human Aortic Smooth Muscle Cells Express Different Bone Alkaline Phosphatase Isoforms, Including the Novel B1x Isoform. J. Vasc. Res. 2013, 50, 167–174. [Google Scholar] [CrossRef]
- Anh, D.J.; Dimai, H.P.; Hall, S.L.; Farley, J.R. Skeletal Alkaline Phosphatase Activity Is Primarily Released from Human Osteoblasts in an Insoluble Form, and the Net Release Is Inhibited by Calcium and Skeletal Growth Factors. Calcif. Tissue Int. 1998, 62, 332–340. [Google Scholar] [CrossRef]
- Anh, D.J.; Eden, A.; Farley, J.R. Quantitation of Soluble and Skeletal Alkaline Phosphatase, and Insoluble Alkaline Phosphatase Anchor-Hydrolase Activities in Human Serum. Clin. Chim. Acta 2001, 311, 137–148. [Google Scholar] [CrossRef]
- Magnusson, P.; Sharp, C.A.; Farley, J.R. Different Distributions of Human Bone Alkaline Phosphatase Isoforms in Serum and Bone Tissue Extracts. Clin. Chim. Acta 2002, 325, 59–70. [Google Scholar] [CrossRef]
- Low, M.G.; Huang, K.S. Factors Affecting the Ability of Glycosylphosphatidylinositol-Specific Phospholipase D to Degrade the Membrane Anchors of Cell Surface Proteins. Biochem. J. 1991, 279 Pt 2, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Nosjean, O.; Briolay, A.; Roux, B. Mammalian GPI Proteins: Sorting, Membrane Residence and Functions. Biochim. Biophys Acta 1997, 1331, 153–186. [Google Scholar] [CrossRef]
- Rhode, H.; Schulze, M.; Cumme, G.A.; Göhlert, A.; Blume, E.; Bublitz, R.; Schilling, K.; Horn, A. Glycosylphosphatidylinositol-Specific Phospholipase D of Human Serum--Activity Modulation by Naturally Occurring Amphiphiles. Biol. Chem. 2000, 381, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Sowadski, J.M.; Handschumacher, M.D.; Murthy, H.M.; Kundrot, C.E.; Wyckoff, H.W. Crystallographic Observations of the Metal Ion Triple in the Active Site Region of Alkaline Phosphatase. J. Mol. Biol. 1983, 170, 575–581. [Google Scholar] [CrossRef]
- Stec, B.; Holtz, K.M.; Kantrowitz, E.R. A Revised Mechanism for the Alkaline Phosphatase Reaction Involving Three Metal Ions. J. Mol. Biol. 2000, 299, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E.; Stura, E.; Lia-Baldini, A.S.; Stigbrand, T.; Ménez, A.; Le Du, M.H. Structural Evidence for a Functional Role of Human Tissue Nonspecific Alkaline Phosphatase in Bone Mineralization. J. Biol. Chem. 2001, 276, 31171–31178. [Google Scholar] [CrossRef] [PubMed]
- Hoylaerts, M.F.; Van Kerckhoven, S.; Kiffer-Moreira, T.; Sheen, C.; Narisawa, S.; Millán, J.L. Functional Significance of Calcium Binding to Tissue-Nonspecific Alkaline Phosphatase. PLoS ONE 2015, 10, e0119874. [Google Scholar] [CrossRef]
- Magnusson, P.; Degerblad, M.; Sääf, M.; Larsson, L.; Thorén, M. Different Responses of Bone Alkaline Phosphatase Isoforms during Recombinant Insulin-like Growth Factor-I (IGF-I) and during Growth Hormone Therapy in Adults with Growth Hormone Deficiency. J. Bone Miner. Res. 1997, 12, 210–220. [Google Scholar] [CrossRef]
- Haarhaus, M.; Gilham, D.; Kulikowski, E.; Magnusson, P.; Kalantar-Zadeh, K. Pharmacologic Epigenetic Modulators of Alkaline Phosphatase in Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens 2020, 29, 4–15. [Google Scholar] [CrossRef]
- Nizet, A.; Cavalier, E.; Stenvinkel, P.; Haarhaus, M.; Magnusson, P. Bone Alkaline Phosphatase: An Important Biomarker in Chronic Kidney Disease—Mineral and Bone Disorder. Clin. Chim. Acta 2020, 501, 198–206. [Google Scholar] [CrossRef]
- Magnusson, P.; Arlestig, L.; Paus, E.; Di Mauro, S.; Testa, M.P.; Stigbrand, T.; Farley, J.R.; Nustad, K.; Millán, J.L. Monoclonal Antibodies against Tissue-Nonspecific Alkaline Phosphatase. Report of the ISOBM TD9 Workshop. Tumour Biol. 2002, 23, 228–248. [Google Scholar] [CrossRef]
- Sardiwal, S.; Magnusson, P.; Goldsmith, D.J.A.; Lamb, E.J. Bone Alkaline Phosphatase in CKD-Mineral Bone Disorder. Am. J. Kidney Dis. 2013, 62, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Brandenburg, V.; Kalantar-Zadeh, K.; Stenvinkel, P.; Magnusson, P. Alkaline Phosphatase: A Novel Treatment Target for Cardiovascular Disease in CKD. Nat. Rev. Nephrol. 2017, 13, 429–442. [Google Scholar] [CrossRef]
- Magnusson, P.; Sharp, C.A.; Magnusson, M.; Risteli, J.; Davie, M.W.; Larsson, L. Effect of Chronic Renal Failure on Bone Turnover and Bone Alkaline Phosphatase Isoforms. Kidney Int. 2001, 60, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Cavalier, E.; Souberbielle, J.-C.; Gadisseur, R.; Dubois, B.; Krzesinski, J.-M.; Delanaye, P. Inter-Method Variability in Bone Alkaline Phosphatase Measurement: Clinical Impact on the Management of Dialysis Patients. Clin. Biochem. 2014, 47, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, P.; Larsson, L.; Magnusson, M.; Davie, M.W.; Sharp, C.A. Isoforms of Bone Alkaline Phosphatase: Characterization and Origin in Human Trabecular and Cortical Bone. J. Bone Miner. Res. 1999, 14, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.D.A.M.; Eijken, M.; van de Peppel, J.; van Leeuwen, J.P.T.M. Calcifying Vascular Smooth Muscle Cells and Osteoblasts: Independent Cell Types Exhibiting Extracellular Matrix and Biomineralization-Related Mimicries. BMC Genom. 2014, 15, 965. [Google Scholar] [CrossRef]
- Magnusson, P.; Häger, A.; Larsson, L. Serum Osteocalcin and Bone and Liver Alkaline Phosphatase Isoforms in Healthy Children and Adolescents. Pediatr. Res. 1995, 38, 955–961. [Google Scholar] [CrossRef]
- Magnusson, P.; Löfman, O.; Larsson, L. Determination of Alkaline Phosphatase Isoenzymes in Serum by High-Performance Liquid Chromatography with Post-Column Reaction Detection. J. Chromatogr. 1992, 576, 79–86. [Google Scholar] [CrossRef]
- Whyte, M.P.; Ma, N.S.; Mumm, S.; Gottesman, G.S.; McAlister, W.H.; Nenninger, A.R.; Bijanki, V.N.; Ericson, K.L.; Magnusson, P. Persistent Idiopathic Hyperphosphatasemia from Bone Alkaline Phosphatase in a Healthy Boy. Bone 2020, 138, 115459. [Google Scholar] [CrossRef]
- Swolin-Eide, D.; Hansson, S.; Larsson, L.; Magnusson, P. The Novel Bone Alkaline Phosphatase B1x Isoform in Children with Kidney Disease. Pediatr. Nephrol. 2006, 21, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Monier-Faugere, M.-C.; Magnusson, P.; Malluche, H.H. Bone Alkaline Phosphatase Isoforms in Hemodialysis Patients with Low versus Non-Low Bone Turnover: A Diagnostic Test Study. Am. J. Kidney Dis. 2015, 66, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Bover, J.; Ureña, P.; Aguilar, A.; Mazzaferro, S.; Benito, S.; López-Báez, V.; Ramos, A.; daSilva, I.; Cozzolino, M. Alkaline Phosphatases in the Complex Chronic Kidney Disease-Mineral and Bone Disorders. Calcif. Tissue Int. 2018, 103, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Fernström, A.; Magnusson, M.; Magnusson, P. Clinical Significance of Bone Alkaline Phosphatase Isoforms, Including the Novel B1x Isoform, in Mild to Moderate Chronic Kidney Disease. Nephrol. Dial. Transpl. 2009, 24, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Millán, J.L. The Role of Phosphatases in the Initiation of Skeletal Mineralization. Calcif. Tissue Int. 2013, 93, 299–306. [Google Scholar] [CrossRef]
- Mornet, E.; Hofmann, C.; Bloch-Zupan, A.; Girschick, H.; Le Merrer, M. Clinical Utility Gene Card for: Hypophosphatasia–Update 2013. Eur. J. Hum. Genet. 2014, 22, 572. [Google Scholar] [CrossRef]
- Millán, J.L.; Whyte, M.P. Alkaline phosphatase and hypophosphatasia. Calcif. Tissue Int. 2016, 98, 398–416. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia—Aetiology, Nosology, Pathogenesis, Diagnosis and Treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef]
- Johnson, K.A.; Hessle, L.; Vaingankar, S.; Wennberg, C.; Mauro, S.; Narisawa, S.; Goding, J.W.; Sano, K.; Millan, J.L.; Terkeltaub, R. Osteoblast Tissue-Nonspecific Alkaline Phosphatase Antagonizes and Regulates PC-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1365–R1377. [Google Scholar] [CrossRef]
- Halling Linder, C.; Narisawa, S.; Millán, J.L.; Magnusson, P. Glycosylation Differences Contribute to Distinct Catalytic Properties among Bone Alkaline Phosphatase Isoforms. Bone 2009, 45, 987–993. [Google Scholar] [CrossRef]
- Magnusson, P.; Farley, J.R. Differences in Sialic Acid Residues among Bone Alkaline Phosphatase Isoforms: A Physical, Biochemical, and Immunological Characterization. Calcif Tissue Int. 2002, 71, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Hunter, G.K.; Kyle, C.L.; Goldberg, H.A. Modulation of Crystal Formation by Bone Phosphoproteins: Structural Specificity of the Osteopontin-Mediated Inhibition of Hydroxyapatite Formation. Biochem. J. 1994, 300 Pt 3, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millán, J.L. Concerted Regulation of Inorganic Pyrophosphate and Osteopontin by Akp2, Enpp1, and Ank: An Integrated Model of the Pathogenesis of Mineralization Disorders. Am. J. Pathol. 2004, 164, 1199–1209. [Google Scholar] [CrossRef]
- Halling Linder, C.; Ek-Rylander, B.; Krumpel, M.; Norgård, M.; Narisawa, S.; Millán, J.L.; Andersson, G.; Magnusson, P. Bone Alkaline Phosphatase and Tartrate-Resistant Acid Phosphatase: Potential Co-Regulators of Bone Mineralization. Calcif. Tissue Int. 2017, 101, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Shanmugham, L.N.; Petrarca, C.; Castellani, M.L.; Symeonidou, I.; Frydas, S.; Vecchiet, J.; Falasca, K.; Tetè, S.; Conti, P.; Salini, V. IL-1beta Induces Alkaline Phosphatase in Human Phagocytes. Arch. Med. Res. 2007, 38, 39–44. [Google Scholar] [CrossRef]
- Cho, I.-J.; Choi, K.H.; Oh, C.H.; Hwang, Y.C.; Jeong, I.-K.; Ahn, K.J.; Chung, H.-Y. Effects of C-Reactive Protein on Bone Cells. Life Sci. 2016, 145, 1–8. [Google Scholar] [CrossRef]
- Huang, R.-L.; Yuan, Y.; Tu, J.; Zou, G.-M.; Li, Q. Opposing TNF-α/IL-1β- and BMP-2-Activated MAPK Signaling Pathways Converge on Runx2 to Regulate BMP-2-Induced Osteoblastic Differentiation. Cell Death Dis. 2014, 5, e1187. [Google Scholar] [CrossRef]
- Sekaran, S.; Vimalraj, S.; Thangavelu, L. The Physiological and Pathological Role of Tissue Nonspecific Alkaline Phosphatase beyond Mineralization. Biomolecules 2021, 11, 1564. [Google Scholar] [CrossRef]
- Lallès, J.-P. Intestinal Alkaline Phosphatase: Novel Functions and Protective Effects. Nutr. Rev. 2014, 72, 82–94. [Google Scholar] [CrossRef]
- Malo, M.S.; Alam, S.N.; Mostafa, G.; Zeller, S.J.; Johnson, P.V.; Mohammad, N.; Chen, K.T.; Moss, A.K.; Ramasamy, S.; Faruqui, A.; et al. Intestinal Alkaline Phosphatase Preserves the Normal Homeostasis of Gut Microbiota. Gut 2010, 59, 1476–1484. [Google Scholar] [CrossRef]
- Pickkers, P.; Mehta, R.L.; Murray, P.T.; Joannidis, M.; Molitoris, B.A.; Kellum, J.A.; Bachler, M.; Hoste, E.A.J.; Hoiting, O.; Krell, K.; et al. Effect of Human Recombinant Alkaline Phosphatase on 7-Day Creatinine Clearance in Patients With Sepsis-Associated Acute Kidney Injury: A Randomized Clinical Trial. JAMA 2018, 320, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, S.; Masereeuw, R.; Moesker, O.; Bouw, M.P.W.J.M.; van der Hoeven, J.G.; Peters, W.H.M.; Russel, F.G.M.; Pickkers, P.; APSEP Study Group. Alkaline Phosphatase Treatment Improves Renal Function in Severe Sepsis or Septic Shock Patients. Crit. Care Med. 2009, 37, 417–423.e1. [Google Scholar] [CrossRef] [PubMed]
- Pickkers, P.; Heemskerk, S.; Schouten, J.; Laterre, P.-F.; Vincent, J.-L.; Beishuizen, A.; Jorens, P.G.; Spapen, H.; Bulitta, M.; Peters, W.H.M.; et al. Alkaline Phosphatase for Treatment of Sepsis-Induced Acute Kidney Injury: A Prospective Randomized Double-Blind Placebo-Controlled Trial. Crit. Care 2012, 16, R14. [Google Scholar] [CrossRef] [PubMed]
- Kaliannan, K.; Hamarneh, S.R.; Economopoulos, K.P.; Nasrin Alam, S.; Moaven, O.; Patel, P.; Malo, N.S.; Ray, M.; Abtahi, S.M.; Muhammad, N.; et al. Intestinal Alkaline Phosphatase Prevents Metabolic Syndrome in Mice. Proc. Natl. Acad. Sci. USA 2013, 110, 7003–7008. [Google Scholar] [CrossRef]
- Malo, M.S. A High Level of Intestinal Alkaline Phosphatase Is Protective Against Type 2 Diabetes Mellitus Irrespective of Obesity. EBioMedicine 2015, 2, 2016–2023. [Google Scholar] [CrossRef]
- Bulum, T.; Kolarić, B.; Duvnjak, M.; Duvnjak, L. Alkaline Phosphatase Is Independently Associated with Renal Function in Normoalbuminuric Type 1 Diabetic Patients. Ren. Fail. 2014, 36, 372–377. [Google Scholar] [CrossRef]
- Zhao, L.; Li, L.; Ren, H.; Zou, Y.; Zhang, R.; Wang, S.; Xu, H.; Zhang, J.; Liu, F. Association between Serum Alkaline Phosphatase and Renal Outcome in Patients with Type 2 Diabetes Mellitus. Ren. Fail. 2020, 42, 818–828. [Google Scholar] [CrossRef]
- Pfleiderer, G.; Baier, M.; Mondorf, A.W.; Stefanescu, T.; Scherberich, J.E.; Müller, H. Change in Alkaline Phosphatase Isoenzyme Pattern in Urine as Possible Marker for Renal Disease. Kidney Int. 1980, 17, 242–249. [Google Scholar] [CrossRef]
- Oh, S.W.; Han, K.H.; Han, S.Y. Associations between Renal Hyperfiltration and Serum Alkaline Phosphatase. PLoS ONE 2015, 10, e0122921. [Google Scholar] [CrossRef]
- Kellett, K.A.B.; Hooper, N.M. The Role of Tissue Non-Specific Alkaline Phosphatase (TNAP) in Neurodegenerative Diseases: Alzheimer’s Disease in the Focus. Subcell. Biochem. 2015, 76, 363–374. [Google Scholar] [CrossRef]
- Boccardi, V.; Bubba, V.; Murasecco, I.; Pigliautile, M.; Monastero, R.; Cecchetti, R.; Scamosci, M.; Bastiani, P.; Mecocci, P.; ReGAL 2.0 Study Group. Serum Alkaline Phosphatase Is Elevated and Inversely Correlated with Cognitive Functions in Subjective Cognitive Decline: Results from the ReGAl 2.0 Project. Aging Clin. Exp. Res. 2021, 33, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Vardy, E.R.L.C.; Kellett, K.A.B.; Cocklin, S.L.; Hooper, N.M. Alkaline Phosphatase Is Increased in Both Brain and Plasma in Alzheimer’s Disease. Neurodegener. Dis. 2012, 9, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kellett, K.A.; Williams, J.; Vardy, E.R.; Smith, A.D.; Hooper, N.M. Plasma Alkaline Phosphatase Is Elevated in Alzheimer’s Disease and Inversely Correlates with Cognitive Function. Int. J. Mol. Epidemiol. Genet. 2011, 2, 114–121. [Google Scholar] [PubMed]
- Loohuis, L.M.; Albersen, M.; de Jong, S.; Wu, T.; Luykx, J.J.; Jans, J.J.M.; Verhoeven-Duif, N.M.; Ophoff, R.A. The Alkaline Phosphatase (ALPL) Locus Is Associated with B6 Vitamer Levels in CSF and Plasma. Genes 2018, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Fonta, C.; Négyessy, L.; Renaud, L.; Barone, P. Areal and Subcellular Localization of the Ubiquitous Alkaline Phosphatase in the Primate Cerebral Cortex: Evidence for a Role in Neurotransmission. Cereb. Cortex 2004, 14, 595–609. [Google Scholar] [CrossRef]
- Nwafor, D.C.; Brown, C.M. A Novel Role for Tissue-Nonspecific Alkaline Phosphatase at the Blood-Brain Barrier during Sepsis. Neural. Regen. Res. 2021, 16, 99–100. [Google Scholar] [CrossRef]
- Tang, W.; Huang, J.; Huang, X.; Han, X.; Tang, W.; Ke, G.; Xu, Q. Effect of Alkaline Phosphatase on Sepsis-Associated Acute Kidney Injury Patients: A Systematic Review and Meta-Analysis. Medicine 2020, 99, e18788. [Google Scholar] [CrossRef]
- Brichacek, A.L.; Benkovic, S.A.; Chakraborty, S.; Nwafor, D.C.; Wang, W.; Jun, S.; Dakhlallah, D.; Geldenhuys, W.J.; Pinkerton, A.B.; Millán, J.L.; et al. Systemic Inhibition of Tissue-Nonspecific Alkaline Phosphatase Alters the Brain-Immune Axis in Experimental Sepsis. Sci. Rep. 2019, 9, 18788. [Google Scholar] [CrossRef]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological Role of Vascular Smooth Muscle Alkaline Phosphatase in Medial Artery Calcification. J. Bone Miner. Res. 2015, 30, 824–836. [Google Scholar] [CrossRef]
- Zhu, D.; Mackenzie, N.C.W.; Millán, J.L.; Farquharson, C.; MacRae, V.E. The Appearance and Modulation of Osteocyte Marker Expression during Calcification of Vascular Smooth Muscle Cells. PLoS ONE 2011, 6, e19595. [Google Scholar] [CrossRef]
- Anderson, H.C.; Sipe, J.B.; Hessle, L.; Dhanyamraju, R.; Atti, E.; Camacho, N.P.; Millán, J.L.; Dhamyamraju, R. Impaired Calcification around Matrix Vesicles of Growth Plate and Bone in Alkaline Phosphatase-Deficient Mice. Am. J. Pathol. 2004, 164, 841–847. [Google Scholar] [CrossRef]
- Moe, S.M.; O’Neill, K.D.; Duan, D.; Ahmed, S.; Chen, N.X.; Leapman, S.B.; Fineberg, N.; Kopecky, K. Medial Artery Calcification in ESRD Patients Is Associated with Deposition of Bone Matrix Proteins. Kidney Int. 2002, 61, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Salam, S.; Gallagher, O.; Gossiel, F.; Paggiosi, M.; Eastell, R.; Khwaja, A. Vascular Calcification Relationship to Vascular Biomarkers and Bone Metabolism in Advanced Chronic Kidney Disease. Bone 2021, 143, 115699. [Google Scholar] [CrossRef] [PubMed]
- Shantouf, R.; Kovesdy, C.P.; Kim, Y.; Ahmadi, N.; Luna, A.; Luna, C.; Rambod, M.; Nissenson, A.R.; Budoff, M.J.; Kalantar-Zadeh, K. Association of Serum Alkaline Phosphatase with Coronary Artery Calcification in Maintenance Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Schultz-Hector, S.; Balz, K.; Böhm, M.; Ikehara, Y.; Rieke, L. Cellular Localization of Endothelial Alkaline Phosphatase Reaction Product and Enzyme Protein in the Myocardium. J. Histochem. Cytochem. 1993, 41, 1813–1821. [Google Scholar] [CrossRef]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.; Millán, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Heart Assoc. 2015, 4, e002499. [Google Scholar] [CrossRef]
- Guo, W.; Li, X.; Wu, J.; Zhu, W.; Lu, J.; Qin, P.; Diao, Q.; Xu, N.; Zhang, Q. Serum Alkaline Phosphatase Is Associated with Arterial Stiffness and 10-Year Cardiovascular Disease Risk in a Chinese Population. Eur. J. Clin. Investig. 2021, 51, e13560. [Google Scholar] [CrossRef]
- Perticone, F.; Perticone, M.; Maio, R.; Sciacqua, A.; Andreucci, M.; Tripepi, G.; Corrao, S.; Mallamaci, F.; Sesti, G.; Zoccali, C. Serum Alkaline Phosphatase Negatively Affects Endothelium-Dependent Vasodilation in Naïve Hypertensive Patients. Hypertension 2015, 66, 874–880. [Google Scholar] [CrossRef]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Vigili de Kreutzenberg, S.; Agostini, C.; Cabrelle, A.; Binotto, G.; Rattazzi, M.; Bertacco, E.; et al. Widespread Increase in Myeloid Calcifying Cells Contributes to Ectopic Vascular Calcification in Type 2 Diabetes. Circ. Res. 2011, 108, 1112–1121. [Google Scholar] [CrossRef]
- Gao, L.; Wang, L.-Y.; Liu, Z.-Q.; Jiang, D.; Wu, S.-Y.; Guo, Y.-Q.; Tao, H.-M.; Sun, M.; You, L.-N.; Qin, S.; et al. TNAP Inhibition Attenuates Cardiac Fibrosis Induced by Myocardial Infarction through Deactivating TGF-Β1/Smads and Activating P53 Signaling Pathways. Cell Death Dis. 2020, 11, 44. [Google Scholar] [CrossRef]
- Tani, T.; Fujiwara, M.; Orimo, H.; Shimizu, A.; Narisawa, S.; Pinkerton, A.B.; Millán, J.L.; Tsuruoka, S. Inhibition of Tissue-Nonspecific Alkaline Phosphatase Protects against Medial Arterial Calcification and Improves Survival Probability in the CKD-MBD Mouse Model. J. Pathol. 2020, 250, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, S.; Cianciolo, G.; De Pascalis, A.; Guglielmo, C.; Urena Torres, P.A.; Bover, J.; Tartaglione, L.; Pasquali, M.; La Manna, G. Bone, Inflammation and the Bone Marrow Niche in Chronic Kidney Disease: What Do We Know? Nephrol. Dial. Transpl. 2018, 33, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Dixon, I.M.C. Tissue Non-Specific Alkaline Phosphatase (TNAP): A Player in Post-MI Cardiac Fibrosis. EBioMedicine 2021, 68, 103430. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, L.; Wen, X.; Gao, L.; Li, G.; Chang, G.; Qin, S.; Zhang, D. TNAP Is a Novel Regulator of Cardiac Fibrosis after Myocardial Infarction by Mediating TGF-β/Smads and ERK1/2 Signaling Pathways. EBioMedicine 2021, 67, 103370. [Google Scholar] [CrossRef]
- Lin, H.; Angeli, M.; Chung, K.J.; Ejimadu, C.; Rosa, A.R.; Lee, T. SFRP2 Activates Wnt/β-Catenin Signaling in Cardiac Fibroblasts: Differential Roles in Cell Growth, Energy Metabolism, and Extracellular Matrix Remodeling. Am. J. Physiol. Cell Physiol 2016, 311, C710–C719. [Google Scholar] [CrossRef]
- Martin, S.; Lin, H.; Ejimadu, C.; Lee, T. Tissue-Nonspecific Alkaline Phosphatase as a Target of SFRP2 in Cardiac Fibroblasts. Am. J. Physiol. Cell Physiol. 2015, 309, C139–C147. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Bakker, S.J.L.; Kootstra-Ros, J.E.; Gansevoort, R.T.; Gregson, J.; Dullaart, R.P.F. Serum Alkaline Phosphatase and Risk of Incident Cardiovascular Disease: Interrelationship with High Sensitivity C-Reactive Protein. PLoS ONE 2015, 10, e0132822. [Google Scholar] [CrossRef]
- Wannamethee, S.G.; Sattar, N.; Papcosta, O.; Lennon, L.; Whincup, P.H. Alkaline Phosphatase, Serum Phosphate, and Incident Cardiovascular Disease and Total Mortality in Older Men. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1070–1076. [Google Scholar] [CrossRef]
- Park, J.-B.; Kang, D.-Y.; Yang, H.-M.; Cho, H.-J.; Park, K.W.; Lee, H.-Y.; Kang, H.-J.; Koo, B.-K.; Kim, H.-S. Serum Alkaline Phosphatase Is a Predictor of Mortality, Myocardial Infarction, or Stent Thrombosis after Implantation of Coronary Drug-Eluting Stent. Eur. Heart J. 2013, 34, 920–931. [Google Scholar] [CrossRef]
- Malo, J.; Alam, M.J.; Shahnaz, M.; Kaliannan, K.; Chandra, G.; Aziz, T.; Sarker, T.; Bala, M.; Paul, R.; Saha, C.K.; et al. Intestinal Alkaline Phosphatase Deficiency Is Associated with Ischemic Heart Disease. Dis. Markers 2019, 2019, 8473565. [Google Scholar] [CrossRef] [PubMed]
- Iba, K.; Takada, J.; Yamashita, T. The Serum Level of Bone-Specific Alkaline Phosphatase Activity Is Associated with Aortic Calcification in Osteoporosis Patients. J. Bone Miner. Metab. 2004, 22, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Fahrleitner-Pammer, A.; Herberth, J.; Browning, S.R.; Obermayer-Pietsch, B.; Wirnsberger, G.; Holzer, H.; Dobnig, H.; Malluche, H.H. Bone Markers Predict Cardiovascular Events in Chronic Kidney Disease. J. Bone Miner. Res. 2008, 23, 1850–1858. [Google Scholar] [CrossRef]
- Barreto, D.V.; Barreto, F.d.C.; de Carvalho, A.B.; Cuppari, L.; Draibe, S.A.; Dalboni, M.A.; Moyses, R.M.A.; Neves, K.R.; Jorgetti, V.; Miname, M.; et al. Association of Changes in Bone Remodeling and Coronary Calcification in Hemodialysis Patients: A Prospective Study. Am. J. Kidney Dis. 2008, 52, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, L.; Zhang, M.; Cai, H.; Ni, Z. Circulating Bone-Specific Alkaline Phosphatase and Abdominal Aortic Calcification in Maintenance Hemodialysis Patients. Biomark. Med. 2018, 12, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Fernström, A.; Qureshi, A.R.; Magnusson, P. The Novel Bone Alkaline Phosphatase Isoform B1x Is Associated with Improved 5-Year Survival in Chronic Kidney Disease. Nutrients 2021, 13, 4402. [Google Scholar] [CrossRef]
- Drechsler, C.; Verduijn, M.; Pilz, S.; Krediet, R.T.; Dekker, F.W.; Wanner, C.; Ketteler, M.; Boeschoten, E.W.; Brandenburg, V.; NECOSAD Study Group. Bone Alkaline Phosphatase and Mortality in Dialysis Patients. Clin. J. Am. Soc. Nephrol. 2011, 6, 1752–1759. [Google Scholar] [CrossRef]
- Filipowicz, R.; Greene, T.; Wei, G.; Cheung, A.K.; Raphael, K.L.; Baird, B.C.; Beddhu, S. Associations of Serum Skeletal Alkaline Phosphatase with Elevated C-Reactive Protein and Mortality. Clin. J. Am. Soc. Nephrol. 2013, 8, 26–32. [Google Scholar] [CrossRef]
- Rhee, C.M.; Molnar, M.Z.; Lau, W.L.; Ravel, V.; Kovesdy, C.P.; Mehrotra, R.; Kalantar-Zadeh, K. Comparative Mortality-Predictability Using Alkaline Phosphatase and Parathyroid Hormone in Patients on Peritoneal Dialysis and Hemodialysis. Perit. Dial. Int. 2014, 34, 732–748. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, X.; Han, F.; Xie, X.; Hua, Z.; Huang, X.; Lindholm, B.; Haarhaus, M.; Stenvinkel, P.; Qureshi, A.R.; et al. High Alkaline Phosphatase and Low Intact Parathyroid Hormone Associate with Worse Clinical Outcome in Peritoneal Dialysis Patients. Perit. Dial. Int. 2021, 41, 236–243. [Google Scholar] [CrossRef]
- Lau, W.L.; Kalantar-Zadeh, K.; Kovesdy, C.P.; Mehrotra, R. Alkaline Phosphatase: Better than PTH as a Marker of Cardiovascular and Bone Disease? Hemodial. Int. 2014, 18, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Molnar, M.Z.; Kovesdy, C.P.; Mucsi, I.; Salusky, I.B.; Kalantar-Zadeh, K. Association of Pre-Kidney Transplant Markers of Mineral and Bone Disorder with Post-Transplant Outcomes. Clin. J. Am. Soc. Nephrol. 2012, 7, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Nickolas, T.L.; Stein, E.; Cohen, A.; Thomas, V.; Staron, R.B.; McMahon, D.J.; Leonard, M.B.; Shane, E. Bone Mass and Microarchitecture in CKD Patients with Fracture. J. Am. Soc. Nephrol. 2010, 21, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Frost, M.L.; Compston, J.E.; Goldsmith, D.; Moore, A.E.; Blake, G.M.; Siddique, M.; Skingle, L.; Fogelman, I. (18)F-Fluoride Positron Emission Tomography Measurements of Regional Bone Formation in Hemodialysis Patients with Suspected Adynamic Bone Disease. Calcif. Tissue Int. 2013, 93, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, P.; Nickolas, T.L. Management of Osteoporosis in CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Dalle Carbonare, L.; Valenti, M.T.; Giannini, S.; Gallieni, M.; Stefani, F.; Ciresa, R.; Politi, C.; Fusaro, M. Bone Biopsy for Histomorphometry in Chronic Kidney Disease (CKD): State-of-the-Art and New Perspectives. J. Clin. Med. 2021, 10, 4617. [Google Scholar] [CrossRef]
- Hughes-Austin, J.M.; Katz, R.; Semba, R.D.; Kritchevsky, S.B.; Bauer, D.C.; Sarnak, M.J.; Ginsberg, C.; Shlipak, M.G.; Lima, F.; Malluche, H.H.; et al. Biomarkers of Bone Turnover Identify Subsets of Chronic Kidney Disease Patients at Higher Risk for Fracture. J. Clin. Endocrinol. Metab. 2020, 105, dgaa317. [Google Scholar] [CrossRef]
- Evenepoel, P.; Cunningham, J.; Ferrari, S.; Haarhaus, M.; Javaid, M.K.; Lafage-Proust, M.-H.; Prieto-Alhambra, D.; Torres, P.U.; Cannata-Andia, J.; European Renal Osteodystrophy (EUROD) Workgroup, an Initiative of the CKD-MBD Working Group of the ERA-EDTA, and the Committee of Scientific Advisors and National Societies of the IOF. European Consensus Statement on the Diagnosis and Management of Osteoporosis in Chronic Kidney Disease Stages G4-G5D. Nephrol. Dial. Transpl. 2021, 36, 42–59. [Google Scholar] [CrossRef]
- Coen, G.; Ballanti, P.; Bonucci, E.; Calabria, S.; Centorrino, M.; Fassino, V.; Manni, M.; Mantella, D.; Mazzaferro, S.; Napoletano, I.; et al. Bone Markers in the Diagnosis of Low Turnover Osteodystrophy in Haemodialysis Patients. Nephrol. Dial. Transpl. 1998, 13, 2294–2302. [Google Scholar] [CrossRef]
- Behets, G.J.; Spasovski, G.; Sterling, L.R.; Goodman, W.G.; Spiegel, D.M.; De Broe, M.E.; D’Haese, P.C. Bone Histomorphometry before and after Long-Term Treatment with Cinacalcet in Dialysis Patients with Secondary Hyperparathyroidism. Kidney Int. 2015, 87, 846–856. [Google Scholar] [CrossRef]
- Moore, C.; Yee, J.; Malluche, H.; Rao, D.S.; Monier-Faugere, M.-C.; Adams, E.; Daramola-Ogunwuyi, O.; Fehmi, H.; Bhat, S.; Osman-Malik, Y. Relationship between Bone Histology and Markers of Bone and Mineral Metabolism in African-American Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, A.R.J.; Spasovski, G.B.; Behets, G.J.; Dams, G.; Polenakovic, M.H.; Zafirovska, K.; Van Hoof, V.O.; De Broe, M.E.; D’Haese, P.C. Useful Biochemical Markers for Diagnosing Renal Osteodystrophy in Predialysis End-Stage Renal Failure Patients. Am. J. Kidney Dis. 2003, 41, 997–1007. [Google Scholar] [CrossRef]
- Ureña, P.; Hruby, M.; Ferreira, A.; Ang, K.S.; de Vernejoul, M.C. Plasma Total versus Bone Alkaline Phosphatase as Markers of Bone Turnover in Hemodialysis Patients. J. Am. Soc. Nephrol. 1996, 7, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Ureña, P.; De Vernejoul, M.C. Circulating Biochemical Markers of Bone Remodeling in Uremic Patients. Kidney Int. 1999, 55, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Bover, J.; Ureña Torres, P. Parathyroid Hormone Metabolism and Signaling in Health and Chronic Kidney Disease. Kidney Int. 2016, 90, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Taniguchi, M.; Kazama, J.J.; Yokoyama, K.; Hosoya, T.; Yokoo, T.; Shigematsu, T.; Iseki, K.; Tsubakihara, Y. A Higher Serum Alkaline Phosphatase Is Associated with the Incidence of Hip Fracture and Mortality among Patients Receiving Hemodialysis in Japan. Nephrol. Dial. Transpl. 2014, 29, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Iimori, S.; Mori, Y.; Akita, W.; Kuyama, T.; Takada, S.; Asai, T.; Kuwahara, M.; Sasaki, S.; Tsukamoto, Y. Diagnostic Usefulness of Bone Mineral Density and Biochemical Markers of Bone Turnover in Predicting Fracture in CKD Stage 5D Patients—A Single-Center Cohort Study. Nephrol. Dial. Transpl. 2012, 27, 345–351. [Google Scholar] [CrossRef]
- Atsumi, K.; Kushida, K.; Yamazaki, K.; Shimizu, S.; Ohmura, A.; Inoue, T. Risk Factors for Vertebral Fractures in Renal Osteodystrophy. Am. J. Kidney Dis. 1999, 33, 287–293. [Google Scholar] [CrossRef]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Executive Summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: What’s Changed and Why It Matters. Kidney Int. 2017, 92, 26–36. [Google Scholar] [CrossRef]
- National Kidney Foundation. K/DOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease. Am. J. Kidney Dis. 2003, 42, S1–S201. [Google Scholar] [CrossRef]
- Jørgensen, H.S.; Behets, G.; Viaene, L.; Bammens, B.; Claes, K.; Meijers, B.; Naesens, M.; Sprangers, B.; Kuypers, D.; Cavalier, E.; et al. Diagnostic Accuracy of Noninvasive Bone Turnover Markers in Renal Osteodystrophy. Am. J. Kidney Dis. 2022, 79, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, C.; Ix, J.H. Diagnosis and Management of Osteoporosis in Advanced Kidney Disease: A Review. Am. J. Kidney Dis. 2022, 79, 427–436. [Google Scholar] [CrossRef]
- Salam, S.; Gallagher, O.; Gossiel, F.; Paggiosi, M.; Khwaja, A.; Eastell, R. Diagnostic Accuracy of Biomarkers and Imaging for Bone Turnover in Renal Osteodystrophy. J. Am. Soc. Nephrol. 2018, 29, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Bellorin-Font, E.; Jorgetti, V.; Carvalho, A.B.; Malluche, H.H.; Ferreira, A.; D’Haese, P.C.; Drüeke, T.B.; Du, H.; Manley, T.; et al. Diagnostic Accuracy of Bone Turnover Markers and Bone Histology in Patients With CKD Treated by Dialysis. Am. J. Kidney Dis. 2016, 67, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Evenepoel, P. Differentiating the Causes of Adynamic Bone in Advanced Chronic Kidney Disease Informs Osteoporosis Treatment. Kidney Int. 2021, 100, 546–558. [Google Scholar] [CrossRef]
- Fusaro, M.; Cianciolo, G.; Brandi, M.L.; Ferrari, S.; Nickolas, T.L.; Tripepi, G.; Plebani, M.; Zaninotto, M.; Iervasi, G.; La Manna, G.; et al. Vitamin K and Osteoporosis. Nutrients 2020, 12, 3625. [Google Scholar] [CrossRef]
- Fusaro, M.; Cianciolo, G.; Evenepoel, P.; Schurgers, L.; Plebani, M. Vitamin K in CKD Bone Disorders. Calcif. Tissue Int. 2021, 108, 476–485. [Google Scholar] [CrossRef]
- Caluwé, R.; Verbeke, F.; De Vriese, A.S. Evaluation of Vitamin K Status and Rationale for Vitamin K Supplementation in Dialysis Patients. Nephrol. Dial. Transpl. 2020, 35, 23–33. [Google Scholar] [CrossRef]
- Fusaro, M.; Gallieni, M.; Porta, C.; Nickolas, T.L.; Khairallah, P. Vitamin K Effects in Human Health: New Insights beyond Bone and Cardiovascular Health. J. Nephrol. 2020, 33, 239–249. [Google Scholar] [CrossRef]
- Tabb, M.M.; Sun, A.; Zhou, C.; Grün, F.; Errandi, J.; Romero, K.; Pham, H.; Inoue, S.; Mallick, S.; Lin, M.; et al. Vitamin K2 Regulation of Bone Homeostasis Is Mediated by the Steroid and Xenobiotic Receptor SXR. J. Biol. Chem. 2003, 278, 43919–43927. [Google Scholar] [CrossRef]
- Ichikawa, T.; Horie-Inoue, K.; Ikeda, K.; Blumberg, B.; Inoue, S. Steroid and Xenobiotic Receptor SXR Mediates Vitamin K2-Activated Transcription of Extracellular Matrix-Related Genes and Collagen Accumulation in Osteoblastic Cells. J. Biol. Chem. 2006, 281, 16927–16934. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Suhara, Y. New Aspects of Vitamin K Research with Synthetic Ligands: Transcriptional Activity via SXR and Neural Differentiation Activity. Int. J. Mol. Sci. 2019, 20, 3006. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Inoue, S. Multiple Modes of Vitamin K Actions in Aging-Related Musculoskeletal Disorders. Int. J. Mol. Sci. 2019, 20, 2844. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Marumo, K. Collagen Cross-Links as a Determinant of Bone Quality: A Possible Explanation for Bone Fragility in Aging, Osteoporosis, and Diabetes Mellitus. Osteoporos Int. 2010, 21, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Kazama, J.J.; Iwasaki, Y.; Fukagawa, M. Uremic Osteoporosis. Kidney Int. Suppl. 2013, 3, 446–450. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Weitzmann, M.N. Vitamin K2 Stimulates Osteoblastogenesis and Suppresses Osteoclastogenesis by Suppressing NF-ΚB Activation. Int. J. Mol. Med. 2011, 27, 3–14. [Google Scholar] [CrossRef]
- Noda, S.; Yamada, A.; Tanabe, R.; Nakaoka, K.; Hosoi, T.; Goseki-Sone, M. Menaquinone-4 (Vitamin K2) up-Regulates Expression of Human Intestinal Alkaline Phosphatase in Caco-2 Cells. Nutr. Res. 2016, 36, 1269–1276. [Google Scholar] [CrossRef]
- Fusaro, M.; D’Alessandro, C.; Noale, M.; Tripepi, G.; Plebani, M.; Veronese, N.; Iervasi, G.; Giannini, S.; Rossini, M.; Tarroni, G.; et al. Low Vitamin K1 Intake in Haemodialysis Patients. Clin. Nutr. 2017, 36, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Ziemińska, M.; Sieklucka, B.; Pawlak, K. Vitamin K and D Supplementation and Bone Health in Chronic Kidney Disease-Apart or Together? Nutrients 2021, 13, 809. [Google Scholar] [CrossRef]
- Wei, F.-F.; Trenson, S.; Verhamme, P.; Vermeer, C.; Staessen, J.A. Vitamin K-Dependent Matrix Gla Protein as Multifaceted Protector of Vascular and Tissue Integrity. Hypertension 2019, 73, 1160–1169. [Google Scholar] [CrossRef]
- Newman, B.; Gigout, L.I.; Sudre, L.; Grant, M.E.; Wallis, G.A. Coordinated Expression of Matrix Gla Protein Is Required during Endochondral Ossification for Chondrocyte Survival. J. Cell Biol. 2001, 154, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Julien, M.; Khoshniat, S.; Lacreusette, A.; Gatius, M.; Bozec, A.; Wagner, E.F.; Wittrant, Y.; Masson, M.; Weiss, P.; Beck, L.; et al. Phosphate-Dependent Regulation of MGP in Osteoblasts: Role of ERK1/2 and Fra-1. J. Bone Miner. Res. 2009, 24, 1856–1868. [Google Scholar] [CrossRef] [PubMed]
- Koshihara, Y.; Hoshi, K.; Okawara, R.; Ishibashi, H.; Yamamoto, S. Vitamin K Stimulates Osteoblastogenesis and Inhibits Osteoclastogenesis in Human Bone Marrow Cell Culture. J. Endocrinol. 2003, 176, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, S.; Liu, J.; Liu, Y.; Liang, Q. Vitamin K2 Stimulates MC3T3-E1 Osteoblast Differentiation and Mineralization through Autophagy Induction. Mol. Med. Rep. 2019, 19, 3676–3684. [Google Scholar] [CrossRef]
- Regidor, D.L.; Kovesdy, C.P.; Mehrotra, R.; Rambod, M.; Jing, J.; McAllister, C.J.; Van Wyck, D.; Kopple, J.D.; Kalantar-Zadeh, K. Serum Alkaline Phosphatase Predicts Mortality among Maintenance Hemodialysis Patients. J. Am. Soc. Nephrol. 2008, 19, 2193–2203. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Kuwae, N.; Regidor, D.L.; Kovesdy, C.P.; Kilpatrick, R.D.; Shinaberger, C.S.; McAllister, C.J.; Budoff, M.J.; Salusky, I.B.; Kopple, J.D. Survival Predictability of Time-Varying Indicators of Bone Disease in Maintenance Hemodialysis Patients. Kidney Int. 2006, 70, 771–780. [Google Scholar] [CrossRef]
- Floege, J.; Kim, J.; Ireland, E.; Chazot, C.; Drueke, T.; de Francisco, A.; Kronenberg, F.; Marcelli, D.; Passlick-Deetjen, J.; Schernthaner, G.; et al. Serum IPTH, Calcium and Phosphate, and the Risk of Mortality in a European Haemodialysis Population. Nephrol. Dial. Transpl. 2011, 26, 1948–1955. [Google Scholar] [CrossRef]
- Soohoo, M.; Feng, M.; Obi, Y.; Streja, E.; Rhee, C.M.; Lau, W.L.; Wang, J.; Ravel, V.A.; Brunelli, S.; Kovesdy, C.P.; et al. Changes in Markers of Mineral and Bone Disorders and Mortality in Incident Hemodialysis Patients. Am. J. Nephrol. 2016, 43, 85–96. [Google Scholar] [CrossRef]
- Haarhaus, M.; Ray, K.K.; Nicholls, S.J.; Schwartz, G.G.; Kulikowski, E.; Johansson, J.O.; Sweeney, M.; Halliday, C.; Lebioda, K.; Wong, N.; et al. Apabetalone Lowers Serum Alkaline Phosphatase and Improves Cardiovascular Risk in Patients with Cardiovascular Disease. Atherosclerosis 2019, 290, 59–65. [Google Scholar] [CrossRef]
- Kulikowski, E.; Halliday, C.; Johansson, J.; Sweeney, M.; Lebioda, K.; Wong, N.; Haarhaus, M.; Brandenburg, V.; Beddhu, S.; Tonelli, M.; et al. Apabetalone Mediated Epigenetic Modulation Is Associated with Favorable Kidney Function and Alkaline Phosphatase Profile in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2018, 43, 449–457. [Google Scholar] [CrossRef]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Conery, A.R.; Sims, R.J. Bromodomains: A New Target Class for Drug Development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) Reduces Vascular Inflammation in Vitro and in CVD Patients by a BET-Dependent Epigenetic Mechanism. Clin. Epigenetics 2019, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Wasiak, S.; Gilham, D.; Tsujikawa, L.M.; Halliday, C.; Calosing, C.; Jahagirdar, R.; Johansson, J.; Sweeney, M.; Wong, N.C.; Kulikowski, E. Downregulation of the Complement Cascade In Vitro, in Mice and in Patients with Cardiovascular Disease by the BET Protein Inhibitor Apabetalone (RVX-208). J. Cardiovasc. Transl. Res. 2017, 10, 337–347. [Google Scholar] [CrossRef]
- Martin, K.J.; González, E.; Lindberg, J.S.; Taccetta, C.; Amdahl, M.; Malhotra, K.; Llach, F. Paricalcitol Dosing According to Body Weight or Severity of Hyperparathyroidism: A Double-Blind, Multicenter, Randomized Study. Am. J. Kidney Dis. 2001, 38, S57–S63. [Google Scholar] [CrossRef]
- Palmer, S.C.; McGregor, D.O.; Macaskill, P.; Craig, J.C.; Elder, G.J.; Strippoli, G.F.M. Meta-Analysis: Vitamin D Compounds in Chronic Kidney Disease. Ann. Intern. Med. 2007, 147, 840–853. [Google Scholar] [CrossRef]
- Rix, M.; Eskildsen, P.; Olgaard, K. Effect of 18 Months of Treatment with Alfacalcidol on Bone in Patients with Mild to Moderate Chronic Renal Failure. Nephrol. Dial. Transpl. 2004, 19, 870–876. [Google Scholar] [CrossRef]
- Barreto, D.V.; Barreto, F.d.C.; de Carvalho, A.B.; Cuppari, L.; Draibe, S.A.; Dalboni, M.A.; Moyses, R.M.A.; Neves, K.R.; Jorgetti, V.; Miname, M.; et al. Phosphate Binder Impact on Bone Remodeling and Coronary Calcification—Results from the BRiC Study. Nephron. Clin. Pract. 2008, 110, c273–c283. [Google Scholar] [CrossRef]
- Raggi, P.; James, G.; Burke, S.K.; Bommer, J.; Chasan-Taber, S.; Holzer, H.; Braun, J.; Chertow, G.M. Decrease in Thoracic Vertebral Bone Attenuation with Calcium-Based Phosphate Binders in Hemodialysis. J. Bone Miner. Res. 2005, 20, 764–772. [Google Scholar] [CrossRef]
- Belozeroff, V.; Goodman, W.G.; Ren, L.; Kalantar-Zadeh, K. Cinacalcet Lowers Serum Alkaline Phosphatase in Maintenance Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 673–679. [Google Scholar] [CrossRef]
- Yuan, F.; Chen, X.; Wang, C.; Li, Z.; Liu, H. Effect of Cinacalcet Combined with Calcitriol on the Clinical Outcome and Bone Metabolism in Patients on Hemodialysis with Severe Secondary Hyperparathyroidism. Blood Purif. 2018, 45, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Merkel, S.; Leitolf, H.; Haller, H. The Effect of Cinacalcet on Bone Remodeling and Renal Function in Transplant Patients with Persistent Hyperparathyroidism. Transplantation 2011, 91, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Peacock, M.; Bolognese, M.A.; Borofsky, M.; Scumpia, S.; Sterling, L.R.; Cheng, S.; Shoback, D. Cinacalcet Treatment of Primary Hyperparathyroidism: Biochemical and Bone Densitometric Outcomes in a Five-Year Study. J. Clin. Endocrinol. Metab. 2009, 94, 4860–4867. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Chew, C.K.; Monroe, D.G.; Farr, J.N.; Atkinson, E.J.; Geske, J.R.; Eckhardt, B.; Thicke, B.; Ruan, M.; Tweed, A.J.; et al. Identification of Osteoclast-Osteoblast Coupling Factors in Humans Reveals Links between Bone and Energy Metabolism. Nat. Commun. 2020, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R.; Lewiecki, E.M.; Geller, M.L.; Bolognese, M.A.; Peacock, M.; Weinstein, R.L.; Ding, B.; Rockabrand, E.; Wagman, R.B.; Miller, P.D. Effect of Denosumab on Bone Mineral Density and Biochemical Markers of Bone Turnover: 8-Year Results of a Phase 2 Clinical Trial. Osteoporos Int. 2013, 24, 227–235. [Google Scholar] [CrossRef]
- Chen, C.-L.; Chen, N.-C.; Hsu, C.-Y.; Chou, K.-J.; Lee, P.-T.; Fang, H.-C.; Renn, J.-H. An Open-Label, Prospective Pilot Clinical Study of Denosumab for Severe Hyperparathyroidism in Patients with Low Bone Mass Undergoing Dialysis. J. Clin. Endocrinol. Metab. 2014, 99, 2426–2432. [Google Scholar] [CrossRef]
- Iseri, K.; Watanabe, M.; Yoshikawa, H.; Mitsui, H.; Endo, T.; Yamamoto, Y.; Iyoda, M.; Ryu, K.; Inaba, T.; Shibata, T. Effects of Denosumab and Alendronate on Bone Health and Vascular Function in Hemodialysis Patients: A Randomized, Controlled Trial. J. Bone Miner. Res. 2019, 34, 1014–1024. [Google Scholar] [CrossRef]
- Bonani, M.; Frey, D.; Brockmann, J.; Fehr, T.; Mueller, T.F.; Saleh, L.; von Eckardstein, A.; Graf, N.; Wüthrich, R.P. Effect of Twice-Yearly Denosumab on Prevention of Bone Mineral Density Loss in De Novo Kidney Transplant Recipients: A Randomized Controlled Trial. Am. J. Transpl. 2016, 16, 1882–1891. [Google Scholar] [CrossRef]
- Makras, P.; Polyzos, S.A.; Papatheodorou, A.; Kokkoris, P.; Chatzifotiadis, D.; Anastasilakis, A.D. Parathyroid Hormone Changes Following Denosumab Treatment in Postmenopausal Osteoporosis. Clin. Endocrinol. 2013, 79, 499–503. [Google Scholar] [CrossRef]
- Cianciolo, G.; Tondolo, F.; Barbuto, S.; Iacovella, F.; Zavatta, G.; Altieri, P.; Grandinetti, V.; Comai, G.; Cozzolino, M.; La Manna, G. Denosumab-Induced Hypocalcemia and Hyperparathyroidism in de Novo Kidney Transplant Recipients. Am. J. Nephrol. 2021, 52, 611–619. [Google Scholar] [CrossRef]
- Samelson, E.J.; Miller, P.D.; Christiansen, C.; Daizadeh, N.S.; Grazette, L.; Anthony, M.S.; Egbuna, O.; Wang, A.; Siddhanti, S.R.; Cheung, A.M.; et al. RANKL Inhibition with Denosumab Does Not Influence 3-Year Progression of Aortic Calcification or Incidence of Adverse Cardiovascular Events in Postmenopausal Women with Osteoporosis and High Cardiovascular Risk. J. Bone Miner. Res. 2014, 29, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.G.G. Bisphosphonates: The First 40 Years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Reid, I.R.; Miller, P.D.; Brown, J.P.; Kendler, D.L.; Fahrleitner-Pammer, A.; Valter, I.; Maasalu, K.; Bolognese, M.A.; Woodson, G.; Bone, H.; et al. Effects of Denosumab on Bone Histomorphometry: The FREEDOM and STAND Studies. J. Bone Miner. Res. 2010, 25, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.M. Pharmacology of Bisphosphonates in Patients with Chronic Kidney Disease. Semin. Dial. 2015, 28, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Hamano, T.; Mikami, S.; Nagasawa, Y.; Isaka, Y.; Moriyama, T.; Horio, M.; Imai, E.; Hori, M.; Ito, T. Risedronate, an Effective Treatment for Glucocorticoid-Induced Bone Loss in CKD Patients with or without Concomitant Active Vitamin D (PRIUS-CKD). Nephrol. Dial. Transpl. 2007, 22, 1601–1607. [Google Scholar] [CrossRef]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Bisphosphonates Alendronate and Ibandronate Inhibit Artery Calcification at Doses Comparable to Those That Inhibit Bone Resorption. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 817–824. [Google Scholar] [CrossRef]
- Hildebrand, S.; Cunningham, J. Is There a Role for Bisphosphonates in Vascular Calcification in Chronic Kidney Disease? Bone 2021, 142, 115751. [Google Scholar] [CrossRef]
- Seeto, A.H.; Abrahamsen, B.; Ebeling, P.R.; Rodríguez, A.J. Cardiovascular Safety of Denosumab Across Multiple Indications: A Systematic Review and Meta-Analysis of Randomized Trials. J. Bone Miner. Res. 2021, 36, 24–40. [Google Scholar] [CrossRef]
- Miller, P.D.; Schwartz, E.N.; Chen, P.; Misurski, D.A.; Krege, J.H. Teriparatide in Postmenopausal Women with Osteoporosis and Mild or Moderate Renal Impairment. Osteoporos Int. 2007, 18, 59–68. [Google Scholar] [CrossRef]
- Cejka, D.; Kodras, K.; Bader, T.; Haas, M. Treatment of Hemodialysis-Associated Adynamic Bone Disease with Teriparatide (PTH1-34): A Pilot Study. Kidney Blood Press. Res. 2010, 33, 221–226. [Google Scholar] [CrossRef]
- Sumida, K.; Ubara, Y.; Hoshino, J.; Mise, K.; Hayami, N.; Suwabe, T.; Kawada, M.; Imafuku, A.; Hiramatsu, R.; Hasegawa, E.; et al. Once-Weekly Teriparatide in Hemodialysis Patients with Hypoparathyroidism and Low Bone Mass: A Prospective Study. Osteoporos Int. 2016, 27, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.-S.; Cheng, S.-L.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. Teriparatide (Human Parathyroid Hormone (1-34)) Inhibits Osteogenic Vascular Calcification in Diabetic Low Density Lipoprotein Receptor-Deficient Mice. J. Biol. Chem. 2003, 278, 50195–50202. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.J.; Lu, J.; Umar, S.; Lee, J.T.; Kulkarni, R.P.; Ding, Y.; Chang, C.-C.; Hsiai, T.K.; Hokugo, A.; Gkouveris, I.; et al. Effects of Teriparatide on Morphology of Aortic Calcification in Aged Hyperlipidemic Mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1203–H1213. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, A.; Clements, J.N. Abaloparatide: A New Pharmacological Option for Osteoporosis. Am. J. Health Syst. Pharm. 2019, 76, 130–135. [Google Scholar] [CrossRef]
- Brandenburg, V.M.; Verhulst, A.; Babler, A.; D’Haese, P.C.; Evenepoel, P.; Kaesler, N. Sclerostin in Chronic Kidney Disease-Mineral Bone Disorder Think First before You Block It! Nephrol. Dial. Transplant. 2019, 34, 408–414. [Google Scholar] [CrossRef]
- Maré, A.D.; D’Haese, P.C.; Verhulst, A. The Role of Sclerostin in Bone and Ectopic Calcification. Int. J. Mol. Sci. 2020, 21, 3199. [Google Scholar] [CrossRef]
- Catalano, A.; Bellone, F.; Morabito, N.; Corica, F. Sclerostin and Vascular Pathophysiology. Int. J. Mol. Sci. 2020, 21, 4779. [Google Scholar] [CrossRef]
- Asadipooya, K.; Weinstock, A. Cardiovascular Outcomes of Romosozumab and Protective Role of Alendronate. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1343–1350. [Google Scholar] [CrossRef]
- Sato, M.; Inaba, M.; Yamada, S.; Emoto, M.; Ohno, Y.; Tsujimoto, Y. Efficacy of Romosozumab in Patients with Osteoporosis on Maintenance Hemodialysis in Japan; an Observational Study. J. Bone Miner. Metab. 2021, 39, 1082–1090. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Model | Results | Reference |
|---|---|---|---|
| “Calcifying human aortic smooth muscle cells express different BALP isoforms, including the novel B1x isoform” | Calcifying human aortic smooth muscle cells (HAoSMC) cultivated for 30 days in a medium containing 5 or 10 mmol/L of glycerophosphate in the presence or absence of the specific inhibitor of ALP (tetramisole). | All bone-specific ALP isoforms (B/I, B1x, B1 and B2) were identified in HAoSMC; calcification was associated with an increase in isoforms B/I, B1x and B2. | [7] |
| “Pathophysiological role of vascular smooth muscle ALP in medial artery calcification” | Mouse model with overexpression of human TNALP in vascular smooth muscle cells. | These mice had vascular calcifications, hypertension, cardiac hypertrophy and early mortality. Administration of ALP inhibitor led to an improvement in cardiovascular outcome and life expectancy. | [69] |
| “Impaired calcification around matrix vesicles of growth plate and bone in ALP-deficient mice” | Knockout mice for the ALP gene (which includes the transcription of BALP). | TNALP Knockout mice showed significant hypomineralization. TNALP is an important promoter of bone mineralization. | [71] |
| “Cellular localization of endothelial ALP reaction product and enzyme protein in the myocardium” | Myocardial tissue samples of different species (human, rat, and pig). | In the myocardium, ALP was localized in all the species studied, mainly in the plasma endothelial membrane and in the pinocytotic vesicles. | [75] |
| “Transgenic overexpression of TNALP in vascular endothelium results in generalized arterial calcification” | Mice with endothelial ALP overexpression. | Mice develops arterial calcifications, increased blood pressure, and compensatory left ventricular hypertrophy. This model demonstrates how ALP positive endothelial cells can also promote vascular calcification. | [76] |
| “Widespread increase in myeloid calcifying cells contributes to ectopic vascular calcification in type 2 diabetes” | Circulating procalcifying cells (osteocalcin and BALP positive) from 100 patients with or without diabetes and CVD. | There is a subpopulation of pro-calcifying cells that come from the myeloid lineage and retain monocyte/macrophages markers (myeloid calcifying cells). They are overrepresented in the blood of patients with type 2 diabetes and in atherosclerotic lesions. | [79] |
| “TNALP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF- β1/Smads and activating p53 signaling pathways” | Sections of heart of patients and rats with myocardial infarction. | Inhibition of TNALP regulated cardiac fibrosis and exerted an antifibrotic effect through AMPK-TGF-β1/Smads and p53 signals. | [80] |
| “Inhibition of TNALP protects against medial arterial calcification and improves survival probability in the CKD-MBD mouse model” | CKD-MBD mouse model. | In mice with inhibited ALP, calcifications were blocked. Survival was 100%, compared to those not treated with the inhibitor (57% survival). | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haarhaus, M.; Cianciolo, G.; Barbuto, S.; La Manna, G.; Gasperoni, L.; Tripepi, G.; Plebani, M.; Fusaro, M.; Magnusson, P. Alkaline Phosphatase: An Old Friend as Treatment Target for Cardiovascular and Mineral Bone Disorders in Chronic Kidney Disease. Nutrients 2022, 14, 2124. https://doi.org/10.3390/nu14102124
Haarhaus M, Cianciolo G, Barbuto S, La Manna G, Gasperoni L, Tripepi G, Plebani M, Fusaro M, Magnusson P. Alkaline Phosphatase: An Old Friend as Treatment Target for Cardiovascular and Mineral Bone Disorders in Chronic Kidney Disease. Nutrients. 2022; 14(10):2124. https://doi.org/10.3390/nu14102124
Chicago/Turabian StyleHaarhaus, Mathias, Giuseppe Cianciolo, Simona Barbuto, Gaetano La Manna, Lorenzo Gasperoni, Giovanni Tripepi, Mario Plebani, Maria Fusaro, and Per Magnusson. 2022. "Alkaline Phosphatase: An Old Friend as Treatment Target for Cardiovascular and Mineral Bone Disorders in Chronic Kidney Disease" Nutrients 14, no. 10: 2124. https://doi.org/10.3390/nu14102124
APA StyleHaarhaus, M., Cianciolo, G., Barbuto, S., La Manna, G., Gasperoni, L., Tripepi, G., Plebani, M., Fusaro, M., & Magnusson, P. (2022). Alkaline Phosphatase: An Old Friend as Treatment Target for Cardiovascular and Mineral Bone Disorders in Chronic Kidney Disease. Nutrients, 14(10), 2124. https://doi.org/10.3390/nu14102124