
Overview of Beneficial Effects of (Poly)phenol Metabolites in the Context of Neurodegenerative Diseases on Model Organisms

 , , ,
, , ,  , and
, and
Abstract
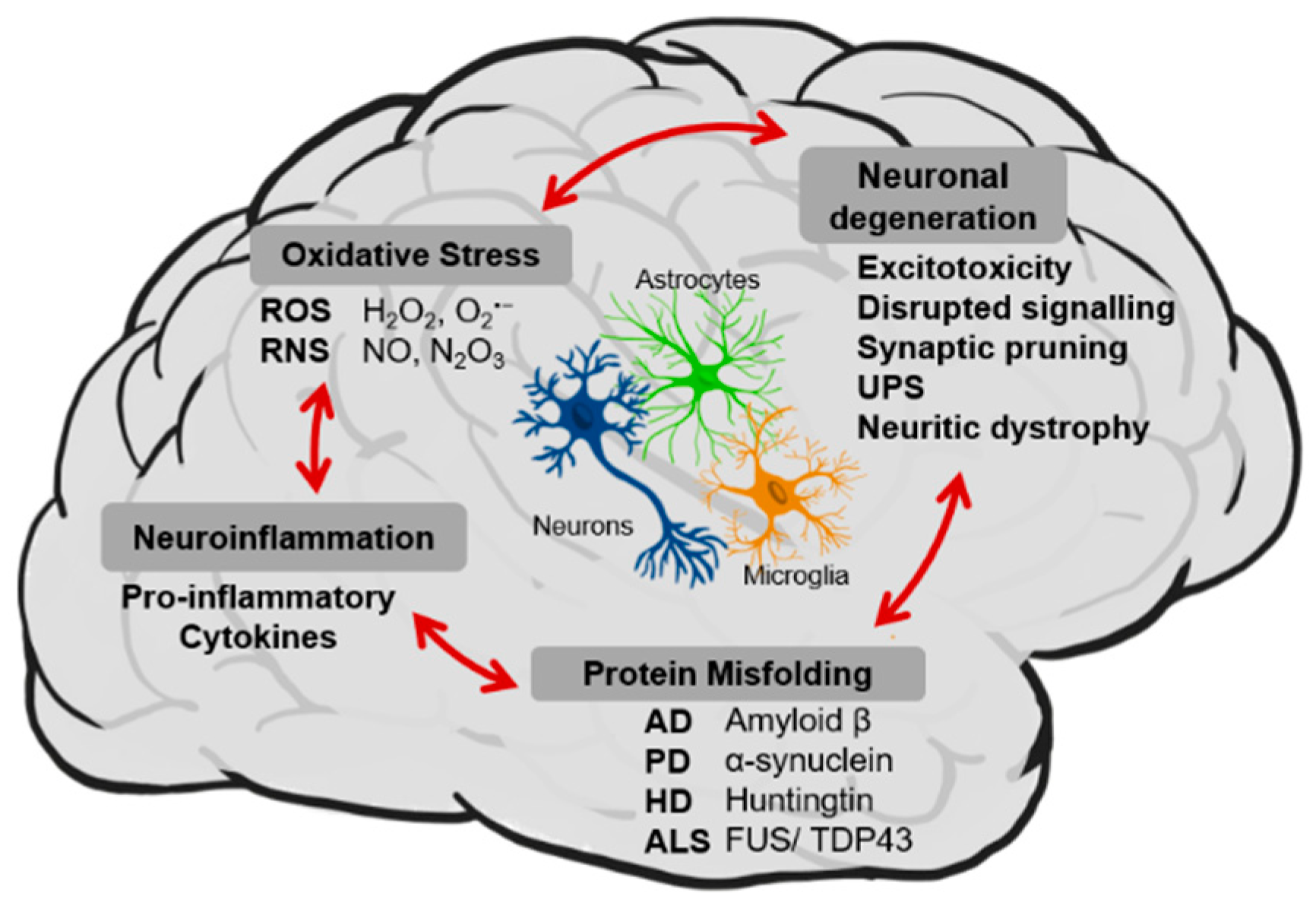
1. Neuroprotective Potential of (Poly)phenol Rich Diets
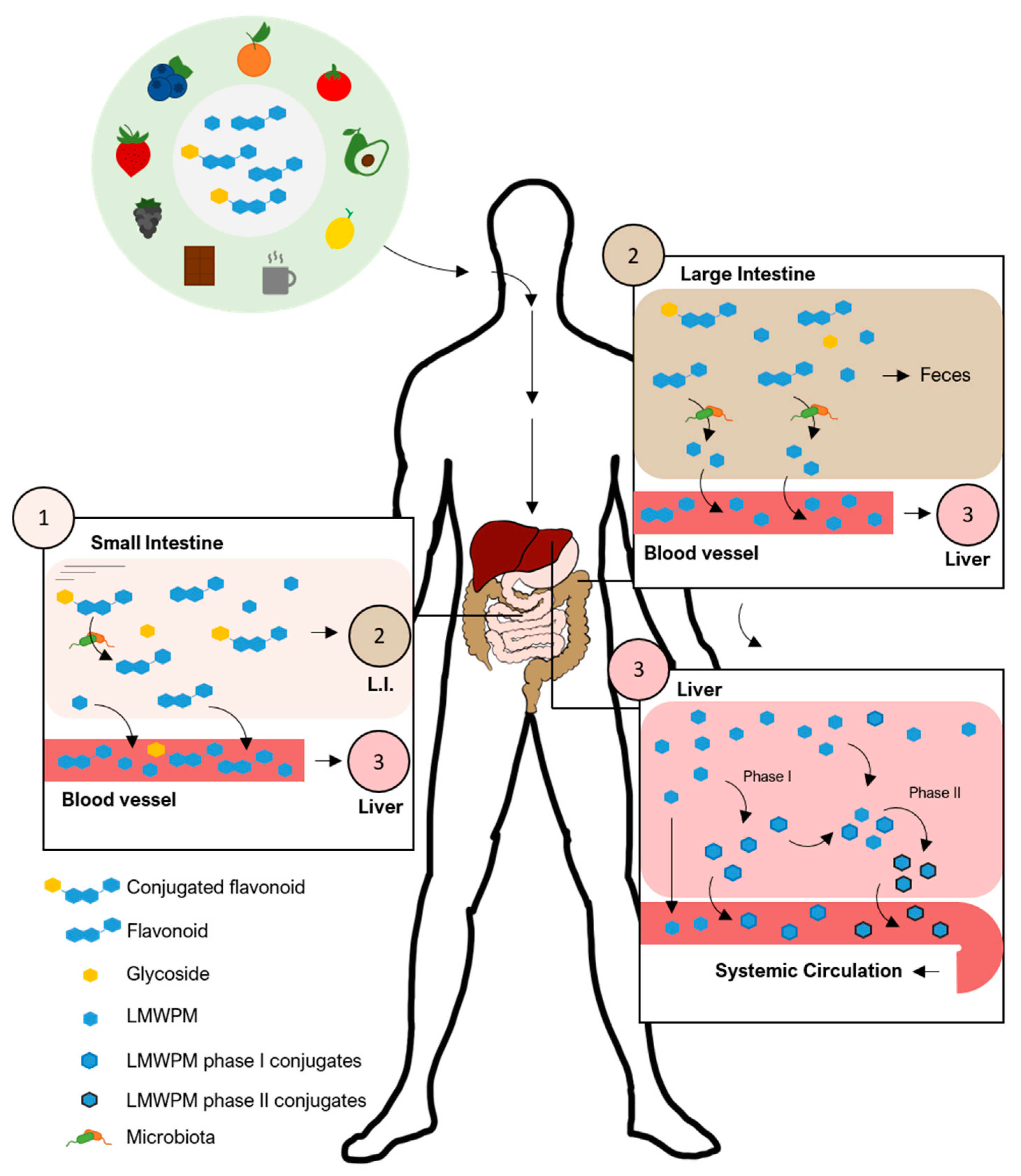
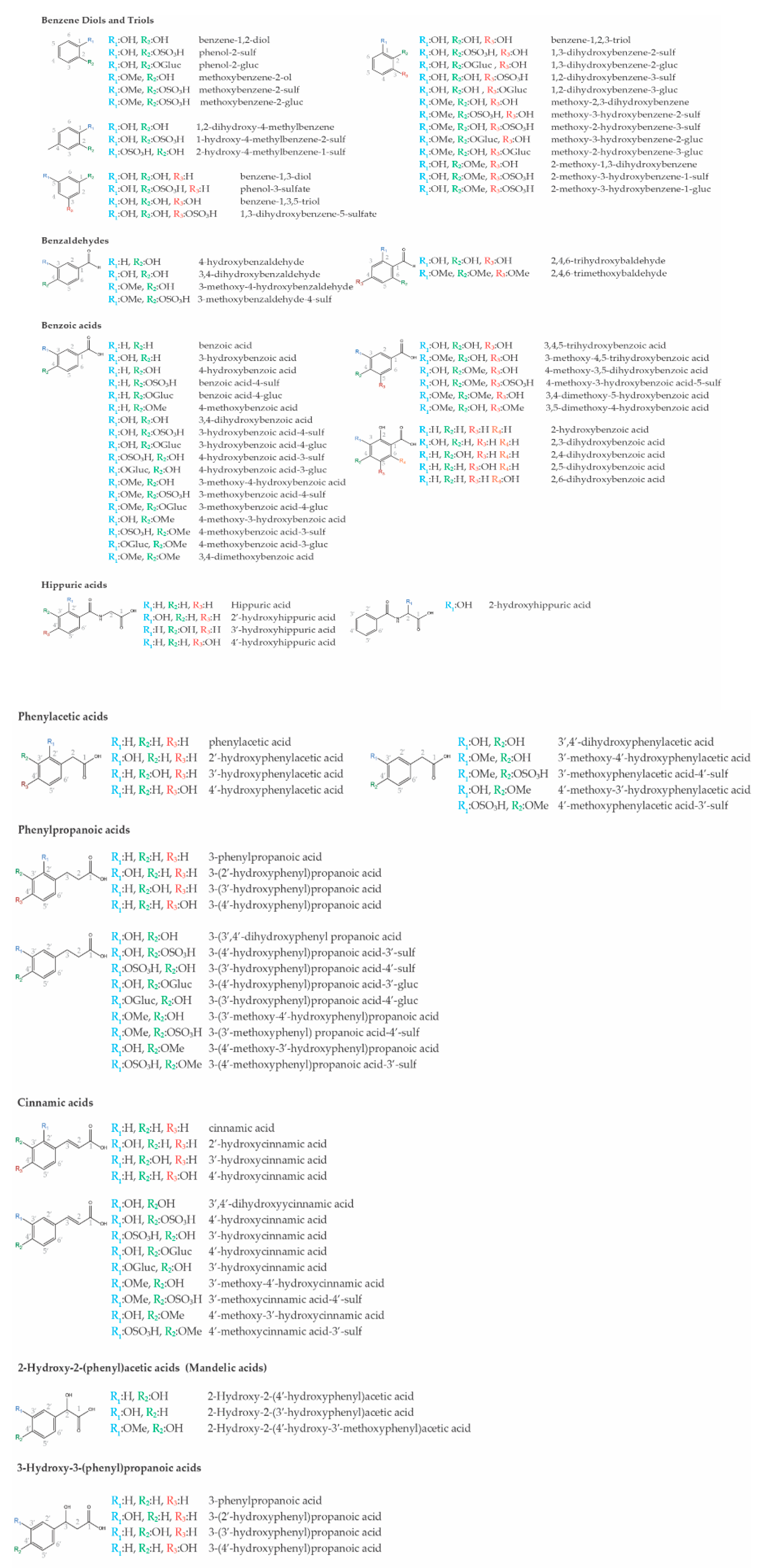
2. Bioavailability of Plant–Derived (Poly)phenols and the Low Molecular Weight (Poly)phenol Metabolites
3. Models for Studying Neurodegenerative Diseases
4. Low Molecular Weight (Poly)phenol Metabolites in Saccharomyces cerevisiae Models of NDDs

{kind=link}
{kind=link}
{kind=link}
| (Poly)phenol Metabolite 1 | Model | Dose and Time | Mechanistic Evidences | Ref. |
|---|---|---|---|---|
| Benzenes | ||||
| Benzene–1,2—Diol (Catechol) | WT and Several Mutants (BY4743) for Oxidative Stress Resistance, Cytoplasmic Thioredoxin System, GSH Synthesis and Recycling, Lipid Metabolism, Vacuole and Endosomes Biogenesis, V–ATPase Assembly and Alpha–Tubulin Folding. | 0.55 mM, 1.1 mM and 2.2 mM for 5 or 15 Generations | ↑ Response Pathways Associated with Oxidative and Cytoskeleton Stress, ER Stress and Vesicular Trafficking | [85] |
| Benzaldehydes | ||||
| 4—Hydroxy–3—Methoxybenzaldehyde (Vanillin) | WT (YPH250) | 4, 6 and 8 mM for 30 min | ↑ ZWF1, GND1 and GND2 Expression Levels | [77] |
| zwf1 Mutant (YPH250) | 2–8 mM for 15 min | ↑ Mitochondrial Fragmentation | ||
| 10–20 mM for 30 min | ↑ P–Body Formation | |||
| WT and Mutants for Chromatin Remodeling, Vesicle Transport, Cell Cycle, DNA Processing and Cellular Transport (BY4743) | 5 mM for 24 h | ↓ Cell Growth | [77] | |
| WT (BY4741) | 4 mM for 6 h | ↓ Translation Activity | [76] | |
| 2–50 mM for 30 min | ↑ P–Body Formation | |||
| 30–50 mM for 30 min | ↑ Stress Granules Formation | |||
| WT (BY4742) | 6–15 mM for 30 min | ↓ Translation Activity | [91] | |
| 6–15 mM for 1 h | ↑ ADH6 and ADH7 mRNA Levels | |||
| adh6 and adh7 Mutants (BY4742) | 8 mM for 36 h | ↓Growth Rate | ||
| Benzoic Acids | ||||
| 3,4,5—Trihydroxybenzoic Acid (Gallic acid) | WT and zrt1 Mutant (BY4741) | 100 µM for 1 h | ↓ Intracellular Zn Content ↑ ZRT2 Transcription | [92] |
| Pdr5 Overexpressing Yeast (AD12456) | 5–200 µM for 1 h | Inhibited the Pdr5 Efflux Pump | [69] | |
| ahp1 Mutant (BY4743) Overexpressing Aβ42_GFP | 50 µM for 6 h | ↓ Protein Aggregation | [81] | |
| 2—Hydroxybenzoic Acid (Salicylic Acid) | H208-3B Carrying PGAPAQ1 Plasmid | 5 mM | ↑ [Ca2+] Cytosolic Levels | [93] |
| yrr1 Mutant (BY4741) | 12.5 mM | ↑ Resistance to 2HBA Mediated by Efflux Pump–Encoding Genes Overexpression | [71] | |
| Benzoic Acid | WT (FY1679–28c) | 0.5 mM for 1 h | ↑ Pdr12 and HSP30 Expression | [70] |
| vac8 Mutant (HAY394) | 0, 0.1, 0.5, and 1 mM for 2 h | ↑ prApe1 Maturation | [75] | |
| 2 mM for 2 h | ↓ Macroautophagy | |||
| WT (HAY75) | 2 mM for 5, 15 and 40 min | ↓ ER–to–Golgi Complex Transport Step | ||
| 2,5—Dihydroxybenzoic Acid | ahp1 Mutant (BY4743) Overexpressing Aβ42_GFP | 50 µM for 6 h | ↓ Protein Aggregation | [81] |
| 2,3—Dihydroxybenzoic Acid | ||||
| 3,4—Dihydroxybenzoic Acid | ||||
| Phenylacetic Acids | ||||
| Phenylacetic Acid | pdr12 Mutant (CEN.PK113–7D) | 2–8 mM for 72 h | ↓Cell Growth | [94] |
| Cinnamic Acids | ||||
| 4—Hydroxy–3—Methoxycinnamic Acid (Ferulic Acid) | WT (BY4741) | 1 mM for 1 h | ↑ YAL005C, YER153C, and YPR125W Expression | [95] |
| ahp1 Mutant (BY4743) Overexpressing Aβ42_GFP | 50 µM for 6 h | ↓ Protein Aggregation | [81] | |
| 2,4—Dihydroxycinnamic Acid | ahp1 Mutant (BY4743) Overexpressing Aβ42_GFP | 50 µM for 6 h | ↓ Protein Aggregation | [81] |
| 3,4—Dihydroxycinnamic Acid | ||||
| 3,4,5—Trimethoxycinnamic Acid |
| (Poly)Phenol Metabolite 1 | Model | Dose and Time | Mechanistic Evidence | Ref. |
|---|---|---|---|---|
| Benzenes | ||||
| Benzene–1,2,3—Triol (Pyrogallol) | WT (BY4741) + 1.5 mM H2O2 for 1 h | 300 μM for 15 min | ↑ Cell Viability ↓ H2O2–Induced Intracellular Oxidation ↓ Protein Carbonylation ↓ Total GSH Levels | [82] |
| sod2 Mutant (BY4741) + 1.5 mM H2O2 for 1 h | 300 μM for 15 min | ↑Oxidative Stress Resistance | ||
| sod2 mutant (BY4741) | 300 μM for 4 Days | =Lifespan | ||
| Benzene–1,3,5—Triol (Phloroglucinol) | WT (BY4741) + 1.5 mM H2O2 for 1 h | 300 μM for 15 min | =Cell Viability =H2O2–Induced Intracellular Oxidation =Protein Carbonylation ↓ Total GSH Levels | [82] |
| sod2 mutant (BY4741) | 300 μM for 4 days | =Lifespan | ||
| Benzaldehydes | ||||
| 4—Hydroxy–3—Methoxybenzaldehyde (Vanillin) | WT (YPH250) | 4, 6 and 8 mM for 30 min | ↑ Expression of Oxidative Stress Response Genes | [84] |
| 8 mM for 15 min | ↑ Mitochondrial Fragmentation | |||
| 8 mM for 30 min | ↑ Oxidation Levels | |||
| Benzoic Acids | ||||
| 3,4—Dihydroxybenzoic Acid (Protocatechuic Acid) | WT (BY4742) + 5 mM H2O2 for 1 h | 13 µM for 30 Days | ↓ Oxidative Stress ↑ Yeast Lifespan | [83] |
| sir2 Mutant (BY4742) + 5 mM H2O2 for 1 h | 13 µM for 17 Days | ↓ ROS Accumulation | ||
| 13 µM until OD600 0.6 | ↑ Yeast Growth | |||
| tor1, sch9, rim15, msn2, msn4 and asg1 Mutants (BY4742) + 5 mM H2O2 for 1 h | 13 µM for 17 Days | ↓ ROS Accumulation | ||
| 4—Hydroxybenzoic Acid | WT (BY4741) and bna7 Mutant (YKB4649) | 50 mM for 2 h | ↑ Cytoplasmic ROS Levels | [86] |
| 2,5—Dihydroxybenzoic Acid | WT and sod2 Mutant (BY4741) | 3–18 mM for 7 Days | ↓ Cell Growth | [96] |
| WT and sod2 Mutant (BY4741) + 1, 2 and 3 mM H2O2 for 7 days | 1 and 3 mM for 7 Days | Recovery from Oxidative Stress | ||
| WT, glr and sod2 Mutants (BY4741) + 0.1 mM GSH for 7 Days | 1 and 3 mM for 7 Days | Recovery of Cell Growth | ||
| Cinnamic Acids | ||||
| 4—Hydroxy–3—Methoxycinnamic Acid (Ferulic Acid) | WT (BY4741) | 10 mM for 72 h | ↓ Cell Growth | [86] |
| 10 mM for 2 h | ↑ Cytoplasmic ROS Levels | |||
| bna7 Mutant (YKB4649) | 10 mM for 72 h | ↑ Cell Growth | ||
| 10 mM for 2 h | ↑ Cytoplasmic ROS Levels |
5. Low Molecular Weight (Poly)phenol Metabolites in Invertebrate Models of NDDs
5.1. C. elegans
| (Poly)Phenol Metabolite 1 | Model | Dose and Time | Mechanistic Evidence | Ref. |
|---|---|---|---|---|
| Benzaldehydes | ||||
| 2—Hydroxy–4—Methoxybenzaldehyde | BZ555 Worms (dat1::gfp) | 2.5 mM Administered on Food for 24 h | ↓ Dopaminergic Neurodegeneration | [99] |
| Benzoic Acids | ||||
| 3,4,5—Trihydroxybenzoic Acid (Gallic Acid) | Amyloid–Related Neurodegenerative Disease Model Expression of ATX3 in the Nervous System | 0.1 mM Administered to Food Scoring After 24 and 48 h | ↑ Mobility No Rescue in Lifespan | [100] |
| Cinnamic Acids | ||||
| 4—Hydroxy–3—Methoxycinnamic Acid (Ferulic Acid) | Alzheimer’s Model (Muscle–Specific Aβ—Expression) | 50 mM Administered on Food from Embryo | ↑ Survival Rate ↑ Protection against Aβ-Cytotoxicity | [104] |
| 3,4—Dihydroxycinnamic Acid (Caffeic Acid) | Wt Mt(Mev-1), A Strain Hypersensitive To Oxidative Stress | 100 µM to 600 µM (Strongest Effect at 300 µM) | ↑ Lifespan WT and Mt(mev–1) ↓ Body Size Altered Lipid–Metabolism Tendency to Delay Reproductive Time = Number of Offspring ↓ Gene Expression of Hsp–3, Hsp–17, and Hsp–16.41 ↑ Gene Expression of Hsp–12.6 | [102] |
| Cinnamic Acid | Alzheimer’s Model Strain CL4176 Expressing Human Aβ | 40, 80, or 200 μM Throughout Development | ↓ Aβ40 Fibrillogenesis ↓ Aβ40–Induced Cytotoxicity Ameliorate AD–Like Symptoms of Worm Paralysis | [101] |
| 2—Hydroxycinnamic Acid | ||||
| 3—Hydroxycinnamic Acid | ||||
| 2,4—Dihydroxycinnamic | ||||
| 3,4,5—Trihydroxycinnamic Acid |
5.2. Drosophila
6. Effects of Low Molecular Weight (Poly)phenol Metabolites in Zebrafish Models of NDDs
| (Poly)phenol Metabolite 1 | Strain/Start of Exposure | Model without Intervention | Dose and Time | Mechanistic Evidence | Ref. |
|---|---|---|---|---|---|
| Benzoic Acids | |||||
| Benzoic Acid | AB/2 dpf | Neuronal Teratogenesis | 0.07; 0.7; 3.47; 6.94 mM Sodium Benzoate in the Water Medium for 24 h 2 | ↓ Tactile Sensitivity and ↓ Larvae Mobility (6.94 mM) ↑ Misalignment of Muscle Fibers (6.94 mM) Defects on Motor Axons and Neuromuscular Junctions ↑ Axonal Projections and AchR Clusters (post–synapses; >69.4 µM) | [147] |
| AB/2 hpf | DA System/Neuronal Development | 0.28; 0.94 mM SB in the Water Medium for up to 3 dpf 2 | ↓ TH and DAT Expression in DA Neurons | [148] | |
| AB/2 hpf | Locomotor Activity | 0.14; 0.28 mM SB in the Water Medium for 3 Days 2 | ↓ Locomotor Activity | [148] | |
| AB/5 hpf | Anxiety–Like Larval Behavior (Thigmotaxis) | 0.35 mM SB in E3 Medium for 72 h 2 | Thigmotaxis ↓ GSR Expression | [149] | |
| 3,4,5—Trihydroxybenzoic Acid Gallic Acid) | AB/72 hpf | Neuronal Hyperactivity/Motoneuron Hyperexcitability | 0.30 mM in E3 Medium for 30 min 3 | ↓ Glutamate and GABA ↑ fosab Expression in Distinct Areas of the Brain (Forebrain, Olfactory Bulbs and Pallial Area) ↑ Locomotor Function: Hyperactivity | [142] |
| n.d/ 4–6 Month | Neurochemical Content Changes | 29.4, 58.8, 117.60 µM in the Water Medium for 24 and 48 h 3 | ↓ Sulfhydryl Content (117.6 µM) ↓ TBA–RS (117.6 µM) ↓ DCFH Oxidation (117.6 µM) ↓ AChE Activity (117.6 µM) = ChAT activity, Nitrates and Nitrites (117.6 µM) ↑ SOD Activity (29.4–58.8 µM) ↑ Catalase Activity (29.4–117.6 µM) = [GSH] (29.4–117.6 µM) | [143] |
| (Poly)phenol Metabolite 1 | Strain/Start of Exposure | Model with Intervention | Dose and Time | Mechanistic Evidence | Ref. |
|---|---|---|---|---|---|
| Benzoic Acids | |||||
| 3,4,5—Trihydroxybenzoic Acid (Gallic Acid) | AB/72 hpf | Cd–Induced Disruption of the Olfactory System | 293.9 µM in Cd–Treated Larvae in E3 Medium for 30 min 2 | =Locomotion | [142] |
| n.d/ 4–6 Month | Ethanol–Induced Neurochemical Changes | 29.4, 58.8 µM in 0.5% (v/v) Ethanol Solution for 24 h | ↑ ChAT Activity (Restored) ↓ TBA–RS (Restored) ↓ DCFH Oxidation (Restored) ↑ SOD Activity (Restored) | [143] | |
| 3,4—Dihydroxybenzoic Acid (Protocatechuic Acid) | AB/1 dpf | PD Model Chemical Induced by 6—OHDA | 0, 12, 25, 50, 100 µM in the Water Medium for 48 h | = DA Neuron Loss DA Neurons Toxicity at 100 mM | [154] |
| AB/1 dpf | PD Model Chemical Induced by 6—OHDA | PCA (6, 12, 25 µM), Chrysin, or PCA + Chrysin in the Water Medium for 48 h | PCA + Chrysin, but not PCA or Chrysin Individually ↓ DA Neuronal Loss | [154] |
7. Effects of Low Molecular Weight (Poly)phenol Metabolites in Rodent Models of NDDS
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rothwell, J.A.; Perez-Jimenez, J.; Neveu, V.; Medina-Remon, A.; M’hiri, N.; Garcia-Lobato, P.; Manach, C.; Knox, C.; Eisner, R.; Wishart, D.S.; et al. Phenol-Explorer 3.0: A major update of the Phenol-Explorer database to incorporate data on the effects of food processing on polyphenol content. Database 2013, 2013, bat070. [Google Scholar] [CrossRef]
- da Silva, A.; Giacomoni, F.; Pavot, B.; Fillâtre, Y.; Rothwell, J.A.; Sualdea, B.B.; Veyrat, C.; Garcia-Villalba, R.; Gladine, C.; Kopec, R.; et al. PhytoHub V1. 4: A new release for the online database dedicated to food phytochemicals and their human metabolites. In Proceeding of the 1st International Conference on Food Bioactives and Health Conference, Norwich, UK, 13–15 September 2016; pp. 13–15. [Google Scholar]
- Marino, M.; Del Bo’, C.; Martini, D.; Porrini, M.; Riso, P. A Review of Registered Clinical Trials on Dietary (Poly)Phenols: Past Efforts and Possible Future Directions. Foods 2020, 9, 1606. [Google Scholar] [CrossRef]
- Medina-Remón, A.; Tresserra-Rimbau, A.; Pons, A.; Tur, J.A.; Martorell, M.; Ros, E.; Buil-Cosiales, P.; Sacanella, E.; Covas, M.I.; Corella, D.; et al. Effects of total dietary polyphenols on plasma nitric oxide and blood pressure in a high cardiovascular risk cohort. The PREDIMED randomized trial. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 60–67. [Google Scholar] [CrossRef]
- Rodriguez-Mateos, A.; Vauzour, D.; Krueger, C.G.; Shanmuganayagam, D.; Reed, J.; Calani, L.; Mena, P.; Rio, D.D.; Crozier, A. Bioavailability, bioactivity and impact on health of dietary flavonoids and related compounds: An update. Arch. Toxicol. 2014, 88, 1803–1853. [Google Scholar] [CrossRef]
- Nooyens, A.C.J.; Bueno-de-Mesquita, H.B.; van Boxtel, M.P.J.; van Gelder, B.M.; Verhagen, H.; Verschuren, W.M.M. Fruit and vegetable intake and cognitive decline in middle-aged men and women: The Doetinchem Cohort Study. Br. J. Nutr. 2011, 106, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Psaltopoulou, T.; Sergentanis, T.N.; Panagiotakos, D.B.; Sergentanis, I.N.; Kosti, R.; Scarmeas, N. Mediterranean diet, stroke, cognitive impairment, and depression: A meta-analysis. Ann. Neurol. 2013, 74, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.E. Food for thought: The role of dietary flavonoids in enhancing human memory, learning and neuro-cognitive performance. Proc. Nutr. Soc. 2008, 67, 238–252. [Google Scholar] [CrossRef]
- Williams, R.J.; Spencer, J.P.E. Flavonoids, cognition, and dementia: Actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Rendeiro, C.; Rhodes, J.S.; Spencer, J.P.E. The mechanisms of action of flavonoids in the brain: Direct versus indirect effects. Neurochem. Int. 2015, 89, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Mayr, H.L.; Thomas, C.J.; Tierney, A.C.; Kucianski, T.; George, E.S.; Ruiz-Canela, M.; Hebert, J.R.; Shivappa, N.; Itsiopoulos, C. Randomization to 6-month Mediterranean diet compared with a low-fat diet leads to improvement in Dietary Inflammatory Index scores in patients with coronary heart disease: The AUSMED Heart Trial. Nutr. Res. 2018, 55, 94–107. [Google Scholar] [CrossRef]
- Devore, E.E.; Kang, J.H.; Breteler, M.M.B.; Grodstein, F. Dietary intakes of berries and flavonoids in relation to cognitive decline. Ann. Neurol. 2012, 72, 135–143. [Google Scholar] [CrossRef]
- Krikorian, R.; Shidler, M.D.; Nash, T.A.; Kalt, W.; Vinqvist-Tymchuk, M.R.; Shukitt-Hale, B.; Joseph, J.A. Blueberry Supplementation Improves Memory in Older Adults. J. Agric. Food Chem. 2010, 58, 3996–4000. [Google Scholar] [CrossRef]
- Bowtell, J.L.; Aboo-Bakkar, Z.; Conway, M.E.; Adlam, A.-L.R.; Fulford, J. Enhanced task-related brain activation and resting perfusion in healthy older adults after chronic blueberry supplementation. Appl. Physiol. Nutr. Metab. 2017, 42, 773–779. [Google Scholar] [CrossRef]
- Miller, M.G.; Hamilton, D.A.; Joseph, J.A.; Shukitt-Hale, B. Dietary blueberry improves cognition among older adults in a randomized, double-blind, placebo-controlled trial. Eur. J. Nutr. 2017, 57, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Macready, A.L.; Kennedy, O.B.; Ellis, J.A.; Williams, C.M.; Spencer, J.P.E.; Butler, L.T. Flavonoids and cognitive function: A review of human randomized controlled trial studies and recommendations for future studies. Genes Nutr. 2009, 4, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.T.; Head, K.; Morris, P.G.; Macdonald, I.A. The Effect of Flavanol-rich Cocoa on the fMRI Response to a Cognitive Task in Healthy Young People. J. Cardiovasc. Pharmacol. 2006, 47 (Suppl. 2), S215–S220. [Google Scholar] [CrossRef]
- Field, D.T.; Williams, C.M.; Butler, L.T. Consumption of cocoa flavanols results in an acute improvement in visual and cognitive functions. Physiol. Behav. 2011, 103, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Lamport, D.J.; Williams, C.M.; Rendeiro, C. Polyphenols and Cognition in Humans: An Overview of Current Evidence from Recent Systematic Reviews and Meta-Analyses. Brain Plast. 2021, 6, 139–153. [Google Scholar] [CrossRef]
- Day, A.J.; Cañada, F.J.; Díaz, J.C.; Kroon, P.A.; McLauchlan, R.; Faulds, C.B.; Plumb, G.W.; Morgan, M.R.A.; Williamson, G. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. Febs Lett. 2000, 468, 166–170. [Google Scholar] [CrossRef]
- Gonzalez-Barrio, R.; Edwards, C.A.; Crozier, A. Colonic Catabolism of Ellagitannins, Ellagic Acid, and Raspberry Anthocyanins: In Vivo and In Vitro Studies. Drug Metab. Dispos. 2011, 39, 1680–1688. [Google Scholar] [CrossRef]
- Fernandes, I.; de Freitas, V.; Mateus, N. Anthocyanins and human health: How gastric absorption may influence acute human physiology. Nutr. Aging 2014, 2, 1–14. [Google Scholar] [CrossRef]
- Czank, C.; Cassidy, A.i.; Zhang, Q.; Morrison, D.J.; Preston, T.; Kroon, P.A.; Botting, N.P.; Kay, C.D. Human metabolism and elimination of the anthocyanin, cyanidin-3-glucoside: A 13C-tracer study. Am. J. Clin. Nutr. 2013, 97, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- de Ferrars, R.M.; Czank, C.; Zhang, Q.; Botting, N.P.; Kroon, P.A.; Cassidy, A.; Kay, C.D. The pharmacokinetics of anthocyanins and their metabolites in humans. Br. J. Pharmacol. 2014, 171, 3268–3282. [Google Scholar] [CrossRef]
- Ottaviani, J.I.; Borges, G.; Momma, T.Y.; Spencer, J.P.E.; Keen, C.L.; Crozier, A.; Schroeter, H. The metabolome of [2-14C](−)-epicatechin in humans: Implications for the assessment of efficacy, safety and mechanisms of action of polyphenolic bioactives. Sci. Rep. 2016, 6, 29034. [Google Scholar] [CrossRef] [PubMed]
- Actis-Goretta, L.; Lévèques, A.; Giuffrida, F.; Romanov-Michailidis, F.; Viton, F.; Barron, D.; Duenas-Paton, M.; Gonzalez-Manzano, S.; Santos-Buelga, C.; Williamson, G.; et al. Elucidation of (−)-epicatechin metabolites after ingestion of chocolate by healthy humans. Free Radic. Biol. Med. 2012, 53, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Stoupi, S.; Williamson, G.; Viton, F.; Barron, D.; King, L.J.; Brown, J.E.; Clifford, M.N. In Vivo Bioavailability, Absorption, Excretion, and Pharmacokinetics of [14C]Procyanidin B2 in Male Rats. Drug Metab. Dispos. 2010, 38, 287–291. [Google Scholar] [CrossRef]
- Williamson, G. The role of polyphenols in modern nutrition. Nutr. Bull. 2017, 42, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, R.; Istas, G.; Heiss, C.; Rodriguez-Mateos, A. Plasma and Urinary Phenolic Profiles after Acute and Repetitive Intake of Wild Blueberry. Molecules 2016, 21, 1120. [Google Scholar] [CrossRef]
- Feliciano, R.P.; Boeres, A.; Massacessi, L.; Istas, G.; Ventura, M.R.; dos Santos, C.N.; Heiss, C.; Rodriguez-Mateos, A. Identification and quantification of novel cranberry-derived plasma and urinary (poly)phenols. Arch. Biochem. Biophys. 2016, 599, 31–41. [Google Scholar] [CrossRef]
- Carecho, R.; Carregosa, D.; dos Santos, C.N. Low Molecular Weight (poly)Phenol Metabolites Across the Blood-Brain Barrier: The Underexplored Journey. Brain Plast. 2021, 6, 193–214. [Google Scholar] [CrossRef]
- Kay, C.D.; Clifford, M.N.; Mena, P.; McDougall, G.J.; Andres-Lacueva, C.; Cassidy, A.; Del Rio, D.; Kuhnert, N.; Manach, C.; Pereira-Caro, G.; et al. Recommendations for standardizing nomenclature for dietary (poly)phenol catabolites. Am. J. Clin. Nutr. 2020, 112, 1051–1068. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef]
- Zheng, Z.; Lee, J.; Yenari, M. Stroke: Molecular Mechanisms and Potential Targets for Treatment. Curr. Mol. Med. 2003, 3, 361–372. [Google Scholar] [CrossRef]
- Schaffer, S.; Eckert, G.P.; Schmitt-Schillig, S.; Müller, W.E. Plant Foods and Brain Aging: A Critical Appraisal. In Local Mediterranean Food Plants and Nutraceuticals; Karger Publishers: Basel, Switzerland, 2006; pp. 86–115. [Google Scholar]
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of microglia. Glia 1993, 7, 111–118. [Google Scholar] [CrossRef]
- McGeer, P.; McGeer, E. The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 1995, 21, 195–218. [Google Scholar] [CrossRef]
- Giulian, D.; Corpuz, M. Microglial secretion products and their impact on the nervous system. Adv. Neurol. 1993, 59, 315–320. [Google Scholar] [PubMed]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.-K.; Ischiropoulos, H.; Przedborski, S. Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P.; Lane, A.; Sleiman, P.M.A. The coenzyme Q10 status of the brain regions of Parkinson’s disease patients. Neurosci. Lett. 2008, 447, 17–19. [Google Scholar] [CrossRef]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair. 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Lindsay, J. Risk Factors for Alzheimer’s Disease: A Prospective Analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef]
- Ghosh, M.; Aguirre, V.; Wai, K.; Felfly, H.; Dietrich, W.D.; Pearse, D.D. The Interplay between Cyclic AMP, MAPK, and NF-κB Pathways in Response to Proinflammatory Signals in Microglia. BioMed Res. Int. 2015, 2015, 308461. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.; Zamboni, P.; Mahajan, R. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar]
- Esposito, E.; Rotilio, D.; Dimatteo, V.; Digiulio, C.; Cacchio, M.; Algeri, S. A review of specific dietary antioxidants and the effects on biochemical mechanisms related to neurodegenerative processes. Neurobiol. Aging 2002, 23, 719–735. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Sulaiman, A.; Alhaddad, H.; Alhadidi, Q. Natural polyphenols: Influence on membrane transporters. J. Intercult. Ethnopharmacol. 2016, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxidative Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, H.; Zhang, G.; Meng, J.; Deng, K.; Zhou, W.; Wang, H.; Wang, Z.; Hu, N.; Suo, Y. Anthocyanins from Lycium ruthenicum Murr. Ameliorated d-Galactose-Induced Memory Impairment, Oxidative Stress, and Neuroinflammation in Adult Rats. J. Agric. Food Chem. 2019, 67, 3140–3149. [Google Scholar] [CrossRef]
- Bredesen, D.E.; Rao, R.V.; Mehlen, P. Cell death in the nervous system. Nature 2006, 443, 796–802. [Google Scholar] [CrossRef]
- Mandel, S.; Youdim, M.B.H. Catechin polyphenols: Neurodegeneration and neuroprotection in neurodegenerative diseases. Free Radic. Biol. Med. 2004, 37, 304–317. [Google Scholar] [CrossRef]
- Loncarevic-Vasiljkovic, N.; Carregosa, D.; Santos, C.N.d. Neuroprotective mechanisms of berry bioavailable polyphenol metabolites. In Berries and Berry Bioactive Compounds in Promoting Health, 1st ed.; Klimis-Zacas, D., Rodriguez-Mateos, A., Eds.; Royal Society of Chemistry: London, UK, 2022. [Google Scholar]
- García-Aguilar, A.; Palomino, O.; Benito, M.; Guillén, C. Dietary Polyphenols in Metabolic and Neurodegenerative Diseases: Molecular Targets in Autophagy and Biological Effects. Antioxidants 2021, 10, 142. [Google Scholar] [CrossRef]
- Novick, P.; Field, C.; Schekman, R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 1980, 21, 205–215. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Moulton, M.J.; Letsou, A. Modeling congenital disease and inborn errors of development in Drosophila melanogaster. Dis. Models Mech. 2016, 9, 253–269. [Google Scholar] [CrossRef]
- Wangler, M.F.; Yamamoto, S.; Chao, H.-T.; Posey, J.E.; Westerfield, M.; Postlethwait, J.; Hieter, P.; Boycott, K.M.; Campeau, P.M.; Bellen, H.J. Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics 2017, 207, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.V.; Vilaça, R.; Santos, C.N.; Costa, V.; Menezes, R. Exploring the power of yeast to model aging and age-related neurodegenerative disorders. Biogerontology 2016, 18, 3–34. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Ramos, R.; Godinho-Pereira, J.; Figueira, I.; Jardim, C.; Garcia, G.; Menezes, R. Exploring the Benefits of Cellular Models to Uncover Bioactive Polyphenols for Neurodegeneration. Curr. Pharm. Des. 2018, 24, 2076–2106. [Google Scholar] [CrossRef]
- Tenreiro, S.; Outeiro, T.F. Simple is good: Yeast models of neurodegeneration. FEMS Yeast Res. 2010, 10, 970–979. [Google Scholar] [CrossRef]
- Snel, B.; Heinicke, S.; Livstone, M.S.; Lu, C.; Oughtred, R.; Kang, F.; Angiuoli, S.V.; White, O.; Botstein, D.; Dolinski, K. The Princeton Protein Orthology Database (P-POD): A Comparative Genomics Analysis Tool for Biologists. PLoS ONE 2007, 2, e766. [Google Scholar] [CrossRef]
- Tardiff, D.F.; Khurana, V.; Chung, C.Y.; Lindquist, S. From yeast to patient neurons and back again: Powerful new discovery platforms. Mov. Disord. 2014, 29, 1231–1240. [Google Scholar] [CrossRef]
- Jha, N.K.; Kar, R.; Niranjan, R. ABC Transporters in Neurological Disorders: An Important Gateway for Botanical Compounds Mediated Neuro-Therapeutics. Curr. Top. Med. Chem. 2019, 19, 795–811. [Google Scholar] [CrossRef]
- Hoosain, F.G.; Choonara, Y.E.; Tomar, L.K.; Kumar, P.; Tyagi, C.; du Toit, L.C.; Pillay, V. Bypassing P-Glycoprotein Drug Efflux Mechanisms: Possible Applications in Pharmacoresistant Schizophrenia Therapy. BioMed Res. Int. 2015, 2015, 484963. [Google Scholar] [CrossRef]
- Pereira Rangel, L.; Fritzen, M.; Yunes, R.A.; Leal, P.C.; Creczynski-Pasa, T.B.; Ferreira-Pereira, A. Inhibitory effects of gallic acid ester derivatives on Saccharomyces cerevisiaemultidrug resistance protein Pdr5p. FEMS Yeast Res. 2010, 10, 244–251. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hatzixanthis, K.; Mollapour, M.; Seymour, I.; Bauer, B.E.; Krapf, G.; Schüller, C.; Kuchler, K.; Piper, P.W. Moderately lipophilic carboxylate compounds are the selective inducers of the Saccharomyces cerevisiae Pdr12p ATP-binding cassette transporter. Yeast 2003, 20, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Kodo, N.; Matsuda, T.; Doi, S.; Munakata, H. Salicylic acid resistance is conferred by a novel YRR1 mutation in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2013, 434, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Piehler, A.P.; Özcürümez, M.; Kaminski, W.E. A-Subclass ATP-Binding Cassette Proteins in Brain Lipid Homeostasis and Neurodegeneration. Front. Psychiatry 2012, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Lehmkuhl, E.M.; Zarnescu, D.C. Lost in Translation: Evidence for Protein Synthesis Deficits in ALS/FTD and Related Neurodegenerative Diseases. Adv. Neurobiol. 2018, 20, 283–301. [Google Scholar]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; Lummertz da Rocha, E.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef]
- Hazan, R.; Levine, A.; Abeliovich, H. Benzoic Acid, a Weak Organic Acid Food Preservative, Exerts Specific Effects on Intracellular Membrane Trafficking Pathways in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2004, 70, 4449–4457. [Google Scholar] [CrossRef] [PubMed]
- Granneman, S.; Iwaki, A.; Ohnuki, S.; Suga, Y.; Izawa, S.; Ohya, Y. Vanillin Inhibits Translation and Induces Messenger Ribonucleoprotein (mRNP) Granule Formation in Saccharomyces cerevisiae: Application and Validation of High-Content, Image-Based Profiling. PLoS ONE 2013, 8, e61748. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Kitajima, S.; Izawa, S. Importance of glucose-6-phosphate dehydrogenase (G6PDH) for vanillin tolerance in Saccharomyces cerevisiae. J. Biosci. Bioeng. 2014, 118, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Bagriantsev, S.; Liebman, S. Modulation of Aβ42low-n oligomerization using a novel yeast reporter system. BMC Biol. 2006, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Barrientos, A. Developing Yeast Models of Human Neurodegenerative Disorders. Methods Mol. Biol. 2011, 793, 113–127. [Google Scholar] [PubMed]
- Tuite, M.F. Yeast models of neurodegenerative diseases. Prog. Mol. Biol. Transl. Sci. 2019, 168, 351–379. [Google Scholar] [PubMed]
- Porzoor, A.; Alford, B.; Hügel, H.; Grando, D.; Caine, J.; Macreadie, I. Anti-Amyloidogenic Properties of Some Phenolic Compounds. Biomolecules 2015, 5, 505–527. [Google Scholar] [CrossRef]
- Mendes, V.; Vilaça, R.; de Freitas, V.; Ferreira, P.M.; Mateus, N.; Costa, V. Effect of Myricetin, Pyrogallol, and Phloroglucinol on Yeast Resistance to Oxidative Stress. Oxidative Med. Cell. Longev. 2015, 2015, 782504. [Google Scholar] [CrossRef]
- Sunthonkun, P.; Palajai, R.; Somboon, P.; Suan, C.L.; Ungsurangsri, M.; Soontorngun, N. Life-span extension by pigmented rice bran in the model yeast Saccharomyces cerevisiae. Sci. Rep. 2019, 9, 18061. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Iwaki, A.; Ohya, Y.; Izawa, S. Vanillin causes the activation of Yap1 and mitochondrial fragmentation in Saccharomyces cerevisiae. J. Biosci. Bioeng. 2014, 117, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Polymenis, M.; North, M.; Tandon, V.J.; Thomas, R.; Loguinov, A.; Gerlovina, I.; Hubbard, A.E.; Zhang, L.; Smith, M.T.; Vulpe, C.D. Genome-Wide Functional Profiling Reveals Genes Required for Tolerance to Benzene Metabolites in Yeast. PLoS ONE 2011, 6, e24205. [Google Scholar] [CrossRef]
- Fletcher, E.; Gao, K.; Mercurio, K.; Ali, M.; Baetz, K. Yeast chemogenomic screen identifies distinct metabolic pathways required to tolerate exposure to phenolic fermentation inhibitors ferulic acid, 4-hydroxybenzoic acid and coniferyl aldehyde. Metab. Eng. 2019, 52, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, F.; Guidetti, P.; Nguyen, Q.; Bennett, S.C.; Muchowski, P.J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005, 37, 526–531. [Google Scholar] [CrossRef]
- Mohammadi, S.; Saberidokht, B.; Subramaniam, S.; Grama, A. Scope and limitations of yeast as a model organism for studying human tissue-specific pathways. BMC Syst. Biol. 2015, 9, 96. [Google Scholar] [CrossRef]
- Braun, R.J.; Büttner, S.; Ring, J.; Kroemer, G.; Madeo, F. Nervous yeast: Modeling neurotoxic cell death. Trends Biochem. Sci. 2010, 35, 135–144. [Google Scholar] [CrossRef]
- Khurana, V.; Lindquist, S. Modelling neurodegeneration in Saccharomyces cerevisiae: Why cook with baker’s yeast? Nat. Rev. Neurosci. 2010, 11, 436–449. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Iwaki, A.; Izawa, S. The ADH7 Promoter of Saccharomyces cerevisiae is Vanillin-Inducible and Enables mRNA Translation Under Severe Vanillin Stress. Front. Microbiol. 2015, 6, 1390. [Google Scholar] [CrossRef]
- Ruta, L.L.; Farcasanu, I.C. Interaction between Polyphenolic Antioxidants and Saccharomyces cerevisiae Cells Defective in Heavy Metal Transport across the Plasma Membrane. Biomolecules 2020, 10, 1512. [Google Scholar] [CrossRef] [PubMed]
- Mori, I.C.; Iida, H.; Tsuji, F.I.; Isobe, M.; Uozumi, N.; Muto, S. Salicylic Acid Induces a Cytosolic Ca2+Elevation in Yeast. Biosci. Biotechnol. Biochem. 2014, 62, 986–989. [Google Scholar] [CrossRef]
- Hazelwood, L.A.; Tai, S.L.; Boer, V.M.; de Winde, J.H.; Pronk, J.T.; Daran, J.M. A new physiological role for Pdr12p in Saccharomyces cerevisiae: Export of aromatic and branched-chain organic acids produced in amino acid catabolism. FEMS Yeast Res. 2006, 6, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Sundström, L.; Larsson, S.; Jönsson, L.J. Identification of Saccharomyces cerevisiae Genes Involved in the Resistance to Phenolic Fermentation Inhibitors. Appl. Biochem. Biotechnol. 2009, 161, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Campbell, B.C.; Mahoney, N.; Chan, K.L.; Molyneux, R.J.; May, G.S. Enhancement of fludioxonil fungicidal activity by disrupting cellular glutathione homeostasis with 2,5-dihydroxybenzoic acid. FEMS Microbiol. Lett. 2007, 270, 284–290. [Google Scholar] [CrossRef]
- Piper, M.D.W.; Partridge, L. Drosophila as a model for ageing. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2707–2717. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barclay, J.W.; Burgoyne, R.D.; Morgan, A. Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem. Cent. J. 2015, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Kamireddy, K.; Chinnu, S.; Priyanka, P.S.; Rajini, P.S.; Giridhar, P. Neuroprotective effect of Decalepis hamiltonii aqueous root extract and purified 2-hydroxy-4-methoxy benzaldehyde on 6-OHDA induced neurotoxicity in Caenorhabditis elegans. Biomed. Pharmacother. 2018, 105, 997–1005. [Google Scholar] [CrossRef]
- Visentin, C.; Pellistri, F.; Natalello, A.; Vertemara, J.; Bonanomi, M.; Gatta, E.; Penco, A.; Relini, A.; De Gioia, L.; Airoldi, C.; et al. Epigallocatechin-3-gallate and related phenol compounds redirect the amyloidogenic aggregation pathway of ataxin-3 towards non-toxic aggregates and prevent toxicity in neural cells and Caenorhabditis elegans animal model. Hum. Mol. Genet. 2017, 26, 3271–3284. [Google Scholar] [CrossRef]
- Hao, S.; Li, X.; Han, A.; Yang, Y.; Luo, X.; Fang, G.; Wang, H.; Liu, J.; Wang, S. Hydroxycinnamic Acid from Corncob and Its Structural Analogues Inhibit Aβ40 Fibrillation and Attenuate Aβ40-Induced Cytotoxicity. J. Agric. Food Chem. 2020, 68, 8788–8796. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, K.; Saul, N.; Chakrabarti, S.; Stürzenbaum, S.R.; Menzel, R.; Steinberg, C.E.W. Hormetins, antioxidants and prooxidants: Defining quercetin-, caffeic acid- and rosmarinic acid-mediated life extension in C. elegans. Biogerontology 2011, 12, 329–347. [Google Scholar] [CrossRef]
- Gutierrez-Zetina, S.M.; González-Manzano, S.; Ayuda-Durán, B.; Santos-Buelga, C.; González-Paramás, A.M. Caffeic and Dihydrocaffeic Acids Promote Longevity and Increase Stress Resistance in Caenorhabditis elegans by Modulating Expression of Stress-Related Genes. Molecules 2021, 26, 1517. [Google Scholar] [CrossRef]
- Jagota, S.; Rajadas, J. Effect of Phenolic Compounds Against Aβ Aggregation and Aβ-Induced Toxicity in Transgenic C. elegans. Neurochem. Res. 2011, 37, 40–48. [Google Scholar] [CrossRef]
- Nagpal, I.; Abraham, S.K. Ameliorative effects of gallic acid, quercetin and limonene on urethane-induced genotoxicity and oxidative stress inDrosophila melanogaster. Toxicol. Mech. Methods 2017, 27, 286–292. [Google Scholar] [CrossRef]
- Jimenez-Del-Rio, M.; Guzman-Martinez, C.; Velez-Pardo, C. The Effects of Polyphenols on Survival and Locomotor Activity in Drosophila melanogaster Exposed to Iron and Paraquat. Neurochem. Res. 2009, 35, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Arellano, H.F.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Life Span and Locomotor Activity Modification by Glucose and Polyphenols in Drosophila melanogaster Chronically Exposed to Oxidative Stress-stimuli: Implications in Parkinson’s Disease. Neurochem. Res. 2011, 36, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Bonilla-Ramirez, L.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Low doses of paraquat and polyphenols prolong life span and locomotor activity in knock-down parkin Drosophila melanogaster exposed to oxidative stress stimuli: Implication in autosomal recessive juvenile Parkinsonism. Gene 2013, 512, 355–363. [Google Scholar] [CrossRef]
- Ogunsuyi, O.B.; Oboh, G.; Oluokun, O.O.; Ademiluyi, A.O.; Ogunruku, O.O. Gallic acid protects against neurochemical alterations in transgenic Drosophila model of Alzheimer’s disease. Adv. Tradit. Med. 2019, 20, 89–98. [Google Scholar] [CrossRef]
- Li, J.-q.; Fang, J.-s.; Qin, X.-m.; Gao, L. Metabolomics profiling reveals the mechanism of caffeic acid in extending lifespan in Drosophila melanogaster. Food Funct. 2020, 11, 8202–8213. [Google Scholar] [CrossRef]
- Peng, C.; Chan, H.Y.E.; Huang, Y.; Yu, H.; Chen, Z.-Y. Apple Polyphenols Extend the Mean Lifespan of Drosophila melanogaster. J. Agric. Food Chem. 2011, 59, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zuo, Y.; Kwan, K.M.; Liang, Y.; Ma, K.Y.; Chan, H.Y.E.; Huang, Y.; Yu, H.; Chen, Z.-Y. Blueberry extract prolongs lifespan of Drosophila melanogaster. Exp. Gerontol. 2012, 47, 170–178. [Google Scholar] [CrossRef]
- Zuo, Y.; Peng, C.; Liang, Y.; Ma, K.Y.; Yu, H.; Edwin Chan, H.Y.; Chen, Z.-Y. Black rice extract extends the lifespan of fruit flies. Food Funct. 2012, 3, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Chan, H.Y.E.; Huang, Y.; Chen, Z.Y. Green tea catechins upregulate superoxide dismutase and catalase in fruit flies. Mol. Nutr. Food Res. 2007, 51, 546–554. [Google Scholar] [CrossRef]
- Varga, J.; Dér, N.P.; Zsindely, N.; Bodai, L. Green tea infusion alleviates neurodegeneration induced by mutant Huntingtin in Drosophila. Nutr. Neurosci. 2018, 23, 183–189. [Google Scholar] [CrossRef]
- Kim, S.J.; Han, D.; Ahn, B.-H.; Rhee, J.S. Effect of Glutathione, Catechin, and Epicatechin on the Survival of Drosophila melanogasterunder Paraquat Treatment. Biosci. Biotechnol. Biochem. 2014, 61, 225–229. [Google Scholar] [CrossRef]
- Si, H.; Fu, Z.; Babu, P.V.A.; Zhen, W.; LeRoith, T.; Meaney, M.P.; Voelker, K.A.; Jia, Z.; Grange, R.W.; Liu, D. Dietary Epicatechin Promotes Survival of Obese Diabetic Mice and Drosophila melanogaster. J. Nutr. 2011, 141, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.E.; Piegholdt, S.; Rabe, D.; Baenas, N.; Schloesser, A.; Eggersdorfer, M.; Stocker, A.; Rimbach, G. Epigallocatechin gallate affects glucose metabolism and increases fitness and lifespan in Drosophila melanogaster. Oncotarget 2015, 6, 30568–30578. [Google Scholar] [CrossRef] [PubMed]
- Siddique, Y.H.; Jyoti, S.; Naz, F. Effect of Epicatechin Gallate Dietary Supplementation on Transgenic Drosophila Model of Parkinson’s Disease. J. Diet. Suppl. 2014, 11, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Perez, D.A.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Epigallocatechin-3-Gallate Protects and Prevents Paraquat-Induced Oxidative Stress and Neurodegeneration in Knockdown dj-1-β Drosophila melanogaster. Neurotox. Res. 2018, 34, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Carregosa, D.; Carecho, R.; Figueira, I.; N Santos, C. Low-Molecular Weight Metabolites from Polyphenols as Effectors for Attenuating Neuroinflammation. J. Agric. Food Chem. 2020, 7, 1790–1807. [Google Scholar] [CrossRef]
- Mueller, T.; Wullimann, M.F. An Evolutionary Interpretation of Teleostean Forebrain Anatomy. Brainbehav. Evol. 2009, 74, 30–42. [Google Scholar] [CrossRef]
- Wullimann, M.F.; Rupp, B.; Reichert, H. Neuroanatomy of the Zebrafish Brain; Springer: Berlin/Heidelberg, Germany, 1996. [Google Scholar]
- Kily, L.J.M.; Cowe, Y.C.M.; Hussain, O.; Patel, S.; McElwaine, S.; Cotter, F.E.; Brennan, C.H. Gene expression changes in a zebrafish model of drug dependency suggest conservation of neuro-adaptation pathways. J. Exp. Biol. 2008, 211, 1623–1634. [Google Scholar] [CrossRef]
- Mueller, T.; Vernier, P.; Wullimann, M.F. The adult central nervous cholinergic system of a neurogenetic model animal, the zebrafish Danio rerio. Brain Res. 2004, 1011, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Lee, Y.; Kim, Y.; Lee, C.-J. Cholinergic modulation of neural activity in the telencephalon of the zebrafish. Neurosci. Lett. 2008, 439, 79–83. [Google Scholar] [CrossRef]
- Agetsuma, M.; Aizawa, H.; Aoki, T.; Nakayama, R.; Takahoko, M.; Goto, M.; Sassa, T.; Amo, R.; Shiraki, T.; Kawakami, K.; et al. The habenula is crucial for experience-dependent modification of fear responses in zebrafish. Nat. Neurosci. 2010, 13, 1354–1356. [Google Scholar] [CrossRef]
- Mahabir, S.; Chatterjee, D.; Buske, C.; Gerlai, R. Maturation of shoaling in two zebrafish strains: A behavioral and neurochemical analysis. Behav. Brain Res. 2013, 247, 1–8. [Google Scholar] [CrossRef]
- Qin, M.; Wong, A.; Seguin, D.; Gerlai, R. Induction of Social Behavior in Zebrafish: Live Versus Computer Animated Fish as Stimuli. Zebrafish 2014, 11, 185–197. [Google Scholar] [CrossRef]
- Valente, A.; Huang, K.H.; Portugues, R.; Engert, F. Ontogeny of classical and operant learning behaviors in zebrafish. Learn. Mem. 2012, 19, 170–177. [Google Scholar] [CrossRef]
- Fontana, B.D.; Mezzomo, N.J.; Kalueff, A.V.; Rosemberg, D.B. The developing utility of zebrafish models of neurological and neuropsychiatric disorders: A critical review. Exp. Neurol. 2018, 299, 157–171. [Google Scholar] [CrossRef]
- Sager, J.J.; Bai, Q.; Burton, E.A. Transgenic zebrafish models of neurodegenerative diseases. Brain Struct. Funct. 2010, 214, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Saleem, S.; Kannan, R.R. Zebrafish: An emerging real-time model system to study Alzheimer’s disease and neurospecific drug discovery. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Vaz, R.L.; Outeiro, T.F.; Ferreira, J.J. Zebrafish as an Animal Model for Drug Discovery in Parkinson’s Disease and Other Movement Disorders: A Systematic Review. Front. Neurol. 2018, 9, 347. [Google Scholar] [CrossRef]
- Ahrens, M.B.; Orger, M.B.; Robson, D.N.; Li, J.M.; Keller, P.J. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 2013, 10, 413–420. [Google Scholar] [CrossRef]
- Peri, F.; Nüsslein-Volhard, C. Live Imaging of Neuronal Degradation by Microglia Reveals a Role for v0-ATPase a1 in Phagosomal Fusion In Vivo. Cell 2008, 133, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Sieger, D.; Moritz, C.; Ziegenhals, T.; Prykhozhij, S.; Peri, F. Long-Range Ca2+ Waves Transmit Brain-Damage Signals to Microglia. Dev. Cell 2012, 22, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Duan, X.; Jacobs, C.; Ullmann, J.; Chan, C.-Y.; Chen, S.; Cheng, S.-H.; Zhao, W.-N.; Poduri, A.; Wang, X.; et al. High-throughput brain activity mapping and machine learning as a foundation for systems neuropharmacology. Nat. Commun. 2018, 9, 5142. [Google Scholar] [CrossRef] [PubMed]
- Burgess, H.A.; Liu, H.; Chen, S.; Huang, K.; Kim, J.; Mo, H.; Iovine, R.; Gendre, J.; Pascal, P.; Li, Q.; et al. A High-Content Larval Zebrafish Brain Imaging Method for Small Molecule Drug Discovery. PLoS ONE 2016, 11, e0164645. [Google Scholar] [CrossRef]
- Silva, N.-V.; Carregosa, D.; Gonçalves, C.; Vieira, O.V.; Nunes dos Santos, C.; Jacinto, A.; Crespo, C.L. A Dietary Cholesterol-Based Intestinal Inflammation Assay for Improving Drug-Discovery on Inflammatory Bowel Diseases. Front. Cell Dev. Biol. 2021, 9, 674749. [Google Scholar] [CrossRef] [PubMed]
- Pitchai, A.; Rajaretinam, R.K.; Freeman, J.L. Zebrafish as an Emerging Model for Bioassay-Guided Natural Product Drug Discovery for Neurological Disorders. Medicines 2019, 6, 61. [Google Scholar] [CrossRef]
- Annona, G.; Tarallo, A.; Nittoli, V.; Varricchio, E.; Sordino, P.; D’Aniello, S.; Paolucci, M. Short-term exposure to the simple polyphenolic compound gallic acid induces neuronal hyperactivity in zebrafish larvae. Eur. J. Neurosci. 2020, 53, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Agostini, J.F.; Santo, G.D.; Baldin, S.L.; Bernardo, H.T.; de Farias, A.C.S.; Rico, E.P.; Wanderley, A.G. Gallic Acid Reverses Neurochemical Changes Induced by Prolonged Ethanol Exposure in the Zebrafish Brain. Neuroscience 2021, 455, 251–262. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, G.; Szeto, S.S.W.; Chong, C.M.; Quan, Q.; Huang, C.; Cui, W.; Guo, B.; Wang, Y.; Han, Y.; et al. Examining the neuroprotective effects of protocatechuic acid and chrysin on in vitro and in vivo models of Parkinson disease. Free Radic. Biol. Med. 2015, 84, 331–343. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Cheang, L.C.V.; Wang, M.-W.; Li, G.-H.; Chu, I.K.; Lin, Z.-X.; Lee, S.M.Y. Ethanolic Extract of Fructus Alpinia oxyphylla Protects Against 6-Hydroxydopamine-Induced Damage of PC12 Cells In Vitro and Dopaminergic Neurons in Zebrafish. Cell. Mol. Neurobiol. 2011, 32, 27–40. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Han, X.; Cao, M.; Tan, Z.; Liu, W. Design, Synthesis, and Evaluation of Novel Ferulic Acid Derivatives as Multi-Target-Directed Ligands for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 10, 1008–1024. [Google Scholar] [CrossRef]
- Tsay, H.-J.; Wang, Y.-H.; Chen, W.-L.; Huang, M.-Y.; Chen, Y.-H. Treatment with sodium benzoate leads to malformation of zebrafish larvae. Neurotoxicol. Teratol. 2007, 29, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Huang, N.-N.; Huang, J.-T.; Chen, S.; Fan, J.; Li, C.; Xie, F.-K. Sodium benzoate exposure downregulates the expression of tyrosine hydroxylase and dopamine transporter in dopaminergic neuronsin developing zebrafish. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2009, 86, 85–91. [Google Scholar] [CrossRef]
- Gaur, H.; Purushothaman, S.; Pullaguri, N.; Bhargava, Y.; Bhargava, A. Sodium benzoate induced developmental defects, oxidative stress and anxiety-like behaviour in zebrafish larva. Biochem. Biophys. Res. Commun. 2018, 502, 364–369. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Chen, L.C.V.; Wang, M.W. Quercetin exerts a neuroprotective effect through inhibition of the iNOS/NO system and pro-inflammation gene expression in PC12 cells and in zebrafish. Int. J. Mol. Med. 2011, 27, 195–203. [Google Scholar] [CrossRef]
- Komorowska, J.; Wątroba, M.; Szukiewicz, D. Review of beneficial effects of resveratrol in neurodegenerative diseases such as Alzheimer’s disease. Adv. Med Sci. 2020, 65, 415–423. [Google Scholar] [CrossRef]
- Peñalver, P.; Belmonte-Reche, E.; Adán, N.; Caro, M.; Mateos-Martín, M.L.; Delgado, M.; González-Rey, E.; Morales, J.C. Alkylated resveratrol prodrugs and metabolites as potential therapeutics for neurodegenerative diseases. Eur. J. Med. Chem. 2018, 146, 123–138. [Google Scholar] [CrossRef]
- Juvekar, A.; Khatri, D. Abrogation of locomotor impairment in a rotenone-induced Drosophila melanogaster and zebrafish model of Parkinson′s disease by ellagic acid and curcumin. Int. J. Nutr. Pharmacol. Neurol. Dis. 2016, 6, 90–96. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, Z.; Xia, J.; Zhang, X.; Liu, K.; Sik, A.; Jin, M. Anti-Parkinson’s disease activity of phenolic acids from Eucommia ulmoides Oliver leaf extracts and their autophagy activation mechanism. Food Funct. 2020, 11, 1425–1440. [Google Scholar] [CrossRef]
- Song, Y.; Cui, T.; Xie, N.; Zhang, X.; Qian, Z.; Liu, J. Protocatechuic acid improves cognitive deficits and attenuates amyloid deposits, inflammatory response in aged AβPP/PS1 double transgenic mice. Int. Immunopharmacol. 2014, 20, 276–281. [Google Scholar] [CrossRef]
- Yu, M.; Chen, X.; Liu, J.; Ma, Q.; Zhuo, Z.; Chen, H.; Zhou, L.; Yang, S.; Zheng, L.; Ning, C.; et al. Gallic acid disruption of Aβ1–42 aggregation rescues cognitive decline of APP/PS1 double transgenic mouse. Neurobiol. Dis. 2019, 124, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Ali, T.; Alam, S.I.; Ullah, R.; Zeb, A.; Lee, K.W.; Rutten, B.P.F.; Kim, M.O. Ferulic Acid Rescues LPS-Induced Neurotoxicity via Modulation of the TLR4 Receptor in the Mouse Hippocampus. Mol. Neurobiol. 2018, 56, 2774–2790. [Google Scholar] [CrossRef] [PubMed]
- Ojha, S.; Javed, H.; Azimullah, S.; Abul Khair, S.B.; Haque, E. Neuroprotective potential of ferulic acid in the rotenone model of Parkinson’s disease. Drug Des. Dev. Ther. 2015, 9, 5499–5510. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Sarkaki, A.; Khorsandi, L.; Winlow, W.; Badavi, M.; Moghaddam, H.F.; Farbood, Y. Vanillic acid attenuates effects of transient bilateral common carotid occlusion and reperfusion in rats. Biomed. Pharmacother. 2017, 96, 667–674. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Sarkaki, A.; Rashno, M.; Farbood, Y. Memory deficits and hippocampal inflammation in cerebral hypoperfusion and reperfusion in male rats: Neuroprotective role of vanillic acid. Life Sci. 2018, 211, 126–132. [Google Scholar] [CrossRef]
- Tsai, S.-j.; Yin, M.-c. Anti-glycative and anti-inflammatory effects of protocatechuic acid in brain of mice treated by d-galactose. Food Chem. Toxicol. 2012, 50, 3198–3205. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.; Choi, B.; Lee, S.; Hong, D.; Lee, S.; Jeong, J.; Park, K.-H.; Song, H.; Choi, H.; Suh, S. Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. Int. J. Mol. Sci. 2018, 19, 1420. [Google Scholar] [CrossRef]
- Amin, F.U.; Shah, S.A.; Kim, M.O. Vanillic acid attenuates Aβ1-42-induced oxidative stress and cognitive impairment in mice. Sci. Rep. 2017, 7, 753. [Google Scholar] [CrossRef]
- Kim, M.-J.; Seong, A.-R.; Yoo, J.-Y.; Jin, C.-H.; Lee, Y.-H.; Kim, Y.J.; Lee, J.; Jun, W.J.; Yoon, H.-G. Gallic acid, a histone acetyltransferase inhibitor, suppresses β-amyloid neurotoxicity by inhibiting microglial-mediated neuroinflammation. Mol. Nutr. Food Res. 2011, 55, 1798–1808. [Google Scholar] [CrossRef]
- Adefegha, S.A.; Oboh, G.; Omojokun, O.S.; Adefegha, O.M. Alterations of Na+/K+-ATPase, cholinergic and antioxidant enzymes activity by protocatechuic acid in cadmium-induced neurotoxicity and oxidative stress in Wistar rats. Biomed. Pharmacother. 2016, 83, 559–568. [Google Scholar] [CrossRef]
- Shi, G.-F.; An, L.-J.; Jiang, B.; Guan, S.; Bao, Y.-M. Alpinia protocatechuic acid protects against oxidative damage in vitro and reduces oxidative stress in vivo. Neurosci. Lett. 2006, 403, 206–210. [Google Scholar] [CrossRef]
- Mansouri, M.T.; Farbood, Y.; Sameri, M.J.; Sarkaki, A.; Naghizadeh, B.; Rafeirad, M. Neuroprotective effects of oral gallic acid against oxidative stress induced by 6-hydroxydopamine in rats. Food Chem. 2013, 138, 1028–1033. [Google Scholar] [CrossRef]
- Naghizadeh, B.; Mansouri, M. Protective Effects of Gallic Acid against Streptozotocin-induced Oxidative Damage in Rat Striatum. Drug Res. 2014, 65, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.C.H.; Kakalij, R.M.; Kshirsagar, R.P.; Kumar, B.H.; Komakula, S.S.B.; Diwan, P.V. Cognitive effects of vanillic acid against streptozotocin-induced neurodegeneration in mice. Pharm. Biol. 2014, 53, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Zhang, R.; Li, Y.; Li, Y.; Yang, Z.; Yang, H. Ferulic acid exerts neuroprotective effects against cerebral ischemia/reperfusion-induced injury via antioxidant and anti-apoptotic mechanisms in vitro and in vivo. Int. J. Mol. Med. 2017, 40, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.H.; Phopin, K.; Suwanjang, W.; Songtawee, N.; Ruankham, W.; Wongchitrat, P.; Prachayasittikul, S.; Prachayasittikul, V. Neuroprotective Effects of Phenolic and Carboxylic Acids on Oxidative Stress-Induced Toxicity in Human Neuroblastoma SH-SY5Y Cells. Neurochem. Res. 2018, 43, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Rekha, K.R.; Selvakumar, G.P.; Sivakamasundari, R.I. Effects of syringic acid on chronic MPTP/probenecid induced motor dysfunction, dopaminergic markers expression and neuroinflammation in C57BL/6 mice. Biomed. Aging Pathol. 2014, 4, 95–104. [Google Scholar] [CrossRef]
- Prorok, T.; Jana, M.; Patel, D.; Pahan, K. Cinnamic Acid Protects the Nigrostriatum in a Mouse Model of Parkinson’s Disease via Peroxisome Proliferator-Activated Receptorα. Neurochem. Res. 2019, 44, 751–762. [Google Scholar] [CrossRef]
- Chandra, S.; Roy, A.; Jana, M.; Pahan, K. Cinnamic acid activates PPARα to stimulate Lysosomal biogenesis and lower Amyloid plaque pathology in an Alzheimer’s disease mouse model. Neurobiol. Dis. 2019, 124, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Zaitone, S.A.; Ahmed, E.; Elsherbiny, N.M.; Mehanna, E.T.; El-Kherbetawy, M.K.; ElSayed, M.H.; Alshareef, D.M.; Moustafa, Y.M. Caffeic acid improves locomotor activity and lessens inflammatory burden in a mouse model of rotenone-induced nigral neurodegeneration: Relevance to Parkinson’s disease therapy. Pharmacol. Rep. 2019, 71, 32–41. [Google Scholar] [CrossRef]
- Güven, M.; Aras, A.B.; Topaloğlu, N.; Özkan, A.; Şen, H.M.; Kalkan, Y.; Okuyucu, A.; Akbal, A.; Gökmen, F.; Coşar, M. The protective effect of syringic acid on ischemia injury in rat brain. Turk. J. Med Sci. 2015, 45, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Khoshnam, S.E.; Farbood, Y.; Fathi Moghaddam, H.; Sarkaki, A.; Badavi, M.; Khorsandi, L. Vanillic acid attenuates cerebral hyperemia, blood-brain barrier disruption and anxiety-like behaviors in rats following transient bilateral common carotid occlusion and reperfusion. Metab. Brain Dis. 2018, 33, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Huang, D.; Lo, Y.M.; Tee, Q.; Kuo, P.; Wu, J.S.; Huang, W.; Shen, S. Protective Effect of Caffeic Acid against Alzheimer’s Disease Pathogenesis via Modulating Cerebral Insulin Signaling, β-Amyloid Accumulation, and Synaptic Plasticity in Hyperinsulinemic Rats. J. Agric. Food Chem. 2019, 67, 7684–7693. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Y.; Yue Zhang, S. Vanillic Acid Improve Neural Function after Focal Cerebral Ischemia-reperfusion Rats. Int. J. Pharmacol. 2018, 14, 488–494. [Google Scholar] [CrossRef]
- Huang, H.-L.; Lin, C.-C.; Jeng, K.-C.G.; Yao, P.-W.; Chuang, L.-T.; Kuo, S.-L.; Hou, C.-W. Fresh Green Tea and Gallic Acid Ameliorate Oxidative Stress in Kainic Acid-Induced Status Epilepticus. J. Agric. Food Chem. 2012, 60, 2328–2336. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Li, J.; Hua, L.; Han, B.; Zhang, Y.; Yang, X.; Zeng, Z.; Bai, H.; Yin, H.; et al. Effects of caffeic acid on learning deficits in a model of Alzheimer’s disease. Int. J. Mol. Med. 2016, 38, 869–875. [Google Scholar] [CrossRef]



| (Poly)phenol Metabolite 1 | Strain | Model | Dose and Time | Mechanistic Evidences | Ref. |
|---|---|---|---|---|---|
| Benzoic Acid | |||||
| 3,4,5—Trihydroxybenzoic Acid (Gallic Acid) | WT Canton–S | PD Chemical Model Induced by PQ | 0.1 mM in 0.1% Sucrose | ↑ Lifespan ↑ Locomotor Activity | [106] |
| Tg (TH—GAL4: UAS—Dmp53 RNAi)Canton–S Background | PD Chemical Model Induced by PQ | 0.1 mM | ↑ Life Span ↑ Locomotor Activity | [107] | |
| Parkin Knockout Tg (TH—Gal4: UAS—ParkinRNAi) | PD Genetic Model Chronically Exposed to PQ | 0.1 or 0.5 mM Alone or With PQ | No Rescue in Lifespan or Locomotor Activity | [108] | |
| Tg (UAS:hAPP, UAS:BACE1) | AD Genetic Model | 50 or 100 µM | ↑ Catalase ↓Malonaldehyde ↓AChE | [109] | |
| WT Oregon–K | Urethane Exposure Genotoxic Environmental Carcinogen Model | 5.88 M + Urethaen 20 mM in 10% Sucrose for 72 h 2 | ↑ Antigenotoxic and Antioxidant Role ↑ Glutathione S–Transferase ↑ SOD ↑ Catalase | [105] | |
| Cinnamic Acid | |||||
| 4—Hydroxy–3—Methoxycinnamic Acid (Ferulic Acid) | wt Canton–S | pd Chemical Mode l20 mm pq Exposure for 24 h | 0.1 mM in 0.1% Sucrose | ↑ Lifespan ↑ Locomotor Activity | [106] |
| 3,4—Dihydroxycinnamic Acid (Caffeic Acid) | wt Canton–S | pd Chemical Model of pq Exposure for 24 h or 48 h | 0.5 and 1 mM in 0.1% Sucrose | ↑ Lifespan ↑ Locomotive Activity | [106] |
| WT W1118 | 1.1 mM to 5.55 mM 3 | ↑ Lifespan ↑ Climbing Behavior ↑ Stress Resistance (Heat and Starvation). ↑ Mitochondrial Function ↑ Antioxidant Capacity | [110] | ||
| (Poly)phenol Metabolite 1 | Species | Model | Dose and Time | Mechanistic Evidence | Ref. |
|---|---|---|---|---|---|
| Benzoic acids | |||||
| 4—hydroxy–3—methoxybenzoic Acid (Vanillic Acid) | Sprague-Dawley rat | Cerebral Ischemia-Reperfusion (I/R) | 50 and 100 mg/kg Gavage for 14 Days | ↓ NF—κB p65 Protein Levels ↓ IL—6, IL—1β and TNF—α | [179] |
| C57BL/6N Mice | Intracerebroventricular (i.c.v.) Injection of Aβ1-42 | 30 mg/kg i.p. for 3 Weeks | ↓ Number of Activated Microglia and Astrocytes Cells ↓ iNOS Expression ↓ NF—kB | [163] | |
| Wistar Rat | Carotid Artery Occlusion and Reperfusion in Rat | 100 mg/kg, Gavage for 14 Days | ↓ IL—6, TNF—α and TUNEL Positive Cells. ↑ IL—10 Levels in the Hippocampus | [177] | |
| Swiss Albino Mice | Streptozotocin (STZ)–Induced Neurodegeneration | 25, 50, and 100 mg/kg Orally for 28 Days | ↑ Spatial Learning and Memory Retention ↓ Oxidative Stress ↓ AChE, Corticosterone, TNF—α ↑ SOD, CAT and GPx Levels | [169] | |
| Wistar Rat | Transient Bilateral Common Carotid Artery Occlusion and Reperfusion | 10, 30, 100 mg/kg Gavage for 2 Weeks | Attenuation of Reactive Hyperemia and BBB Disruption. ↑ Sensory Motor Signs and Anxiolytic Behavior. | [177] | |
| Wistar Rat | Transient Bilateral Common Carotid Artery Occlusion and Reperfusion | 100 mg/kg Gavage for 2 Weeks | ↑ Locomotion and Memory ↓ Cell Death of CA1 Neurons | [159] | |
| Wistar Rat | Transient Bilateral Common Carotid Artery Occlusion and Reperfusion | 100 mg/kg Gavage for 2 Weeks | Restoration of the Spatial Memory ↓ IL—6, TNF—α and TUNEL Positive Cells ↑ IL—10 Levels in the Hippocampus | [160] | |
| Sprague-Dawley | Middle Cerebral Artery Occlusion | 50 and 100 mg/kg Gavage for 14 Days | ↓ Levels of Lipid Peroxidation ↓ Malondialdehyde ↑ SOD and CAT | [179] | |
| 3,4—Dihydroxybenzoic Acid (Protocatechuic Acid) | AβPP/PS1 | Animal Transgenic Model | 100 mg/kg Orally for 4 Weeks | ↓ Aβ Deposition ↓ Tnf—A, Il—1β, Il—6 And Il—8 Expression. ↑ Bdnf ↑ Learning and Memory Performance | [155] |
| Balb/cA mice | D—Galactose | 0.5%, 1% or 2% Enriched Food Content for 8 Weeks | ↓ IL—1β, TNF—α, IL—6 and Prostaglandin E2 ↓ COX—2 Activity and Expression ↓ mRNA Expression and Protein Production of NF—kB p65 | [161] | |
| Sprague-Dawley rat | Ischemia–Induced Hippocampal Neuronal Death | 30 mg/kg/day Orally for 7 Days | ↓ Microglial Activation ↓ Astroglial Activation | [162] | |
| Wistar rat | Cadmium–Induced Neurotoxicity | 10 and 20 mg/kg Orally for 21 Days | ↓ Na+/K+–ATPase Activity ↓ Acetylcholinesterase, Butyrylcholinesterase and Endogenous Antioxidant Enzymes | [165] | |
| Sprague-Dawley rat | Ischemia–Induced Hippocampal Neuronal Death | 30 mg/kg/day Orally for 7 Days | ↓ Neuronal Cell Death ↓ Oxidative Stress ↓ BBB Disruption | [162] | |
| AβPP/PS1 mice | Animal Transgenic Model | 100 mg/kg orally for 4 Weeks | ↓ Aβ Deposition in Hippocampal Tissue ↓ APP Expression | [155] | |
| 3,4,5—Trihydroxybenzoic Acid (Gallic Acid) | FVB Mice | Kainate-Induced Neuronal Injury | 1 mg/kg by Gavage for 3 Days | ↓ Lipid Peroxidation In Vivo ↓ Ca2+ Release, ROS, Lipid Peroxidation, Expression of COX—2 and p38 MAPK | [180] |
| Wistar Rat | Oxidative Stress Induced by 6—OHDA | 50, 100 and 200 mg/kg Oral Gavage for 10 Days | ↑ Levels of GSH, SOD and CAT ↓ NO Concentration. ↓ TNF—α, IL—1β and IL—6. | [167] | |
| Wistar Albino Rat | Intracerebroventricular Injection of Streptozotocin (STZ) | 30 mg/kg Oral Gavage for 26 Days | ↑ Total Thiol Content ↑ GSH–Px, SOD and CAT Enzyme Activities | [168] | |
| APP/PS1 Mouse Model of AD | Animal transgenic model | 30 mg/kg/day administration through gavage for 30 days | ↓ Aβ1–42 Plaque Size in Hippocampus and Cortex | [156] | |
| 4—Hydroxy–3,5—Dimethoxybenzoic Acid (Syringic Acid) | C57BL/6 Mice | MPTP and probencid (MPTP/p) | 20 mg/kg orally or oral gavage for 35 days | ↑ Motor Functions ↑ TH, DAT and VMAT2 in SN | [172] |
| C57BL/6 Mice | Artery occlusion ischemic injury | 10 mg/kg i.p. injection at the time of occlusion | ↑ SOD ↑ Nuclear Respiratory Factor 1 Levels ↓ Caspase—3 and—9 Levels | [176] | |
| Cinnamic acids | |||||
| Cinnamic acid | C57BL/6 Mice WT and PPARα (-/-) | MPTP Induced Parkinson’s Disease | 100 mg/ kg Orally For 7 Days | ↓ Neuronal Cell Death ↑ Dopamine ↑ Locomotion ↑ PPARα | [173] |
| B6SJL-Tg (5xFAD) | Trangenic Model of Ahlzeimer‘s Disease | 100 mg/kg | ↑ Locomotion and Memory ↓ Plaque Formation and Aβ1–40 ↑ Lysosomal Biogenesis | [174] | |
| 4—Hydroxy–3—Methoxycinnamic Acid (Ferulic Acid) | C57BL/6N mice | LPS | 20 mg/kg Orally for 11 Days | ↓ Glial Cell Activation ↓ p—JNK, p—NF—κB ↓ iNOS, COX—2, TNF—α, and IL—1β in the Mouse Hippocampus and BV2 Microglial Cells | [157] |
| Sprague-Dawley Rat | Hypoxia/Ischemia-Induced Cell Injury | 28, 56 and 112 mg/kg for 5 Days | ↓ Apoptosis ↓ Cleaved Caspase—3 and Bax ↑ Bcl—2 ↓ ROS Generation ↓ Ca2+ Influx ↓ Superoxide Anion (O2−) ↓ Malondialdehyde ↑ SOD and GSH-Px Activity | [170] | |
| Wistar Rat | Rotenone Induced Parkinson’s Disease | 50 mg/kg i.p. for 4 Weeks | ↓ Cell Death ↓ Lipid Peroxidation ↓ Malondialdehyde Content ↑ SOD, CAT and GSH | [158] | |
| 3,4—dihydroxycinnamic acid (Caffeic Acid) | Sprague- Dawley Rat | High—Fat Diet—Induced Hyperinsulinemia | 30 mg/kg/Daily Oraly for 30 Weeks | ↑ Sod And Gsk3β ↓ P—Tau ↓ App, Bace And Aβ 1−42 ↑ Levels Synaptic Proteins ↓ Memory Deficits | [178] |
| Swiss Albino Mice | Rotenone Induced Parkinson’s Disease | 2.5, 5 or 10 mg/kg | ↑ Dopamine Concentration ↓ COX—2, TNF—α, and IL—1β, CD11b ↓ iNOS, NF—κB ↓ Neuronal Cell Death | [175] | |
| Sprague-Dawley Rat | Intracranial Injection of Aβ1-40 | 100 mg/kg Injection for 2 Weeks | ↑ Learning ↓ AChe, ROS ↑ CAT, GSH ↓ TNF—α, IL—6 ↓ NF—κB, Caspase—3, p—53 | [181] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carregosa, D.; Mota, S.; Ferreira, S.; Alves-Dias, B.; Loncarevic-Vasiljkovic, N.; Crespo, C.L.; Menezes, R.; Teodoro, R.; Santos, C.N.d. Overview of Beneficial Effects of (Poly)phenol Metabolites in the Context of Neurodegenerative Diseases on Model Organisms. Nutrients 2021, 13, 2940. https://doi.org/10.3390/nu13092940
Carregosa D, Mota S, Ferreira S, Alves-Dias B, Loncarevic-Vasiljkovic N, Crespo CL, Menezes R, Teodoro R, Santos CNd. Overview of Beneficial Effects of (Poly)phenol Metabolites in the Context of Neurodegenerative Diseases on Model Organisms. Nutrients. 2021; 13(9):2940. https://doi.org/10.3390/nu13092940
Chicago/Turabian StyleCarregosa, Diogo, Sara Mota, Sofia Ferreira, Beatriz Alves-Dias, Natasa Loncarevic-Vasiljkovic, Carolina Lage Crespo, Regina Menezes, Rita Teodoro, and Cláudia Nunes dos Santos. 2021. "Overview of Beneficial Effects of (Poly)phenol Metabolites in the Context of Neurodegenerative Diseases on Model Organisms" Nutrients 13, no. 9: 2940. https://doi.org/10.3390/nu13092940
APA StyleCarregosa, D., Mota, S., Ferreira, S., Alves-Dias, B., Loncarevic-Vasiljkovic, N., Crespo, C. L., Menezes, R., Teodoro, R., & Santos, C. N. d. (2021). Overview of Beneficial Effects of (Poly)phenol Metabolites in the Context of Neurodegenerative Diseases on Model Organisms. Nutrients, 13(9), 2940. https://doi.org/10.3390/nu13092940