Fructose, Omega 3 Fatty Acids, and Vitamin E: Involvement in Pediatric Non-Alcoholic Fatty Liver Disease


Abstract
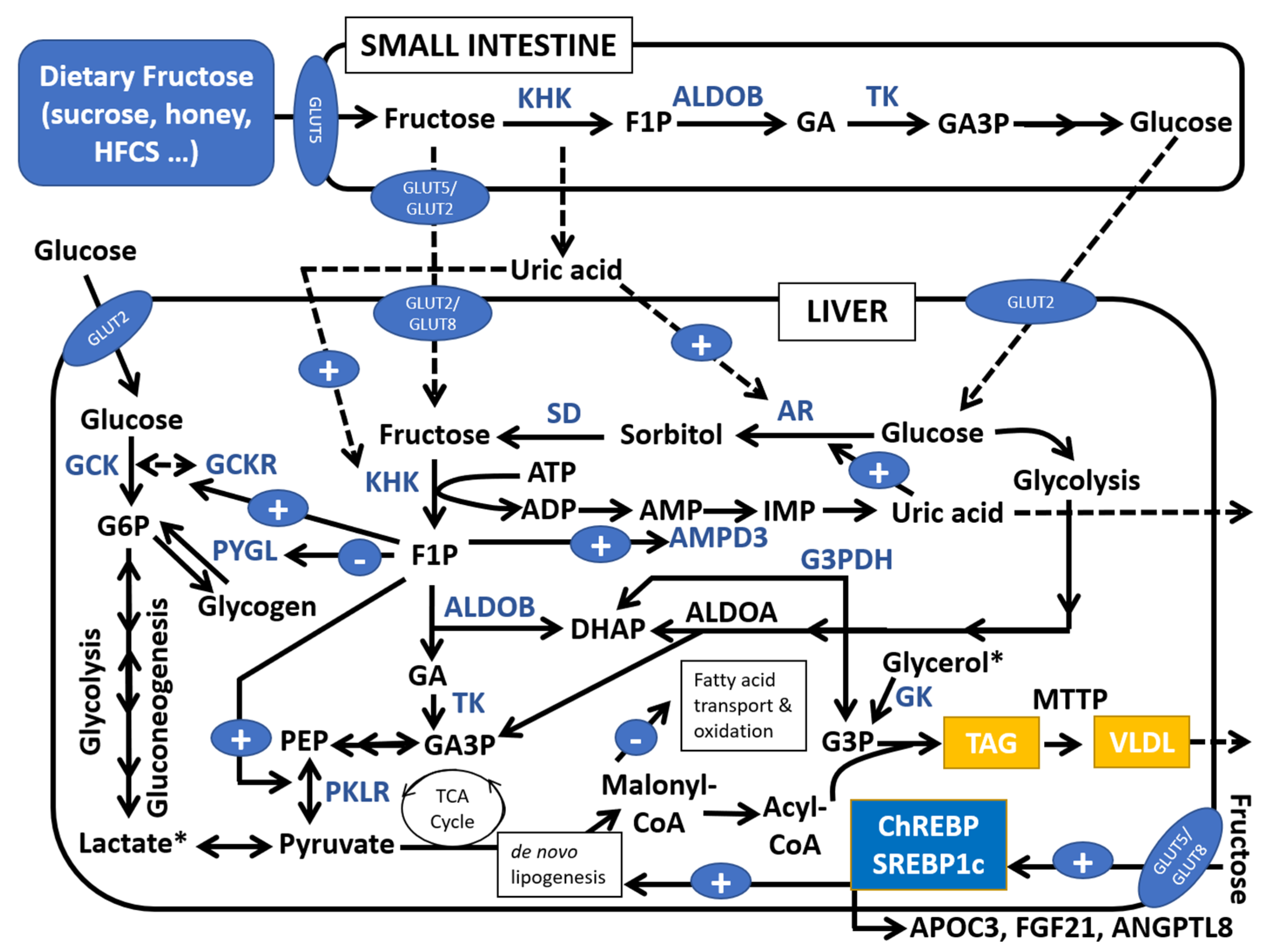
1. Introduction
2. Effects of Fructose, Omega-3 Fatty Acids and Vitamin E on Pediatric NAFLD
2.1. Reduction in Free Sugars and Sugar-Sweetened Beverages (SSBs)
2.2. Omega-3 Long-Chain Polyunsaturated Fatty Acids
2.3. Vitamin E
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Schall, R.A.; Jia, C.; McColgan, B.; et al. The Natural History of Advanced Fibrosis due to Nonalcoholic Steatohepatitis: Data from the Simtuzumab Trials. Hepatology 2019, 30664. [Google Scholar] [CrossRef] [PubMed]
- Kovalic, A.J.; Banerjee, P.; Tran, Q.T.; Singal, A.K.; Satapathy, S.K. Genetic and Epigenetic Culprits in the Pathogenesis of Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2018, 8, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Tricò, D.; Caprio, S.; Rosaria Umano, G.; Pierpont, B.; Nouws, J.; Galderisi, A.; Kim, G.; Mata, M.M.; Santoro, N. Metabolic Features of Nonalcoholic Fatty Liver (NAFL) in Obese Adolescents: Findings from a Multiethnic Cohort. Hepatology 2018, 68, 1376–1390. [Google Scholar] [CrossRef]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.T.; Lawlor, D.A.; Fraser, A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef]
- Braun, H.A.; Faasse, S.A.; Vos, M.B. Advances in Pediatric Fatty Liver Disease: Pathogenesis, Diagnosis, and Treatment. Gastroenterol. Clin. N. Am. 2018, 47, 949–968. [Google Scholar] [CrossRef]
- Kotronen, A.; Yki-Järvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Bedogni, G.; Marcellini, M.; Devito, R.; Ciampalini, P.; Sartorelli, M.R.; Comparcola, D.; Piemonte, F.; Nobili, V. Waist circumference correlates with liver fibrosis in children with non-alcoholic steatohepatitis. Gut 2008, 57, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rivera-Vega, M.; Alsayed, H.M.A.A.; Boesch, C.; Libman, I. Metabolic inflexibility and insulin resistance in obese adolescents with non-alcoholic fatty liver disease. Pediatr. Diabetes 2015, 16, 211–218. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Deutsch, R.; Rauch, J.B.; Behling, C.; Newbury, R.; Lavine, J.E. Obesity, insulin resistance, and other clinicopathological correlates of pediatric nonalcoholic fatty liver disease. J. Pediatr. 2003, 143, 500–505. [Google Scholar] [CrossRef]
- Dharmalingam, M.; Yamasandhi, P. Nonalcoholic fatty liver disease and Type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2018, 22, 421. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Bonora, E.; Targher, G. Nonalcoholic fatty liver disease and risk of incident type 2 diabetes: A meta-analysis. Diabetes Care 2018, 41, 372–382. [Google Scholar] [CrossRef]
- Sao, R.; Aronow, W.S. Association of non-alcoholic fatty liver disease with cardiovascular disease and subclinical atherosclerosis. Arch. Med. Sci. 2018, 14, 1233–1244. [Google Scholar] [CrossRef]
- Madan, S.A.; John, F.; Pyrsopoulos, N.; Pitchumoni, C.S. Nonalcoholic fatty liver disease and carotid artery atherosclerosis in children and adults: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2015, 27, 1237–1248. [Google Scholar] [CrossRef]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Charatcharoenwitthaya, P.; Treeprasertsuk, S.; Benson, J.T.; Enders, F.B.; Angulo, P. The natural history of non-alcoholic fatty liver disease in children: A follow-up study for up to 20 years. Gut 2009, 58, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Vipani, A.; Bresee, C.; Todo, T.; Kim, I.K.; Alkhouri, N.; Setiawan, V.W.; Tran, T.; Ayoub, W.S.; Lu, S.C.; et al. NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications for Liver Transplant and Ethnic and Gender Variances. Am. J. Gastroenterol. 2018, 113, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Benítez, C.; Wolff, R. Current Status and Future Challenges of Liver Transplantation Programs in Chile. Liver Transpl. 2018, 24, 1757–1761. [Google Scholar] [CrossRef]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Sayiner, M.; Otgonsuren, M.; Cable, R.; Younossi, I.; Afendy, M.; Golabi, P.; Henry, L.; Younossi, Z.M. Variables Associated with Inpatient and Outpatient Resource Utilization among Medicare Beneficiaries with Nonalcoholic Fatty Liver Disease with or without Cirrhosis. J. Clin. Gastroenterol. 2017, 51, 254–260. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Zheng, L.; Stepanova, M.; Venkatesan, C.; Mishra, A. Clinical outcomes and resource utilisation in Medicare patients with chronic liver disease: A historical cohort study. BMJ Open 2014, 4, e004318. [Google Scholar] [CrossRef]
- Younossi, Z.M. Patient-Reported Outcomes and the Economic Effects of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: The Value Proposition. Hepatology 2018, 68, 2405–2412. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD). European Association for the Study of Obesity (EASO) EASL–EASD–EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Henry, L.; Bush, H.; Mishra, A. Clinical and Economic Burden of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Syn, W.-K. Current treatment options for nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 2019, 35, 168–176. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Nobili, V.; Manco, M.; Devito, R.; Di Ciommo, V.; Comparcola, D.; Sartorelli, M.R.; Piemonte, F.; Marcellini, M.; Angulo, P. Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: A randomized, controlled trial. Hepatology 2008, 48, 119–128. [Google Scholar] [CrossRef]
- Vos, M.B.; Abrams, S.H.; Barlow, S.E.; Caprio, S.; Daniels, S.R.; Kohli, R.; Mouzaki, M.; Sathya, P.; Schwimmer, J.B.; Sundaram, S.S.; et al. NASPGHAN Clinical Practice Guideline for the Diagnosis and Treatment of Nonalcoholic Fatty Liver Disease in Children: Recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nu. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 319–334. [Google Scholar] [CrossRef]
- Perumpail, B.; Li, A.; John, N.; Sallam, S.; Shah, N.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Therapeutic Implications of the Gut Microbiome and Probiotics in Patients with NAFLD. Diseases 2019, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Fan, X.; Schnabl, B. Role of the intestinal microbiome in liver fibrosis development and new treatment strategies. Transl. Res. 2019. [Google Scholar] [CrossRef]
- Yan, J.-H.; Guan, B.-J.; Gao, H.-Y.; Peng, X.-E. Omega-3 polyunsaturated fatty acid supplementation and non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Medicine 2018, 97, e12271. [Google Scholar] [CrossRef]
- Mann, J.P.; Tang, G.Y.; Nobili, V.; Armstrong, M.J. Evaluations of Lifestyle, Dietary, and Pharmacologic Treatments for Pediatric Nonalcoholic Fatty Liver Disease: A Systematic Review. Clin. Gastroenterol. Hepatol. 2018. [Google Scholar] [CrossRef]
- AlKhater, S.A. Paediatric non-alcoholic fatty liver disease: An overview. Obes. Rev. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alkhouri, N.; Alisi, A.; Della Corte, C.; Fitzpatrick, E.; Raponi, M.; Dhawan, A. Nonalcoholic fatty liver disease: A challenge for pediatricians. JAMA Pediatr. 2015, 169, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Fryar, C.D.; Hales, C.M.; Carroll, M.D.; Aoki, Y.; Freedman, D.S. Differences in obesity Prevalence by Demographics and Urbanization in US Children and Adolescents, 2013–2016. JAMA 2018, 319, 2410–2418. [Google Scholar] [CrossRef] [PubMed]
- Fidler Mis, N.; Braegger, C.; Bronsky, J.; Campoy, C.; Domellöf, M.; Embleton, N.D.; Hojsak, I.; Hulst, J.; Indrio, F.; Lapillonne, A.; et al. Sugar in Infants, Children and Adolescents: A Position Paper of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition Committee on Nutrition. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Kaar, J.L.; Welsh, J.A.; Van Horn, L.V.; Feig, D.I.; Anderson, C.A.M.; Patel, M.J.; Cruz Munos, J.; Krebs, N.F.; Xanthakos, S.A.; et al. Added sugars and cardiovascular disease risk in children: A scientific statement from the American Heart Association. Circulation 2017, 135, e1017–e1034. [Google Scholar] [CrossRef] [PubMed]
- Popkin, B.M.; Hawkes, C. Sweetening of the global diet, particularly beverages: Patterns, trends, and policy responses. Lancet Diabetes Endocrinol. 2016, 4, 174–186. [Google Scholar] [CrossRef]
- Luo, S.; Monterosso, J.R.; Sarpelleh, K.; Page, K.A. Differential effects of fructose versus glucose on brain and appetitive responses to food cues and decisions for food rewards. Proc. Natl. Acad. Sci. USA 2015, 112, 6509–6514. [Google Scholar] [CrossRef]
- Pan, A.; Hu, F.B. Effects of carbohydrates on satiety: Differences between liquid and solid food. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 385–390. [Google Scholar] [CrossRef]
- Sylvetsky, A.C.; Visek, A.J.; Halberg, S.; Rhee, D.K.; Ongaro, Z.; Essel, K.D.; Dietz, W.H.; Sacheck, J. Beyond taste and easy access: Physical, cognitive, interpersonal, and emotional reasons for sugary drink consumption among children and adolescents. Appetite 2020, 155. [Google Scholar] [CrossRef]
- World Health Organization Guideline: Sugars Intake for Adults and Children; World Health Organization: Geneva, Switzerland, 2015.
- U.S. Department of Health and Human Services; U.S. Department of Agriculture. 2015–2020 Dietary Guidelines for Americans. 8th Edition. December 2015. Available online: http://health.gov/dietaryguidelines/2015/guidelines/ (accessed on 12 November 2020).
- Vitolo, M.R. How Much Free Sugars Intake Should Be Recommended for Children Younger Than 2 Years Old? J. Pediatr. Gastroenterol. Nutr. 2018, 66, e87. [Google Scholar] [CrossRef]
- Tondt, J.; Yancy, W.S.; Westman, E.C. Application of Nutrient Essentiality Criteria to Dietary Carbohydrates. Nutr. Res. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H. Fructose: Metabolic, hedonic, and societal parallels with ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Packard, C.J.; Borén, J. Dietary fructose and the metabolic syndrome. Nutrients 2019, 11, 1987. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.T.; Betts, J.A. Dietary Fructose Metabolism by Splanchnic Organs: Size Matters. Cell Metab. 2018, 27, 483–485. [Google Scholar] [CrossRef]
- Chong, M.F.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—What isotopic tracer studies tell us. Nutr. Metab. 2012, 9, 89. [Google Scholar] [CrossRef]
- Chiu, S.; Mulligan, K.; Schwarz, J.M. Dietary carbohydrates and fatty liver disease: De novo lipogenesis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 277–282. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Dushay, J.R.; Toschi, E.; Mitten, E.K.; Fisher, F.M.; Herman, M.A.; Maratos-Flier, E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol. Metab. 2015, 4, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Söderlund, S.; Bogl, L.H.; Hakkarainen, A.; Matikainen, N.; Pietiläinen, K.H.; Räsänen, S.; Lundbom, N.; Björnson, E.; Eliasson, B.; et al. Adverse effects of fructose on cardiometabolic risk factors and hepatic lipid metabolism in subjects with abdominal obesity. J. Intern. Med. 2017, 282, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Zhu, Y.; Rebelo, A.P.; Negri, S.; Courel, S.; Abreu, L.; Bacon, C.J.; Bai, Y.; Bis-Brewer, D.M.; Bugiardini, E.; et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 2020, 52, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Andres-Hernando, A.; Garcia-Arroyo, F.E.; Cicerchi, C.; Li, N.; Kuwabara, M.; Roncal-Jimenez, C.A.; Johnson, R.J.; Lanaspa, M.A. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J. Biol. Chem. 2019, 294, 4272–4281. [Google Scholar] [CrossRef]
- Russo, E.; Leoncini, G.; Esposito, P.; Garibotto, G.; Pontremoli, R.; Viazzi, F. Fructose and uric acid: Major mediators of cardiovascular disease risk starting at pediatric age. Int. J. Mol. Sci. 2020, 21, 4479. [Google Scholar] [CrossRef]
- Nier, A.; Brandt, A.; Conzelmann, I.B.; Özel, Y.; Bergheim, I. Non-alcoholic fatty liver disease in overweight children: Role of fructose intake and dietary pattern. Nutrients 2018, 10, 1329. [Google Scholar] [CrossRef]
- Mosca, A.; Nobili, V.; De Vito, R.; Crudele, A.; Scorletti, E.; Villani, A.; Alisi, A.; Byrne, C.D. Serum uric acid concentrations and fructose consumption are independently associated with NASH in children and adolescents. J. Hepatol. 2017, 66, 1031–1036. [Google Scholar] [CrossRef]
- Qiu, L.; Guo, C. Natural Aldose Reductase Inhibitor: A Potential Therapeutic Agent for Non-Alcoholic Fatty Liver Disease. Curr. Drug Targets 2019, 21, 599–609. [Google Scholar] [CrossRef]
- Lustig, R.H.; Schmidt, L.A.; Brindis, C.D. Public health: The toxic truth about sugar. Nature 2012, 482, 27–29. [Google Scholar] [CrossRef]
- Sievenpiper, J.L.; Tappy, L.; Brouns, F. Fructose as a Driver of Diabetes: An Incomplete View of the Evidence. Mayo Clin. Proc. 2015, 90, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Hu, F.B. Fructose and cardiometabolic Health What the Evidence from Sugar-Sweetened Beverages Tells Us. J. Am. Coll. Cardiol. 2015, 66, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.A.; Frese, M.; Romero, J.; Cunningham, J.H.; Mills, K.E. Chronic fructose substitution for glucose or sucrose in food or beverages has little effect on fasting blood glucose, insulin, or triglycerides: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2017, 106, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.A.; Frese, M.; Romero, J.; Cunningham, J.H.; Mills, K.E. Fructose replacement of glucose or sucrose in food or beverages lowers postprandial glucose and insulin without raising triglycerides: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2017, 106, 506–518. [Google Scholar] [CrossRef]
- Sievenpiper, J.L. Fructose: Back to the future? Am. J. Clin. Nutr. 2017, 106, 439–442. [Google Scholar] [CrossRef]
- Semnani-Azad, Z.; Khan, T.A.; Blanco Mejia, S.; De Souza, R.J.; Leiter, L.A.; Kendall, C.W.C.; Hanley, A.J.; Sievenpiper, J.L. Association of Major Food Sources of Fructose-Containing Sugars with Incident Metabolic Syndrome: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2020, 3, e209993. [Google Scholar] [CrossRef]
- Ebbeling, C.B.; Feldman, H.A.; Chomitz, V.R.; Antonelli, T.A.; Gortmaker, S.L.; Osganian, S.K.; Ludwig, D.S. A randomized trial of sugar-sweetened beverages and adolescent body weight. N. Engl. J. Med. 2012, 367, 1407–1416. [Google Scholar] [CrossRef]
- De Ruyter, J.C.; Olthof, M.R.; Seidell, J.C.; Katan, M.B. A trial of sugar-free or sugar-sweetened beverages and body weight in children. N. Engl. J. Med. 2012, 367, 1397–1406. [Google Scholar] [CrossRef]
- Wehmeyer, M.H.; Zyriax, B.C.; Jagemann, B.; Roth, E.; Windler, E.; Zur Wiesch, J.S.; Lohse, A.W.; Kluwe, J. Nonalcoholic fatty liver disease is associated with excessive calorie intake rather than a distinctive dietary pattern. Medicine 2016, 95. [Google Scholar] [CrossRef]
- Jin, R.; Vos, M.B. Fructose and liver function—Is this behind nonalcoholic liver disease? Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 490–495. [Google Scholar] [CrossRef]
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The multifaceted aspects of fructose metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Perito, E.R.; Rodriguez, L.A.; Lustig, R.H. Dietary treatment of nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2013, 29, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K.; et al. Effects of Dietary Fructose Restriction on Liver Fat, De Novo Lipogenesis, and Insulin Kinetics in Children with Obesity. Gastroenterology 2017, 153, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Welsh, J.A.; Le, N.-A.; Holzberg, J.; Sharma, P.; Martin, D.R.; Vos, M.B. Dietary Fructose Reduction Improves Markers of Cardiovascular Disease Risk in Hispanic-American Adolescents with NAFLD. Nutrients 2014, 6, 3187–3201. [Google Scholar] [CrossRef]
- Goss, A.M.; Dowla, S.; Pendergrass, M.; Ashraf, A.; Bolding, M.; Morrison, S.; Amerson, A.; Soleymani, T.; Gower, B. Effects of a carbohydrate-restricted diet on hepatic lipid content in adolescents with non-alcoholic fatty liver disease: A pilot, randomized trial. Pediatr. Obes. 2020, 15. [Google Scholar] [CrossRef]
- Mosca, A.; Della Corte, C.; Sartorelli, M.R.; Ferretti, F.; Nicita, F.; Vania, A.; Nobili, V. Beverage consumption and paediatric NAFLD. Eat. Weight Disord. 2016, 21, 581–588. [Google Scholar] [CrossRef]
- Panera, N.; Barbaro, B.; Della Corte, C.; Mosca, A.; Nobili, V.; Alisi, A. A review of the pathogenic and therapeutic role of nutrition in pediatric nonalcoholic fatty liver disease. Nutr. Res. 2018, 58, 1–16. [Google Scholar] [CrossRef]
- Sekkarie, A.; Welsh, J.A.; Vos, M.B. Carbohydrates and diet patterns in nonalcoholic fatty liver disease in children and adolescents. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 283–288. [Google Scholar] [CrossRef]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef]
- Jiang, X.; Zheng, J.; Zhang, S.; Wang, B.; Wu, C.; Guo, X. Advances in the Involvement of Gut Microbiota in Pathophysiology of NAFLD. Front. Med. 2020, 7, 361. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Willment, A.; Patel, S.S.; Sun, X.; Song, M.; Mannery, Y.O.; Kosters, A.; McClain, C.J.; Vos, M.B. Fructose Induced Endotoxemia in Pediatric Nonalcoholic Fatty Liver Disease. Int. J. Hepatol. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Jin, R.; Le, N.A.; Liu, S.; Epperson, M.F.; Ziegler, T.R.; Welsh, J.A.; Jones, D.P.; McClain, C.J.; Vos, M.B. Children with NAFLD are more sensitive to the adverse metabolic effects of fructose beverages than children without NAFLD. J. Clin. Endocrinol. Metab. 2012, 97, E1088. [Google Scholar] [CrossRef] [PubMed]
- Kakleas, K.; Christodouli, F.; Karavanaki, K. Nonalcoholic fatty liver disease, insulin resistance, and sweeteners: A literature review. Expert Rev. Endocrinol. Metab. 2020, 15, 83–93. [Google Scholar] [CrossRef]
- Scorletti, E.; Byrne, C.D. Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu. Rev. Nutr. 2013, 33, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P.; DiNicolantonio, J.J. The importance of a balanced ω-6 to ω-3 ratio in the prevention and management of obesity. Open Heart 2016, 3. [Google Scholar] [CrossRef]
- Glaser, C.; Heinrich, J.; Koletzko, B. Role of FADS1 and FADS2 polymorphisms in polyunsaturated fatty acid metabolism. Metabolism 2010, 59, 993–999. [Google Scholar] [CrossRef]
- Scorletti, E.; Byrne, C.D. Omega-3 fatty acids and non-alcoholic fatty liver disease: Evidence of efficacy and mechanism of action. Mol. Asp. Med. 2018, 64, 135–146. [Google Scholar] [CrossRef]
- Lev-Tzion, R.; Griffiths, A.M.; Leder, O.; Turner, D. Omega 3 fatty acids (fish oil) for maintenance of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Dietary Reference Values for the European Union 2019. Available online: https://www.efsa.europa.eu/en/topics/topic/dietary-reference-values (accessed on 27 October 2020).
- Jump, D.B.; Depner, C.M.; Tripathy, S.; Lytle, K.A. Potential for dietary ω-3 fatty acids to prevent nonalcoholic fatty liver disease and reduce the risk of primary liver cancer. Adv. Nutr. 2015, 6, 694–702. [Google Scholar] [CrossRef] [PubMed]
- St-Jules, D.E.; Watters, C.A.; Brunt, E.M.; Wilkens, L.R.; Novotny, R.; Belt, P.; Lavine, J.E.; Abrams, S.H.; Himes, R.; Krisnamurthy, R.; et al. Estimation of fish and ω-3 fatty acid intake in pediatric nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Corte, C.D.; Iasevoli, S.; Strologo, A.D.; Sanseviero, M.; Nobili, V. Omega-3 fatty acids and fatty liver disease in children. In Advances in Food and Nutrition Research; Academic Press: Cambridge, MA, USA, 2018; Volume 85, pp. 59–77. [Google Scholar]
- Nobili, V.; Bedogni, G.; Alisi, A.; Pietrobattista, A.; Risé, P.; Galli, C.; Agostoni, C. Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: Double-blind randomised controlled clinical trial. Arch. Dis. Child. 2011, 96, 350–353. [Google Scholar] [CrossRef]
- Nobili, V.; Alisi, A.; Della Corte, C.; Risé, P.; Galli, C.; Agostoni, C.; Bedogni, G. Docosahexaenoic acid for the treatment of fatty liver: Randomised controlled trial in children. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Carpino, G.; Alisi, A.; De Vito, R.; Franchitto, A.; Alpini, G.; Onori, P.; Gaudio, E. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Boyraz, M.; Pirgon, Ö.; Dündar, B.; Çekmez, F.; Hatipoğlu, N. Long-term treatment with n-3 polyunsaturated fatty acids as a monotherapy in children with nonalcoholic fatty liver disease. JCRPE J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 121–127. [Google Scholar] [CrossRef]
- Pacifico, L.; Bonci, E.; Di Martino, M.; Versacci, P.; Andreoli, G.; Silvestri, L.M.; Chiesa, C. A double-blind, placebo-controlled randomized trial to evaluate the efficacy of docosahexaenoic acid supplementation on hepatic fat and associated cardiovascular risk factors in overweight children with nonalcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 734–741. [Google Scholar] [CrossRef]
- Janczyk, W.; Lebensztejn, D.; Wierzbicka-Rucińska, A.; Mazur, A.; Neuhoff-Murawska, J.; Matusik, P.; Socha, P. Omega-3 fatty acids therapy in children with nonalcoholic fatty liver disease: A randomized controlled trial. J. Pediatr. 2015, 166, 1358–1363. [Google Scholar] [CrossRef]
- Spahis, S.; Alvarez, F.; Ahmed, N.; Dubois, J.; Jalbout, R.; Paganelli, M.; Grzywacz, K.; Delvin, E.; Peretti, N.; Levy, E. Non-alcoholic fatty liver disease severity and metabolic complications in obese children: Impact of omega-3 fatty acids. J. Nutr. Biochem. 2018, 58, 28–36. [Google Scholar] [CrossRef]
- Zöhrer, E.; Alisi, A.; Jahnel, J.; Mosca, A.; Della Corte, C.; Crudele, A.; Fauler, G.; Nobili, V. Efficacy of docosahexaenoic acid–choline–vitamin E in paediatric NASH: A randomized controlled clinical trial. Appl. Physiol. Nutr. Metab. 2017, 42, 948–954. [Google Scholar] [CrossRef]
- Spooner, M.H.; Jump, D.B. Omega-3 fatty acids and nonalcoholic fatty liver disease in adults and children: Where do we stand? Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Musa-Veloso, K.; Venditti, C.; Lee, H.Y.; Darch, M.; Floyd, S.; West, S.; Simon, R. Systematic review and meta-analysis of controlled intervention studies on the effectiveness of long-chain omega-3 fatty acids in patients with nonalcoholic fatty liver disease. Nutr. Rev. 2018, 76, 581–602. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yuan, M.; Wang, L. The effect of omega-3 unsaturated fatty acids on non-alcoholic fatty liver disease: A systematic review and meta-analysis of RCTs. Pak. J. Med. Sci. 2017, 33, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.F.; Yang, B.; Tang, J.; Li, D. Fatty acid and non-alcoholic fatty liver disease: Meta-analyses of case-control and randomized controlled trials. Clin. Nutr. 2018, 37, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “Hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Couillard, C.; Ruel, G.; Archer, W.R.; Pomerleau, S.; Bergeron, J.; Couture, P.; Lamarche, B.; Bergeron, N. Circulating levels of oxidative stress markers and endothelial adhesion molecules in men with abdominal obesity. J. Clin. Endocrinol. Metab. 2005, 90, 6454–6459. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Palmieri, V.O.; Grattagliano, I.; Portincasa, P.; Palasciano, G. Systemic oxidative alterations are associated with visceral adiposity and liver steatosis in patients with metabolic syndrome. J. Nutr. 2006, 136, 3022–3026. [Google Scholar] [CrossRef]
- Lee, G.Y.; Han, S.N. The role of vitamin E in immunity. Nutrients 2018, 10, 1614. [Google Scholar] [CrossRef] [PubMed]
- Nagashimada, M.; Ota, T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB Life 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Colvin, R.; Belt, P.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Schwimmer, J.B.; Tonascia, J.; Unalp, A.; Lavine, J.E.; et al. Correlation of Vitamin E, uric acid and diet composition with histologic features of pediatric nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Mandato, C.; Franzese, A.; Ciccimarra, E.; Lucariello, S.; Savoia, M.; Capuano, G.; Migliaro, F. Vitamin E treatment in pediatric obesity-related liver disease: A randomized study. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 48–55. [Google Scholar] [CrossRef]
- Nobili, V.; Manco, M.; Devito, R.; Ciampalini, P.; Piemonte, F.; Marcellini, M. Effect of vitamin E on aminotransferase levels and insulin resistance in children with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2006, 24, 1553–1561. [Google Scholar] [CrossRef]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of Vitamin e or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents the tonic randomized controlled trial. JAMA J. Am. Med. Assoc. 2011, 305, 1659–1668. [Google Scholar] [CrossRef]
- Cho, T.; Kim, Y.J.; Paik, S.S. The efficacy of pharmacological treatment in pediatric nonalcoholic fatty liver disease. Pediatr. Gastroenterol. Hepatol. Nutr. 2012, 15, 256–265. [Google Scholar] [CrossRef]
- Murer, S.B.; Aeberli, I.; Braegger, C.P.; Gittermann, M.; Hersberger, M.; Leonard, S.W.; Taylor, A.W.; Traber, M.G.; Zimmermann, M.B. Antioxidant supplements reduced oxidative stress and stabilized liver function tests but did not reduce inflammation in a randomized controlled trial in obese children and adolescents. J. Nutr. 2014, 144, 193–201. [Google Scholar] [CrossRef]
- Wang, C.L.; Liang, L.; Fu, J.F.; Zou, C.C.; Hong, F.; Xue, J.Z.; Lu, J.R.; Wu, X.M. Effect of lifestyle intervention on non-alcoholic fatty liver disease in Chinese obese children. World J. Gastroenterol. 2008, 14, 1598–1602. [Google Scholar] [CrossRef]
- Amanullah, I.; Khan, Y.H.; Anwar, I.; Gulzar, A.; Mallhi, T.H.; Raja, A.A. Effect of Vitamin E in non-alcoholic fatty liver disease: A systematic review and meta-analysis of randomised controlled trials. Postgrad. Med. J. 2019, 95, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The selenium and vitamin E cancer prevention trial (SELECT). JAMA J. Am. Med. Assoc. 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Antioxidant supplements and mortality. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Reduction in Fructose Consumption | ||||
|---|---|---|---|---|
| Author, Year, Country | Sample Size and Age Range | Intervention and Duration | Outcomes | Results |
| Jin et al. [88], 2014, USA | 24 overweight adolescents (11–18 years) with hepatic fat >8% on MRS | Calorie-matched study-provided fructose only or glucose only beverages (3 × 8 fl bottles per day), for 4 weeks | (1) Hepatic steatosis (measured by MRS), ALT, AST (2) Associated cardiovascular risk factors (BW, TG, FI, HOMA-IR, FFA, LDL) | No significant change in hepatic fat, liver enzymes or body weight in either group. In the glucose beverage group there was significantly improved HOMA-IR, hs-CRP, and LDL oxidation. |
| Schwarz et al. [87], 2017, USA | Obese Children (9–18 years old; n = 41) with habitual high sugar consumption | 9 days of isocaloric fructose restriction | (1) Liver, visceral and subcutaneous fat (2) de novo lipogenesis (DNL) | Reductions in liver fat, visceral fat and DNL after fructose restriction |
| Jin et al. [96], 2012, USA | Nine children with NAFLD and 10 matched controls without NAFLD (11–18 years old) | 24-h periods of isocaloric macronutrient-balanced meals with either a fructose beverage or a glucose beverage | Plasma glucose, insulin, triglyceride, apolipoprotein B, high-density lipoprotein cholesterol, and nonesterified free fatty acid levels. | Effects of fructose on plasma lipids occurred in both healthy and NAFLD children, being the latter more sensitive to fructose actions |
| Jin et al. [95], 2014, USA | As in Jin et al. [88,96] | As in Jin et al. [88,96] | As in Jin et al. [88,96] | Endotoxin levels in NAFLD increased after fructose beverages compared to healthy children. |
| Omega 3 Fatty Acids | ||||
| Nobili et al. [107,108], 2011, 2013, Italy | 60, <18 years | DHA 500 mg/day, or DHA 250 mg/day, or placebo for 24 months (with evaluations at 6, 12, 18, and 24 months) | (1) Change in liver fat (detected by ultrasonography) (2) TG, ALT, BMI, HOMA-IR | The severity of liver steatosis decreased in both treated groups vs. placebo (OR ≤ 0.02, p ≤ 0.05). TG were lower in both the treated groups. ALT was lower in the treatment groups from month 12 onwards. HOMA was lower in the DHA 250 mg group vs. placebo at 6 and 12 months. |
| Boyraz et al. [110], 2015, Turkey | 108 obese adolescents, 9–17 years | 1000 mg/day omega-3 plus diet (50% carbohydrates, 20% protein and 30% fat) plus lifestyle intervention (exercise three times per week for 1 h and the promotion of self-initiated physical activities) or placebo plus diet plus lifestyle intervention for 12 months | (1) US, ALT, AST (2) BMI, HDL-C, TG, TC, FI, FG, IS, HOMA-IR, SBP | Improvement of steatosis, ALT and AST in both groups. Increase HDL-C, improvement of TG, FI and HOMA-IR. Improvement in BMI in the intervention and control groups. Improvement of SBP in the intervention group. |
| Janczyk et al. [112], 2015, Poland | 76 | 450–1300 mg/day 3:2 DHA:EPA plus individually prescribed diet and PA versus omega 6 sunflower oil for 6 months | (1) ALT, AST, GGT (2) FSG, FI, HOMA- IR, adiponectin, cholesterol, steatosis (US), BMI, WC | Lower ALT levels in both groups the intervention and control (but without statistical difference between the groups). Lower AST and GGT levels in the intervention group. No changes in ALT, steatosis, FSG, FI, HOMA-IR, BMI z score or WC z score in both groups. |
| Pacifico et al. [111], 2015, Italy | 51, <18 years | 250 mg/day DHA plus low-calorie diet plus 60 min PA 5 times a week versus placebo (290 mg/day linoleic acid) for 6 months | (1) Hepatic fat, ALT (2) TG, Z-BMI, FI, HOMA-IR | Lower hepatic fat by MRS, FI, and TG in the intervention group. |
| Vitamin E | ||||
| Vajro et al. [129], 2004, Italy | 28 obese participants, age range NR | Oral a-acetatetocopherol (400 mg/day for 2 months, 100 mg/day for 3 months) plus low-calorie diet plus exercise versus placebo | (1) Liver echogenicity, ALT (2) BMI | Results were stratified based on weight loss: decrease BMI plus vitamin E: improved liver echogenicity, and ALT levels. Stable BMI plus vitamin E: improved the liver echogenicity. |
| Nobili et al. [35,130], 2006, 2008, Italy | 90 participants, 3–18 years | Alpha-tocopherol (600 ID/d) plus ascorbic acid (500 mg/day) plus hypo-/isocaloric diet plus 45 min/day aerobic exercise versus placebo for 24 months | (1) NAFLD Activity Score, ALT, AST (2) Z-BMI, BMI, TG, TC, FSG, FI, HOMA-IR | Improvement in NAFLD activity score, ALT, and AST in both the intervention and control groups (but without statistical difference between the groups). |
| Wang et al. [134], 2008, China | 76 obese children with NAFLD, 10–17 years | Three groups: Vitamin E (100 mg/day) versus life-style interventions (low calorie diet + exercise + reduced caloric intake) versus no intervention | (1) US, ALT, AST (2) Z-BMI, TG, TC, FSG, FI, HOMA | No change to liver echogenicity in any group. Reduction in ALT, AST, BMI, TG, TC, FI, FSG and HOMA- IR in the intervention group. |
| Lavine et al. [131], 2011, USA | 173 participants, 8–17 years | 800 IU of vitamin E (58 patients), 1000 mg of metformin (57 patients), or placebo (58 patients) for 96 weeks | (1) ALT (2) NAFLD activity score, QOL, Z-BMI, BMI, BW, WC, TG, TC, FSG, HOMA-IR, HDL, LDL | No significant improvement in ALT levels between the groups. Improved the NAFLD activity score in the intervention group. Resolution of NASH was significantly greater in the children treated with vitamin E versus the placebo group. |
| Murer et al. [133], 2014, Switzerland | 44 overweight or obese children and adolescents, 10–18 years | Daily antioxidants (vitamin E, 400 IU; vitamin C, 500 mg; selenium, 50 μg) plus lifestyle modifications or placebo for 4 months | (1) Biomarkers of oxidative stress, inflammation, and liver function | Significant treatment effect of antioxidant supplementation on antioxidant status and on ALT. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alberti, G.; Gana, J.C.; Santos, J.L. Fructose, Omega 3 Fatty Acids, and Vitamin E: Involvement in Pediatric Non-Alcoholic Fatty Liver Disease. Nutrients 2020, 12, 3531. https://doi.org/10.3390/nu12113531
Alberti G, Gana JC, Santos JL. Fructose, Omega 3 Fatty Acids, and Vitamin E: Involvement in Pediatric Non-Alcoholic Fatty Liver Disease. Nutrients. 2020; 12(11):3531. https://doi.org/10.3390/nu12113531
Chicago/Turabian StyleAlberti, Gigliola, Juan Cristóbal Gana, and José L. Santos. 2020. "Fructose, Omega 3 Fatty Acids, and Vitamin E: Involvement in Pediatric Non-Alcoholic Fatty Liver Disease" Nutrients 12, no. 11: 3531. https://doi.org/10.3390/nu12113531
APA StyleAlberti, G., Gana, J. C., & Santos, J. L. (2020). Fructose, Omega 3 Fatty Acids, and Vitamin E: Involvement in Pediatric Non-Alcoholic Fatty Liver Disease. Nutrients, 12(11), 3531. https://doi.org/10.3390/nu12113531