Physiopathology of Lifestyle Interventions in Non-Alcoholic Fatty Liver Disease (NAFLD)


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Meal Timing, Frequency and Speed Eating Affect NAFLD
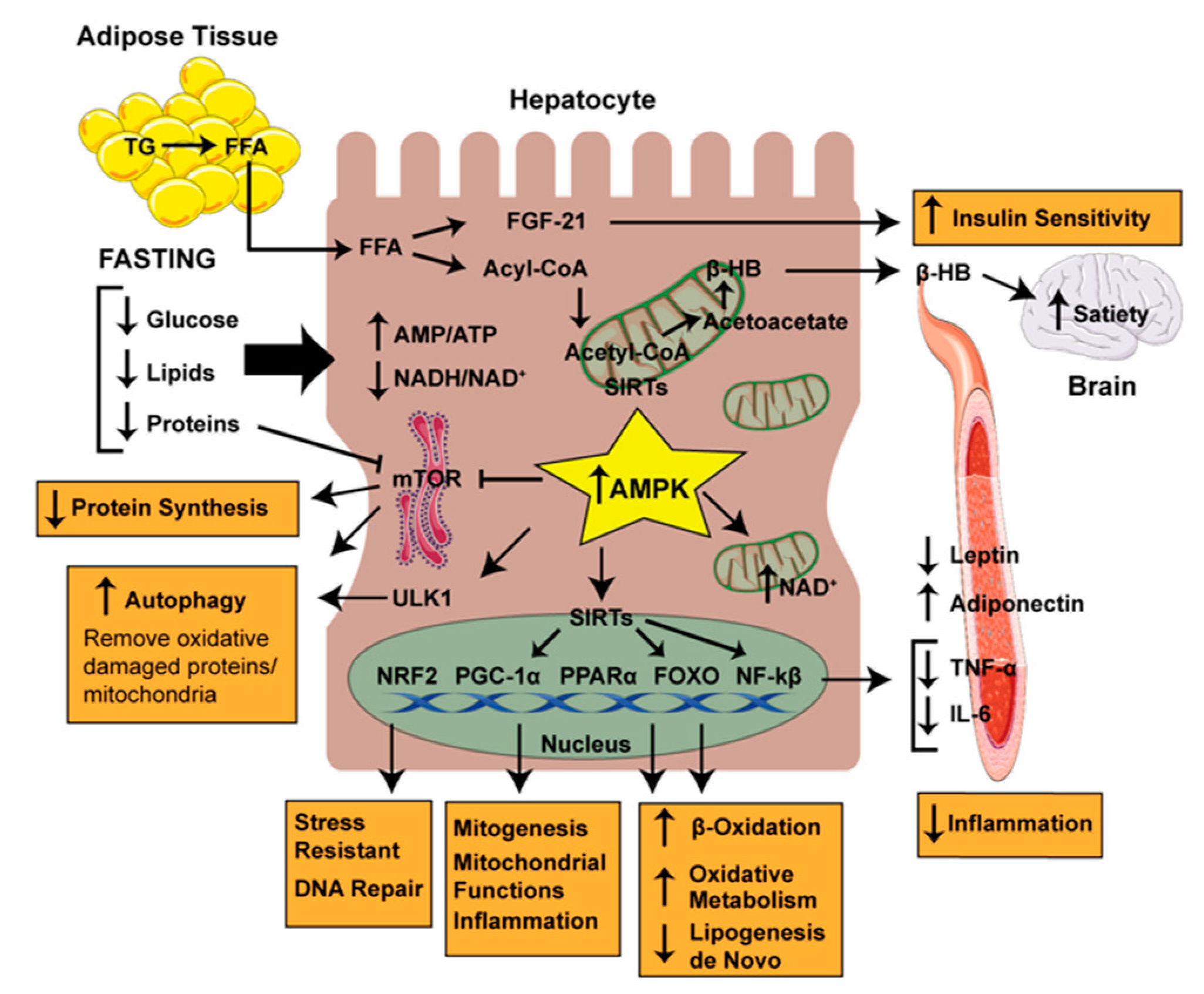
3. Strategies and Interventions Altering Meal Frequency and Timing with Fatty Liver Disease: Molecular Mechanisms
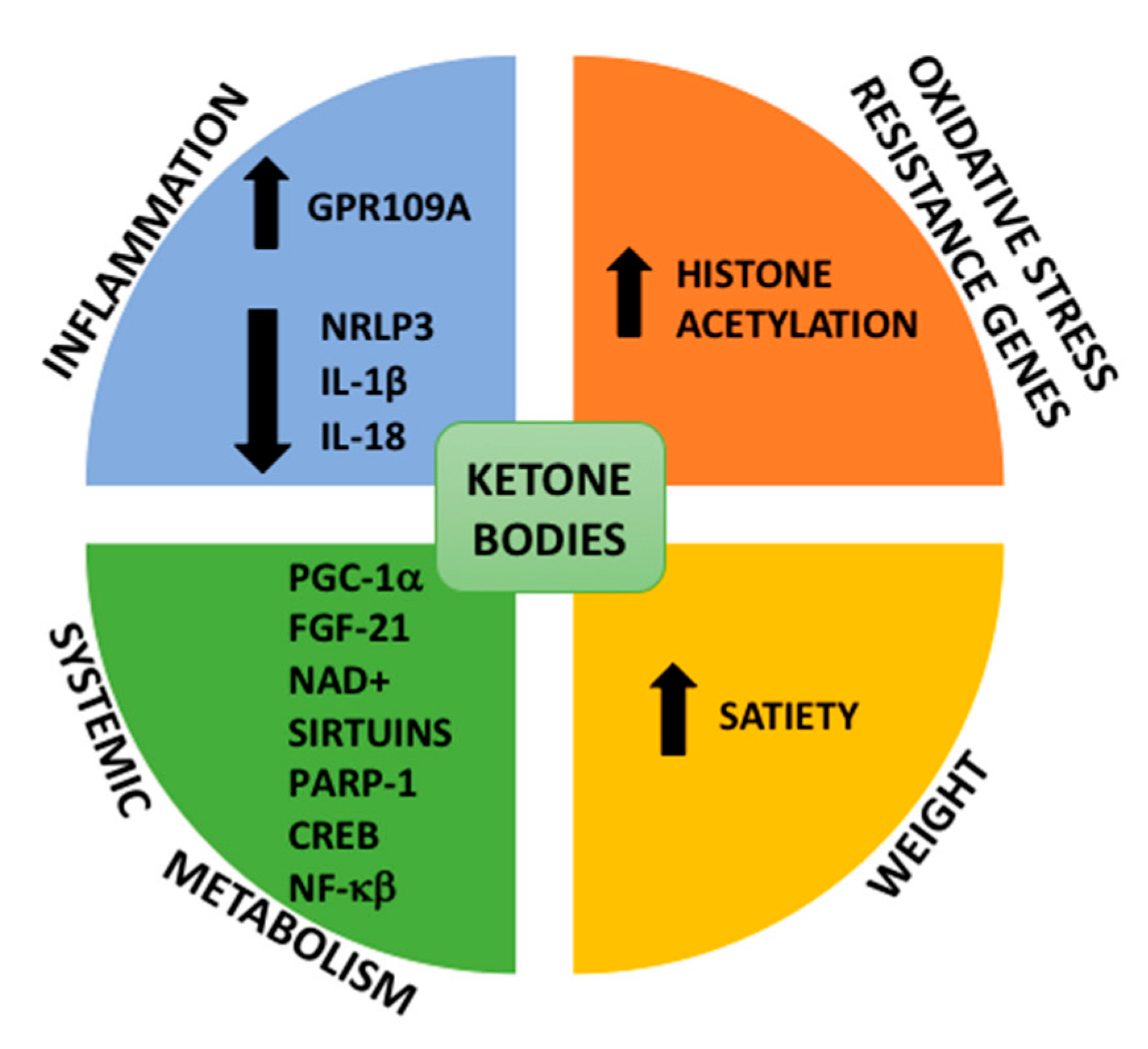
3.1. Metabolic Switching: Ketosis
3.2. Stress Resistance, Mitochondrial Biogenesis, Inflammation and Autophagy
3.3. Fasting-Mimicking Diet
4. Exercise and NAFLD
5. Liver and Skeletal Muscle Are Linked in NAFLD
Unbalance in Insulin Growth Factor-1 (IGF-I) Signaling and NAFLD/Sarcopenia Progression
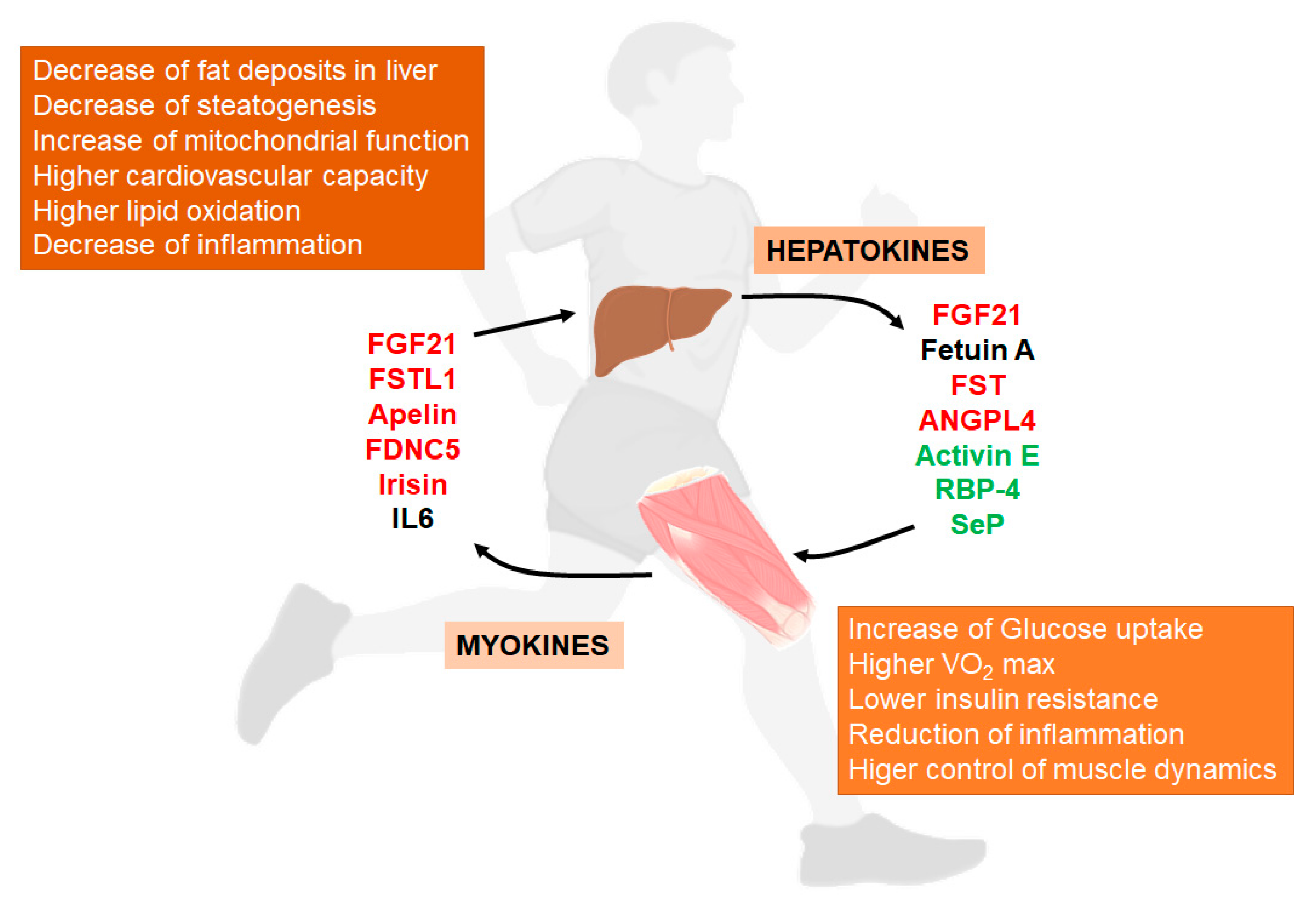
6. Exercise Activates Liver-Muscle Signaling Pathways Involved in NAFLD
6.1. Hepatokines
6.1.1. Fibroblast Growth Factor 21
6.1.2. Fetuin A
6.1.3. Activin and Follistatin (FST)
6.1.4. Retinol-Binding Protein 4
6.1.5. Angiopoietin-Like Protein 4
6.1.6. Selenoprotein P
6.2. Myokines
6.2.1. Follistatin-Like 1 and Apelin
6.2.2. FNDC5 and Irisin
6.2.3. Interleukin-6 (IL-6)
7. The Future of Lifestyle Modification in NAFLD
Author Contributions
Funding
Conflicts of Interest
References
- Rinella, M.E. Nonalcoholic Fatty Liver Disease. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Divella, R.; Mazzocca, A.; Daniele, A.; Sabbà, C.; Paradiso, A. Obesity, Nonalcoholic Fatty Liver Disease and Adipocytokines Network in Promotion of Cancer. Int. J. Biol. Sci. 2019, 15, 610–616. [Google Scholar] [CrossRef]
- Sarwar, R.; Pierce, N.; Koppe, S. Obesity and Nonalcoholic Fatty Liver Disease: Current Perspectives. Diabetes Metab. Syndr. 2018, 11, 533–542. [Google Scholar] [CrossRef]
- Dyson, J.; Day, C. Treatment of Non-Alcoholic Fatty Liver Disease. Dig. Dis. 2014, 32, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Kontogianni, M.D.; Tileli, N.; Margariti, A.; Georgoulis, M.; Deutsch, M.; Tiniakos, D.; Fragopoulou, E.; Zafiropoulou, R.; Manios, Y.; Papatheodoridis, G. Adherence to the Mediterranean Diet Is Associated with the Severity of Non-Alcoholic Fatty Liver Disease. Clin. Nutr. 2014, 33, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Koppe, S.W. Obesity and the Liver: Nonalcoholic Fatty Liver Disease. Transl. Res. 2014, 164, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Çolak, Y.; Tuncer, I.; Şenateş, E.; Ozturk, O.; Doganay, L.; Yilmaz, Y. Nonalcoholic Fatty Liver Disease: A Nutritional Approach. Metab. Syndr. Relat. Disord. 2012, 10, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Blendis, L.; Halpern, Z.; Oren, R. Long Term Nutritional Intake and the Risk for Non-Alcoholic Fatty Liver Disease (NAFLD): A Population Based Study. J. Hepatol. 2007, 47, 711–717. [Google Scholar] [CrossRef]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose Consumption as a Risk Factor for Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef]
- Patel, N.S.; Doycheva, I.; Peterson, M.R.; Hooker, J.; Kisselva, T.; Schnabl, B.; Seki, E.; Sirlin, C.B.; Loomba, R. Effect of Weight Loss on Magnetic Resonance Imaging Estimation of Liver Fat and Volume in Patients with Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2014, 13, 561–568.e1. [Google Scholar] [CrossRef]
- Stefan, N.; Häring, H.-U.; Schulze, M.B. Metabolically Healthy Obesity: The Low-Hanging Fruit in Obesity Treatment? Lancet Diabetes Endocrinol. 2018, 6, 249–258. [Google Scholar] [CrossRef]
- De Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, A.; Di Germanio, C.; Bernier, M.; De Cabo, R. A Time to Fast. Science 2018, 362, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Thoma, C.; Day, C.P.; Trenell, M.I. Lifestyle Interventions for the Treatment of Non-Alcoholic Fatty Liver Disease in Adults: A Systematic Review. J. Hepatol. 2012, 56, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Purslow, L.R.; Sandhu, M.S.; Forouhi, N.G.; Young, E.H.; Luben, R.N.; Welch, A.A.; Khaw, K.-T.; Bingham, S.A.; Wareham, N.J. Energy Intake at Breakfast and Weight Change: Prospective Study of 6,764 Middle-aged Men and Women. Am. J. Epidemiol. 2007, 167, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Esteban, J.P.G.; Rein, L.E.; Szabo, A.; Gawrieh, S.; Saeian, K. Not Just What, but Also When You Eat: Analyzing the Impact of Meal Timing Patterns on Non-Alcoholic Fatty Liver Disease. Hepatology 2016, 64, 17A–18A. [Google Scholar]
- Nishi, T.; Babazono, A.; Maeda, T.; Imatoh, T.; Une, H. Effects of Eating Fast and Eating Before Bedtime on the Development of Nonalcoholic Fatty Liver Disease. Popul. Health Manag. 2016, 19, 279–283. [Google Scholar] [CrossRef]
- Koopman, K.E.; Caan, M.W.A.; Nederveen, A.J.; Pels, A.; Ackermans, M.T.; Fliers, E.; La Fleur, S.E.; Serlie, M.J. Hypercaloric Diets with Increased Meal Frequency, but Not Meal Size, Increase Intrahepatic Triglycerides: A Randomized Controlled Trial. Hepatology 2014, 60, 545–553. [Google Scholar] [CrossRef]
- Okauchi, H.; Hashimoto, C.; Nakao, R.; Oishi, K. Timing of Food Intake Is More Potent Than Habitual Voluntary Exercise to Prevent Diet-Induced Obesity in Mice. Chronobiol. Int. 2018, 36, 57–74. [Google Scholar] [CrossRef]
- Garaulet, M.; Gómez-Abellán, P.; Alburquerque-Béjar, J.J.; Lee, Y.-C.; Ordovás, J.M.; Scheer, F.A.J.L. Timing of Food Intake Predicts Weight Loss Effectiveness. Int. J. Obes. 2013, 37, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Sato, S.; Ohira, T.; Maeda, K.; Noda, H.; Kubota, Y.; Nishimura, S.; Kitamura, A.; Kiyama, M.; Okada, T.; et al. The Joint Impact on Being Overweight of Self Reported Behaviours of Eating Quickly and Eating Until Full: Cross Sectional Survey. BMJ 2008, 337, a2002. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ko, B.-J.; Gong, Y.; Han, K.; Lee, A.; Han, B.-D.; Yoon, Y.J.; Park, S.; Kim, J.-H.; Mantzoros, C.S. Self- Reported Eating Speed in Relation to Non-Alcoholic Fatty Liver Disease in Adults. Eur. J. Nutr. 2015, 55, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Hariya, N.; Miyauchi, R.; Misaki, Y.; Ichikawa, Y.; Goda, T. Self-Reported Faster Eating Associated with Higher Alt Activity in Middle-Aged, Apparently Healthy Japanese Women. Nutrients 2014, 30, 69–74. [Google Scholar] [CrossRef]
- Mochizuki, K.; Miyauchi, R.; Hariya, N.; Misaki, Y.; Kasezawa, N.; Tohyama, K.; Goda, T. Self-Reported Rate of Eating Is Associated with Higher Circulating ALT Activity in Middle-Aged Apparently Healthy Japanese Men. Eur. J. Nutr. 2012, 52, 985–990. [Google Scholar] [CrossRef]
- Ohkuma, T.; Fujii, H.; Iwase, M.; Kikuchi, Y.; Ogata, S.; Idewaki, Y.; Ide, H.; Doi, Y.; Hirakawa, Y.; Mukai, N.; et al. Impact of Eating Rate on Obesity and Cardiovascular Risk Factors According to Glucose Tolerance Status: The Fukuoka Diabetes Registry and the Hisayama Study. Diabetologia 2012, 56, 70–77. [Google Scholar] [CrossRef]
- Yamagishi, K.; Sairenchi, T.; Sawada, N.; Sunou, K.; Sata, M.; Murai, U.; Takizawa, N.; Irie, F.; Watanabe, H.; Iso, H.; et al. Impact of Speed-Eating Habit on Subsequent Body Mass Index and Blood Pressure among Schoolchildren―The Ibaraki Children’s Cohort Study (IBACHIL). Circ. J. 2018, 82, 419–422. [Google Scholar] [CrossRef]
- Cao, X.; Gu, Y.; Bian, S.; Zhang, Q.; Meng, G.; Liu, L.; Wu, H.; Zhang, S.; Wang, Y.; Zhang, T.; et al. Association Between Eating Speed and Newly Diagnosed Nonalcoholic Fatty Liver Disease Among the General Population. Nutr. Res. 2020, 80, 78–88. [Google Scholar] [CrossRef]
- Hameed, S.; Dhillo, W.S.; Bloom, S.R. Gut Hormones and Appetite Control. Oral Dis. 2009, 15, 18–26. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with Diet, Physical Activity and Exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Anton, S.D.; Moehl, K.; Donahoo, W.T.; Marosi, K.; Lee, S.A.; Mainous, A.G.; Leeuwenburgh, C.; Mattson, M.P. Flipping the Metabolic Switch: Understanding and Applying the Health Benefits of Fasting. Obesity 2018, 26, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Eslamparast, T.; Tandon, P.; Raman, M. Dietary Composition Independent of Weight Loss in the Management of Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 800. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Sayiner, M.; Lam, B.; Golabi, P.; Younossi, Z.M. Advances and Challenges in the Management of Advanced Fibrosis in Nonalcoholic Steatohepatitis. Ther. Adv. Gastroenterol. 2018, 11, 1756284818811508. [Google Scholar] [CrossRef]
- Cantero, I.; Abete, I.; Monreal, J.I.; Martinez, J.A.; Zulet, M.A. Fruit Fiber Consumption Specifically Improves Liver Health Status in Obese Subjects under Energy Restriction. Nutrients 2017, 9, 667. [Google Scholar] [CrossRef]
- Ratziu, V. Non-Pharmacological Interventions in Non-Alcoholic Fatty Liver Disease Patients. Liver Int. 2017, 37, 90–96. [Google Scholar] [CrossRef]
- Liao, C.-Y.; Rikke, B.A.; Johnson, T.E.; Diaz, V.; Nelson, J.F. Genetic Variation in the Murine Lifespan Response to Dietary Restriction: From Life Extension to Life Shortening. Aging Cell 2010, 9, 92–95. [Google Scholar] [CrossRef]
- Vitousek, P.M.; Ladefoged, T.N.; Kirch, P.V.; Hartshorn, A.S.; Graves, M.W.; Tuljapurkar, S.; Chadwick, O.A.; Hotchkiss, S.C. Soils, Agriculture, and Society in Precontact Hawaii. Science 2004, 304, 1665–1669. [Google Scholar] [CrossRef]
- Hunt, N.D.; Li, G.D.; Zhu, M.; Levette, A.; Chachich, M.E.; Spangler, E.L.; Allard, J.S.; Hyun, D.-H.; Ingram, D.K.; De Cabo, R. Effect of Calorie Restriction and Refeeding on Skin Wound Healing in the Rat. AGE 2011, 34, 1453–1458. [Google Scholar] [CrossRef]
- Kristan, D.M. Calorie Restriction and Susceptibility to Intact Pathogens. AGE 2008, 30, 147–156. [Google Scholar] [CrossRef]
- Johnson, N.A.; Sachinwalla, T.; Walton, D.W.; Smith, K.; Armstrong, A.; Thompson, M.W.; George, J. Aerobic Exercise Training Reduces Hepatic and Visceral Lipids in Obese Individuals Without Weight Loss. Hepatology 2009, 50, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Hems, R. Fatty Acid Metabolism in the Perfused Rat Liver. Biochem. J. 1970, 119, 525–533. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Foster, D.W. The Regulation of Ketogenesis From Oleic Acid and the Influence of Antiketogenic Agents. J. Biol. Chem. 1971, 246, 6247–6253. [Google Scholar] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Müller-Fielitz, H.; Pokorná, B.; Vollbrandt, T.; Stölting, I.; Nadrowitz, R.; et al. The β-Hydroxybutyrate Receptor HCA2 Activates a Neuroprotective Subset of Macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.J.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome–Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by -Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Gibson, A.A.; Seimon, R.V.; Lee, C.M.Y.; Ayre, J.; Franklin, J.; Markovic, T.P.; Caterson, I.D.; Sainsbury, A. Do Ketogenic Diets Really Suppress Appetite? A Systematic Review and Meta-Analysis. Obes. Rev. 2014, 16, 64–76. [Google Scholar] [CrossRef]
- Yancy, W.S., Jr.; Olsen, M.K.; Guyton, J.R.; Bakst, R.P.; Westman, E.C. A Low-Carbohydrate, Ketogenic Diet versus a Low-Fat Diet to Treat Obesity and Hyperlipidemia: A Randomized, Controlled Trial. Ann. Intern. Med. 2004, 140, 769–777. [Google Scholar] [CrossRef]
- Hite, A.H.; Berkowitz, V.G.; Berkowitz, K. Low-Carbohydrate Diet Review: Shifting the Paradigm. Nutr. Clin. Pract. 2011, 26, 300–308. [Google Scholar] [CrossRef]
- Bruci, A.; Tuccinardi, D.; Tozzi, R.; Balena, A.; Santucci, S.; Frontani, R.; Mariani, S.; Basciani, S.; Spera, G.; Gnessi, L.; et al. Very Low-Calorie Ketogenic Diet: A Safe and Effective Tool for Weight Loss in Patients with Obesity and Mild Kidney Failure. Nutrients 2020, 12, 333. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Bianco, A.; Grimaldi, K.A.; Lodi, A.; Bosco, G. Long Term Successful Weight Loss with a Combination Biphasic Ketogenic Mediterranean Diet and Mediterranean Diet Maintenance Protocol. Nutrients 2013, 5, 5205–5217. [Google Scholar] [CrossRef] [PubMed]
- D’Abbondanza, M.; Ministrini, S.; Pucci, G.; Migliola, E.N.; Martorelli, E.-E.; Gandolfo, V.; Siepi, D.; Lupattelli, G.; Vaudo, G. Very Low-Carbohydrate Ketogenic Diet for the Treatment of Severe Obesity and Associated Non-Alcoholic Fatty Liver Disease: The Role of Sex Differences. Nutrients 2020, 12, 2748. [Google Scholar] [CrossRef] [PubMed]
- Bisschop, P.H.; Velden, M.G.M.D.S.-V.D.; Stellaard, F.; Kuipers, F.; Meijer, A.J.; Sauerwein, H.P.; Romijn, J.A. Dietary Carbohydrate Deprivation Increases 24-Hour Nitrogen Excretion without Affecting Postabsorptive Hepatic or Whole Body Protein Metabolism in Healthy Men. J. Clin. Endocrinol. Metab. 2003, 88, 3801–3805. [Google Scholar] [CrossRef]
- Pozefsky, T.; Felig, P.; Tobin, J.D.; Soeldner, J.S.; Cahill, G.F. Amino Acid Balance Across Tissues of the Forearm in Postabsorptive Man. Effects of Insulin at Two Dose Levels. J. Clin. Investig. 1969, 48, 2273–2282. [Google Scholar] [CrossRef]
- Watanabe, M.; Tozzi, R.; Risi, R.; Tuccinardi, D.; Mariani, S.; Basciani, S.; Spera, G.; Lubrano, C.; Gnessi, L. Beneficial Effects of the Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Comprehensive Review of the Literature. Obes. Rev. 2020, 21. [Google Scholar] [CrossRef]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’Yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805–815.e4. [Google Scholar] [CrossRef]
- Drinda, S.; Grundler, F.; Neumann, T.; Lehmann, T.; Steckhan, N.; Michalsen, A.; De Toledo, F.W. Effects of Periodic Fasting on Fatty Liver Index—A Prospective Observational Study. Nutrients 2019, 11, 2601. [Google Scholar] [CrossRef]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The Effect of Fasting or Calorie Restriction on Autophagy Induction: A Review of the Literature. Ageing Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef]
- Mindikoglu, A.L.; Abdulsada, M.M.; Jain, A.; Choi, J.M.; Jalal, P.K.; Devaraj, S.; Mezzari, M.P.; Petrosino, J.F.; Opekun, A.R.; Jung, S.Y. Intermittent Fasting from Dawn to Sunset for 30 Consecutive Days Is Associated with Anticancer Proteomic Signature and Upregulates Key Regulatory Proteins of Glucose and Lipid Metabolism, Circadian Clock, DNA Repair, Cytoskeleton Remodeling, Immune System and Cognitive Function in Healthy Subjects. J. Proteom. 2020, 217, 103645. [Google Scholar] [CrossRef]
- Pieri, C.; Falasca, M.; Marcheselli, F.; Moroni, F.; Recchioni, R.; Marmocchi, F.; Lupidi, G. Food Restriction in Female Wistar Rats: V. Lipid Peroxidation and Antioxidant Enzymes in the Liver. Arch. Gerontol. Geriatr. 1992, 14, 93–99. [Google Scholar] [CrossRef]
- Poon, H.F.; Shepherd, H.M.; Reed, T.T.; Calabrese, V.; Stella, A.-M.G.; Pennisi, G.; Cai, J.; Pierce, W.M.; Klein, J.B.; Butterfield, D.A. Proteomics Analysis Provides Insight Into Caloric Restriction Mediated Oxidation and Expression of Brain Proteins Associated with Age-Related Impaired Cellular Processes: Mitochondrial Dysfunction, Glutamate Dysregulation and Impaired Protein Synthesis. Neurobiol. Aging 2006, 27, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Aliasghari, F.; Izadi, A.; Gargari, B.P.; Ebrahimi, S. The Effects of Ramadan Fasting on Body Composition, Blood Pressure, Glucose Metabolism, and Markers of Inflammation in NAFLD Patients: An Observational Trial. J. Am. Coll. Nutr. 2017, 36, 640–645. [Google Scholar] [CrossRef]
- Ruderman, N.; Prentki, M. AMP Kinase and Malonyl-CoA: Targets for Therapy of the Metabolic Syndrome. Nat. Rev. Drug Discov. 2004, 3, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; Wong, K.I.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK–Caspase-6 Axis Controls Liver Damage in Nonalcoholic Steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.T.; Kaarniranta, K. AMP- Activated Protein Kinase Inhibits NF-κB Signaling and Inflammation: Impact on Healthspan and Lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Brandhorst, S.; Longo, V.D. Dietary Restrictions and Nutrition in the Prevention and Treatment of Cardiovascular Disease. Circ. Res. 2019, 124, 952–965. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting-Mimicking Diet and Markers/Risk Factors for Aging, Diabetes, Cancer, and Cardiovascular Disease. Sci. Transl. Med. 2017, 9, eaai8700. [Google Scholar] [CrossRef]
- Choi, I.Y.; Piccio, L.; Childress, P.; Bollman, B.; Ghosh, A.; Brandhorst, S.; Suarez, J.; Michalsen, A.; Cross, A.H.; Morgan, T.E.; et al. A Diet Mimicking Fasting Promotes Regeneration and Reduces Autoimmunity and Multiple Sclerosis Symptoms. Cell Rep. 2016, 15, 2136–2146. [Google Scholar] [CrossRef]
- Wei, S.; Han, R.; Zhao, J.; Wang, S.; Huang, M.; Wang, Y.; Chen, Y. Intermittent Administration of a Fasting-Mimicking Diet Intervenes in Diabetes Progression, Restores β Cells and Reconstructs Gut Microbiota in Mice. Nutr. Metab. 2018, 15, 1–12. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/results?cond=fasting+mimicking+diet&term=&cntry=&state=&city=&dist= (accessed on 9 November 2020).
- Miyake, T.; Kumagi, T.; Hirooka, M.; Furukawa, S.; Kawasaki, K.; Koizumi, M.; Todo, Y.; Yamamoto, S.; Nunoi, H.; Tokumoto, Y.; et al. Significance of Exercise in Nonalcoholic Fatty Liver Disease in Men: A Community-Based Large Cross-Sectional Study. J. Gastroenterol. 2014, 50, 230–237. [Google Scholar] [CrossRef]
- Palmer, M.; Schaffner, F. Effect of Weight Reduction on Hepatic Abnormalities in Overweight Patients. Gastroenterology 1990, 99, 1408–1413. [Google Scholar] [CrossRef]
- Keating, S.E.; Hackett, D.A.; Parker, H.M.; O’Connor, H.T.; Gerofi, J.A.; Sainsbury, A.; Baker, M.K.; Chuter, V.; Caterson, I.D.; George, J.; et al. Effect of Aerobic Exercise Training Dose on Liver Fat and Visceral Adiposity. J. Hepatol. 2015, 63, 174–182. [Google Scholar] [CrossRef]
- Hallsworth, K.; Fattakhova, G.; Hollingsworth, K.G.; Thoma, C.; Moore, S.; Taylor, R.; Day, C.P.; Trenell, M.I. Resistance Exercise Reduces Liver Fat and Its Mediators in Non-Alcoholic Fatty Liver Disease Independent of Weight Loss. Gut 2011, 60, 1278–1283. [Google Scholar] [CrossRef]
- Baba, C.S.; Alexander, G.; Kalyani, B.; Pandey, R.; Rastogi, S.; Pandey, A.; Choudhuri, G. Effect of Exercise and Dietary Modification on Serum Aminotransferase Levels in Patients with Nonalcoholic Steatohepatitis. J. Gastroenterol. Hepatol. 2006, 21, 191–198. [Google Scholar] [CrossRef]
- Balducci, S.; Cardelli, P.; Pugliese, L.; D’Errico, V.; Haxhi, J.; Alessi, E.; Iacobini, C.; Menini, S.; Bollanti, L.; Conti, F.G.; et al. Volume-Dependent Effect of Supervised Exercise Training on Fatty Liver and Visceral Adiposity Index in Subjects with Type 2 Diabetes The Italian Diabetes Exercise Study (IDES). Diabetes Res. Clin. Pract. 2015, 109, 355–363. [Google Scholar] [CrossRef]
- Takahashi, A.; Abe, K.; Usami, K.; Imaizumi, H.; Hayashi, M.; Okai, K.; Kanno, Y.; Tanji, N.; Watanabe, H.; Ohira, H. Simple Resistance Exercise helps Patients with Non-alcoholic Fatty Liver Disease. Int. J. Sports Med. 2015, 36, 848–852. [Google Scholar] [CrossRef]
- AbdelBasset, W.K.; Tantawy, S.A.; Kamel, D.M.; Alqahtani, B.A.; Elnegamy, T.E.; Soliman, G.S.; Ibrahim, A.A. Effects of High-Intensity Interval and Moderate-Intensity Continuous Aerobic Exercise on Diabetic Obese Patients with Nonalcoholic Fatty Liver Disease. Medicine 2020, 99, e19471. [Google Scholar] [CrossRef] [PubMed]
- Huber, Y.; Pfirrmann, D.; Gebhardt, I.; Labenz, C.; Gehrke, N.; Straub, B.K.; Ruckes, C.; Bantel, H.; Belda, E.; Clément, K.; et al. Improvement of non-invasive markers of NAFLD from an individualised, web-based exercise program. Aliment. Pharmacol. Ther. 2019, 50, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Bilet, L.; Brouwers, B.; Van Ewijk, P.A.; Hesselink, M.K.C.; Kooi, M.E.; Schrauwen, P.; Schrauwen-Hinderling, V.B. Acute Exercise Does Not Decrease Liver Fat in Men with Overweight or NAFLD. Sci. Rep. 2015, 5, 9709. [Google Scholar] [CrossRef]
- Kullman, E.L.; Kelly, K.R.; Haus, J.M.; Fealy, C.E.; Scelsi, A.R.; Pagadala, M.R.; Flask, C.A.; McCullough, A.J.; Kirwan, J.P. Short-Term Aerobic Exercise Training Improves Gut Peptide Regulation in Nonalcoholic Fatty Liver Disease. J. Appl. Physiol. 2016, 120, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Liong, E.C.; So, K.F.; Fung, M.-L.; Tipoe, G.L. Beneficial Mechanisms of Aerobic Exercise on Hepatic Lipid Metabolism in Non-Alcoholic Fatty Liver Disease. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 139–144. [Google Scholar] [CrossRef]
- Farzanegi, P.; Dana, A.; Ebrahimpoor, Z.; Asadi, M.; Azarbayjani, M.A. Mechanisms of Beneficial Effects of Exercise Training on Non-Alcoholic Fatty Liver Disease (NAFLD): Roles of Oxidative Stress and Inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Prado, C.M.; Purcell, S.A.; Alish, C.; Pereira, S.L.; Deutz, N.E.; Heyland, D.K.; Goodpaster, B.H.; Tappenden, K.A.; Heymsfield, S.B. Implications of Low Muscle Mass Across the Continuum of Care: A Narrative Review. Ann. Med. 2018, 50, 675–693. [Google Scholar] [CrossRef]
- Xu, Y.; Guan, Y.; Jin, W.; Ding, L.; Wu, J. Higher Appendicular Skeletal Muscle Mass Percentage Is an Independent Protective Factor for Non-Alcoholic Steatohepatitis and Significant Fibrosis in Male with NAFLD. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Dasarathy, S. Consilience in Sarcopenia of Cirrhosis. J. Cachex Sarcopenia Muscle 2012, 3, 225–237. [Google Scholar] [CrossRef]
- Baumgartner, R.N. Body Composition in Healthy Aging. Ann. N. Y. Acad. Sci. 2006, 904, 437–448. [Google Scholar] [CrossRef]
- Montano-Loza, A.J.; Angulo, P.; Meza-Junco, J.; Prado, C.M.M.; Sawyer, M.B.; Beaumont, C.; Esfandiari, N.; Ma, M.; Baracos, V.E. Sarcopenic Obesity and Myosteatosis Are Associated with Higher Mortality in Patients with Cirrhosis. J. Cachex Sarcopenia Muscle 2016, 7, 126–135. [Google Scholar] [CrossRef]
- Nachit, M.; Leclercq, I.A. Emerging Awareness on the Importance of Skeletal Muscle in Liver Diseases: Time to Dig Deeper into Mechanisms! Clin. Sci. 2019, 133, 465–481. [Google Scholar] [CrossRef]
- Sinclair, M.; Gow, P.J.; Grossmann, M.; Angus, P.W. Review Article: Sarcopenia in Cirrhosis—Aetiology, Implications and Potential Therapeutic Interventions. Aliment. Pharmacol. Ther. 2016, 43, 765–777. [Google Scholar] [CrossRef]
- Marchesini, G.; Zoli, M.; Angiolini, A.; Dondi, C.; Bianchi, F.B.; Pisi, E. Muscle Protein Breakdown in Liver Cirrhosis and the Role of Altered Carbohydrate Metabolism. Hepatology 1981, 1, 294–299. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Oberbach, A.; Bossenz, Y.; Lehmann, S.; Niebauer, J.; Adams, V.; Paschke, R.; Schön, M.R.; Blüher, M.; Punkt, K. Altered Fiber Distribution and Fiber-Specific Glycolytic and Oxidative Enzyme Activity in Skeletal Muscle of Patients with Type 2 Diabetes. Diabetes Care 2006, 29, 895–900. [Google Scholar] [CrossRef]
- Chalé-Rush, A.; Morris, E.P.; Kendall, T.L.; Brooks, N.E.; Fielding, R.A. Effects of Chronic Overload on Muscle Hypertrophy and mTOR Signaling in Young Adult and Aged Rats. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1232–1239. [Google Scholar] [CrossRef]
- Cabrera, D.; Ruiz, A.; Cabello-Verrugio, C.; Brandan, E.; Estrada, L.; Pizarro, M.; Solis, N.; Torres, J.; Barrera, F.; Arrese, M. Diet-Induced Nonalcoholic Fatty Liver Disease Is Associated with Sarcopenia and Decreased Serum Insulin-Like Growth Factor. Dig. Dis. Sci. 2016, 61, 3190–3198. [Google Scholar] [CrossRef]
- Kim, G.; Lee, S.; Lee, Y.; Jun, J.E.; Ahn, J.; Bae, J.C.; Jin, S.; Hur, K.Y.; Jee, J.H.; Lee, M.; et al. Relationship Between Relative Skeletal Muscle Mass and Nonalcoholic Fatty Liver Disease: A 7-Year Longitudinal Study. Hepatology 2018, 68, 1755–1768. [Google Scholar] [CrossRef]
- Bhanji, R.A.; Narayanan, P.; Allen, A.M.; Malhi, H.; Watt, K.D. Sarcopenia in Hiding: The Risk and Consequence of Underestimating Muscle Dysfunction in Nonalcoholic Steatohepatitis. Hepatology 2017, 66, 2055–2065. [Google Scholar] [CrossRef]
- Hong, H.C.; Hwang, S.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Relationship Between Sarcopenia and Nonalcoholic Fatty Liver Disease: The Korean Sarcopenic Obesity Study. Hepatology 2014, 59, 1772–1778. [Google Scholar] [CrossRef]
- Wang, Y.; Wehling-Henricks, M.; Welc, S.S.; Fisher, A.L.; Zuo, Q.; Tidball, J.G. Aging of the Immune System Causes Reductions in Muscle Stem Cell Populations, Promotes Their Shift to a Fibrogenic Phenotype, and Modulates Sarcopenia. FASEB J. 2019, 33, 1415–1427. [Google Scholar] [CrossRef]
- Machek, S.B. Mechanisms of Sarcopenia: Motor Unit Remodelling and Muscle Fibre Type Shifts with Ageing. J. Physiol. 2018, 596, 3467–3468. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; Macdonald, N.; Mantovani, G.; et al. Definition and Classification of Cancer Cachexia: An International Consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Zhai, Y.; Xiao, Q. The Common Mechanisms of Sarcopenia and NAFLD. BioMed Res. Int. 2017, 2017, 1–5. [Google Scholar] [CrossRef]
- Song, M.; Xia, L.; Liu, Q.; Sun, M.; Wang, F.; Yang, C. Sarcopenia in Liver Disease: Current Evidence and Issues to Be sResolved. In Muscle Atrophy; Springer: Berlin/Heidelberg, Germany, 2018; pp. 413–433. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin Resistance in Non-Diabetic Patients with Non-Alcoholic Fatty Liver Disease: Sites and Mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ—Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Bril, F.; Barb, D.; Portillo-Sanchez, P.; Biernacki, D.; Lomonaco, R.; Suman, A.; Weber, M.H.; Budd, J.T.; Lupi, M.E.; Cusi, K. Metabolic and Histological Implications of Intrahepatic Triglyceride Content in Nonalcoholic Fatty Liver Disease. Hepatology 2017, 65, 1132–1144. [Google Scholar] [CrossRef]
- Stefan, N.; Schick, F.; Häring, H.-U. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab. 2017, 26, 292–300. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Poggiogalle, E.; Lubrano, C.; Gnessi, L.; Mariani, S.; Lenzi, A.; Donini, L.M. Fatty Liver Index Associates with Relative Sarcopenia and GH/IGF-1 Status in Obese Subjects. PLoS ONE 2016, 11, e0145811. [Google Scholar] [CrossRef] [PubMed]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Carulli, L.; Bertolotti, M.; Lonardo, A. Endocrine and Liver Interaction: The Role of Endocrine Pathways in NASH. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 236–247. [Google Scholar] [CrossRef]
- Perrini, S.; Laviola, L.; Carreira, M.C.; Cignarelli, A.; Natalicchio, A.; Giorgino, F. The GH/IGF1 Axis and Signaling Pathways in the Muscle and Bone: Mechanisms Underlying Age-Related Skeletal Muscle Wasting and Osteoporosis. J. Endocrinol. 2010, 205, 201–210. [Google Scholar] [CrossRef]
- Cabrera, D.; Cabello-Verrugio, C.; Solís, N.; Martín, D.S.; Cofré, C.; Pizarro, M.; Arab, J.P.; Ábrigo, J.; Campos, F.; Irigoyen, B.; et al. Somatotropic Axis Dysfunction in Non-Alcoholic Fatty Liver Disease: Beneficial Hepatic and Systemic Effects of Hormone Supplementation. Int. J. Mol. Sci. 2018, 19, 1339. [Google Scholar] [CrossRef]
- Aguirre, G.A.; Ita, J.R.-D.; De La Garza, R.G.; Castilla-Cortázar, I. Insulin-Like Growth Factor-1 Deficiency and Metabolic Syndrome. J. Transl. Med. 2016, 14, 1–23. [Google Scholar] [CrossRef]
- Ennequin, G.; Sirvent, P.; Whitham, M. Role of Exercise-Induced Hepatokines in Metabolic Disorders. Am. J. Physiol. Metab. 2019, 317, E11–E24. [Google Scholar] [CrossRef]
- Severinsen, M.C.K.; Pedersen, B.K. Muscle–Organ Crosstalk: The Emerging Roles of Myokines. Endocr. Rev. 2020, 41, 594–609. [Google Scholar] [CrossRef]
- Weigert, C.; Hoene, M.; Plomgaard, P. Hepatokines—A Novel Group of Exercise Factors. Pflügers Arch. Eur. J. Physiol. 2018, 471, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking Nonalcoholic Fatty Liver Disease and Insulin Resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef]
- Ke, Y.; Xu, C.; Lin, J.; Li, Y. Role of Hepatokines in Non-Alcoholic Fatty Liver Disease. J. Transl. Intern. Med. 2019, 7, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific Expression of βKlotho and Fibroblast Growth Factor (FGF) Receptor Isoforms Determines Metabolic Activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a Novel FGF, FGF-21, Preferentially Expressed in the Liver. Biochim. Biophys. Acta 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Badman, M.K.; Koester, A.; Flier, J.S.; Kharitonenkov, A.; Maratos-Flier, E. Fibroblast Growth Factor 21-Deficient Mice Demonstrate Impaired Adaptation to Ketosis. Endocrinology 2009, 150, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-López, T.; Escolà-Gil, J.C.; Cedó, L.; Zali, M.R.; Molaei, M.; et al. Hepatic Regulation of VLDL Receptor by PPARβ/δ and FGF21 Modulates Non-Alcoholic Fatty Liver Disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 Promotes NASH-HCC Transition via Hepatocyte-TLR4-IL-17A Signaling. Theranostics 2020, 10, 9923–9936. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Bina, H.A.; Ouchi, N.; Akasaki, Y.; Kharitonenkov, A.; Walsh, K. FGF21 is an Akt-Regulated Myokine. FEBS Lett. 2008, 582, 3805–3810. [Google Scholar] [CrossRef]
- Hansen, J.S.; Pedersen, B.K.; Xu, G.; Lehmann, R.; Weigert, C.; Plomgaard, P. Exercise-Induced Secretion of FGF21 and Follistatin Are Blocked by Pancreatic Clamp and Impaired in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 2816–2825. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, W.; Zeng, L.-Q.; Bai, H.; Li, J.; Zhou, J.; Zhou, G.-Y.; Fang, C.-W.; Wang, F.; Qin, X.-J. Exercise and Dietary Intervention Ameliorate High-Fat Diet-Induced NAFLD and Liver Aging by Inducing Lipophagy. Redox Biol. 2020, 36, 101635. [Google Scholar] [CrossRef]
- Xiao, J.; Bei, Y.; Liu, J.; Dimitrova-Shumkovska, J.; Kuang, D.; Zhou, Q.; Li, J.; Yang, Y.; Xiang, Y.; Wang, F.; et al. miR-212 Downregulation Contributes to the Protective Effect of Exercise Against Non-Alcoholic Fatty Liver via Targeting FGF-21. J. Cell. Mol. Med. 2016, 20, 204–216. [Google Scholar] [CrossRef]
- Slusher, A.L.; Whitehurst, M.; Zoeller, R.F.; Mock, J.; Maharaj, M.; Huang, C.-J. Attenuated Fibroblast Growth Factor 21 Response to Acute Aerobic Exercise in Obese Individuals. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Abe, K.; Fujita, M.; Hayashi, M.; Okai, K.; Ohira, H. Simple Resistance Exercise Decreases Cytokeratin 18 and Fibroblast Growth Factor 21 Levels in Patients with Nonalcoholic Fatty Liver Disease. Medicine 2020, 99, e20399. [Google Scholar] [CrossRef] [PubMed]
- Trepanowski, J.F.; Mey, J.T.; Varady, K.A. Fetuin-A: A Novel Link Between Obesity and Related Complications. Int. J. Obes. 2014, 39, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, J.B.; Gitterman, A.; Ryan, A.S.; Prior, S.J. Effects of Exercise Training and Weight Loss on Plasma Fetuin-A Levels and Insulin Sensitivity in Overweight Older Men. J. Diabetes Res. 2017, 2017, 1–7. [Google Scholar] [CrossRef]
- Malin, S.K.; Mulya, A.; Fealy, C.E.; Haus, J.M.; Pagadala, M.R.; Scelsi, A.R.; Huang, H.; Flask, C.A.; McCullough, A.J.; Kirwan, J.P. Fetuin-A Is Linked to Improved Glucose Tolerance After Short-Term Exercise Training in Nonalcoholic Fatty Liver Disease. J. Appl. Physiol. 2013, 115, 988–994. [Google Scholar] [CrossRef]
- Malin, S.K.; Del Rincon, J.P.; Huang, H.; Kirwan, J.P. Exercise-Induced Lowering of Fetuin-A May Increase Hepatic Insulin Sensitivity. Med. Sci. Sports Exerc. 2014, 46, 2085–2090. [Google Scholar] [CrossRef]
- Fang, J.; Wang, S.; Smiley, E.; Bonadio, J. Genes Coding for Mouse Activin βCand βEAre Closely Linked and Exhibit a Liver-Specific Expression Pattern in Adult Tissues. Biochem. Biophys. Res. Commun. 1997, 231, 655–661. [Google Scholar] [CrossRef]
- Yndestad, A.; Haukeland, J.W.; Dahl, T.B.; Bjøro, K.; Gladhaug, I.P.; Berge, C.; Damås, J.K.; Haaland, T.; Løberg, E.M.; Linnestad, P.; et al. A Complex Role of Activin A in Non-Alcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2009, 104, 2196–2205. [Google Scholar] [CrossRef]
- Hashimoto, O.; Funaba, M.; Sekiyama, K.; Doi, S.; Shindo, D.; Satoh, R.; Itoi, H.; Oiwa, H.; Morita, M.; Suzuki, C.; et al. Activin E Controls Energy Homeostasis in Both Brown and White Adipose Tissues as a Hepatokine. Cell Rep. 2018, 25, 1193–1203. [Google Scholar] [CrossRef]
- Ungerleider, N.A.; Bonomi, L.M.; Brown, M.L.; Schneyer, A. Increased Activin Bioavailability Enhances Hepatic Insulin Sensitivity While Inducing Hepatic Steatosis in Male Mice. Endocrinology 2013, 154, 2025–2033. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Anastasilakis, A.D.; Triantafyllou, G.A.; Mantzoros, C.S. Activin A and Follistatin in Patients with Nonalcoholic Fatty Liver Disease. Metabolism 2016, 65, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Walton, K.L.; Qian, H.; Colgan, T.D.; Hagg, A.; Watt, M.J.; Harrison, C.A.; Gregorevic, P. Differential Effects of IL6 and Activin A in the Development of Cancer-Associated Cachexia. Cancer Res. 2016, 76, 5372–5382. [Google Scholar] [CrossRef] [PubMed]
- Loumaye, A.; De Barsy, M.; Nachit, M.; Lause, P.; Frateur, L.; Van Maanen, A.; Tréfois, P.; Gruson, D.; Thissen, J.-P. Role of Activin A and Myostatin in Human Cancer Cachexia. J. Clin. Endocrinol. Metab. 2015, 100, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Patel, K. Follistatin. Int. J. Biochem. Cell Biol. 1998, 30, 1087–1093. [Google Scholar] [CrossRef]
- Hansen, J.; Brandt, C.; Nielsen, A.R.; Hojman, P.; Whitham, M.; Febbraio, M.A.; Pedersen, B.K.; Plomgaard, P. Exercise Induces a Marked Increase in Plasma Follistatin: Evidence That Follistatin Is a Contraction-Induced Hepatokine. Endocrinology 2011, 152, 164–171. [Google Scholar] [CrossRef]
- Hofmann, M.; Schober-Halper, B.; Oesen, S.; Franzke, B.; Tschan, H.; Bachl, N.; Strasser, E.-M.; Quittan, M.; Wagner, K.-H.; Wessner, B. Effects of Elastic Band Resistance Training and Nutritional Supplementation on Muscle Quality and Circulating Muscle Growth and Degradation Factors of Institutionalized Elderly Women: The Vienna Active Ageing Study (VAAS). Eur. J. Appl. Physiol. 2016, 116, 885–897. [Google Scholar] [CrossRef]
- Takebayashi, K.; Aso, Y.; Inukai, T. Role of Retinol-Binding Protein 4 in the Pathogenesis of Type 2 Diabetes. Expert Rev. Endocrinol. Metab. 2008, 3, 161–173. [Google Scholar] [CrossRef]
- Mansouri, M.; Nikooie, R.; Keshtkar, A.; Larijani, B.; Omidfar, K. Effect of Endurance Training on Retinol-Binding Protein 4 Gene Expression and Its Protein Level in Adipose Tissue and the Liver in Diabetic Rats Induced by a High-Fat Diet and Streptozotocin. J. Diabetes Investig. 2014, 5, 484–491. [Google Scholar] [CrossRef]
- Christou, G.; Tselepis, A.; Kiortsis, D. The Metabolic Role of Retinol Binding Protein 4: An Update. Horm. Metab. Res. 2011, 44, 6–14. [Google Scholar] [CrossRef]
- Balagopal, P.; Graham, T.E.; Kahn, B.B.; Altomare, A.; Funanage, V.; George, D. Reduction of Elevated Serum Retinol Binding Protein in Obese Children by Lifestyle Intervention: Association with Subclinical Inflammation. J. Clin. Endocrinol. Metab. 2007, 92, 1971–1974. [Google Scholar] [CrossRef]
- Kersten, S.; Lichtenstein, L.; Steenbergen, E.; Mudde, K.; Hendriks, H.F.; Hesselink, M.K.; Schrauwen, P.; Müller, M. Caloric Restriction and Exercise Increase Plasma ANGPTL4 Levels in Humans via Elevated Free Fatty Acids. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.E.; Lam, L.N.; Yang, K.; Zhou, A.Y.; Koliwad, S.; Wang, J.-C. Angiopoietin-Like 4 (Angptl4) Protein Is a Physiological Mediator of Intracellular Lipolysis in Murine Adipocytes. J. Biol. Chem. 2017, 292, 16135. [Google Scholar] [CrossRef] [PubMed]
- Ingerslev, B.; Hansen, J.S.; Hoffmann, C.; Clemmesen, J.O.; Secher, N.H.; Scheler, M.; De Angelis, M.H.; Häring, H.U.; Pedersen, B.K.; Weigert, C.; et al. Angiopoietin-Like Protein 4 Is an Exercise-Induced Hepatokine in Humans, Regulated by Glucagon and cAMP. Mol. Metab. 2017, 6, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, H.; Ryan, A.S. Skeletal Muscle Angiopoietin-Like Protein 4 and Glucose Metabolism in Older Adults after Exercise and Weight Loss. Metabolites 2020, 10, 354. [Google Scholar] [CrossRef] [PubMed]
- Cullberg, K.B.; Christiansen, T.; Paulsen, S.K.; Bruun, J.M.; Pedersen, S.B.; Richelsen, B. Effect of Weight Loss and Exercise on Angiogenic Factors in the Circulation and in Adipose Tissue in Obese Subjects. Obesity 2013, 21, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Hwang, S.Y.; Lee, C.H.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Increased Selenoprotein P Levels in Subjects with Visceral Obesity and Nonalcoholic Fatty Liver Disease. Diabetes Metab. J. 2013, 37, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, S.; Hashida, R.; Takano, Y.; Bekki, M.; Nakano, D.; Omoto, M.; Nago, T.; Kawaguchi, T.; Matsuse, H.; Torimura, T.; et al. Hybrid Training System Improves Insulin Resistance in Patients with Nonalcoholic Fatty Liver Disease: A Randomized Controlled Pilot Study. Tohoku J. Exp. Med. 2020, 252, 23–32. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscles and Their Myokines. J. Exp. Biol. 2010, 214, 337–346. [Google Scholar] [CrossRef]
- Huh, J.Y. The Role of Exercise-Induced Myokines in Regulating Metabolism. Arch. Pharmacal Res. 2017, 41, 14–29. [Google Scholar] [CrossRef]
- Fan, N.; Sun, H.; Wang, Y.; Wang, Y.; Zhang, L.; Xia, Z.; Peng, L.; Hou, Y.; Shen, W.; Liu, R.; et al. Follistatin-Like 1: A Potential Mediator of Inflammation in Obesity. Mediat. Inflamm. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Xi, Y.; Gong, D.-W.; Tian, Z. FSTL1 as a Potential Mediator of Exercise-Induced Cardioprotection in Post-Myocardial Infarction Rats. Sci. Rep. 2016, 6, 32424. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Ahn, C.W.; Park, J.S.; Kim, Y.; Nam, J.S. Circulating Myokine Levels in Different Stages of Glucose Intolerance. Medicine 2020, 99, e19235. [Google Scholar] [CrossRef] [PubMed]
- Bessepatin, A.; Montastier, E.; Vinel, C.; Castanlaurell, I.; Louche, K.; Dray, C.; Daviaud, D.; Mir, L.M.; Marques, M.-A.; Thalamas, C.; et al. Effect of Endurance Training on Skeletal Muscle Myokine Expression in Obese Men: Identification of Apelin as a Novel Myokine. Int. J. Obes. 2014, 38, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Görgens, S.W.; Raschke, S.; Holven, K.B.; Jensen, J.; Eckardt, K.; Eckel, J. Regulation of Follistatin-Like Protein 1 Expression and Secretion in Primary Human Skeletal Muscle Cells. Arch. Physiol. Biochem. 2013, 119, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kon, M.; Tanimura, Y.; Yoshizato, H. Effects of Acute Endurance Exercise on Follistatin-Like 1 and Apelin in the Circulation and Metabolic Organs in Rats. Arch. Physiol. Biochem. 2020, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kon, M.; Ebi, Y.; Nakagaki, K. Effects of Acute Sprint Interval Exercise on Follistatin-Like 1 and Apelin Secretions. Arch. Physiol. Biochem. 2019, 1–5. [Google Scholar] [CrossRef]
- Bostroem, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostroem, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α- Dependent Myokine That Drives Brown-Fat-Like Development of White Fat and Thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Kurdiova, T.; Balaz, M.; Vician, M.; Maderova, D.; Vlcek, M.; Valkovic, L.; Srbecky, M.; Imrich, R.; Kyselovicova, O.; Belan, V.; et al. Effects of Obesity, Diabetes and Exercise onfndc5gene Expression and Irisin Release in Human Skeletal Muscle and Adipose Tissue: In Vivo and in Vitro studies. J. Physiol. 2014, 592, 1091–1107. [Google Scholar] [CrossRef]
- Cao, R.Y.; Zheng, H.; Redfearn, D.; Yang, J. FNDC5: A Novel Player in Metabolism and Metabolic Syndrome. Biochimie 2019, 158, 111–116. [Google Scholar] [CrossRef]
- Canivet, C.M.; Bonnafous, S.; Rousseau, D.; LeClere, P.S.; Lacas-Gervais, S.; Patouraux, S.; Sans, A.; Luci, C.; Bailly-Maitre, B.; Iannelli, A.; et al. Hepatic FNDC5 is a Potential Local Protective Factor Against Non-Alcoholic Fatty Liver. Biochim. Biophys. Acta 2020, 1866, 165705. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Anastasilakis, A.D.; Geladari, E.V.; Mantzoros, C.S. Irisin in Patients with Nonalcoholic Fatty Liver Disease. Metabolism 2014, 63, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Shanaki, M.; Moradi, N.; Emamgholipour, S.; Fadaei, R.; Poustchi, H. Lower Circulating Irisin Is Associated with Nonalcoholic Fatty Liver Disease and Type 2 Diabetes. Diabetes Metab. Syndr. 2017, 11, S467–S472. [Google Scholar] [CrossRef] [PubMed]
- Waluga, M.; Kukla, M.; Kotulski, R.; Zorniak, M.; Boryczka, G.; Kajor, M.; Lekstan, A.; Olczyk, P.; Waluga, E. Omentin, Vaspin and Irisin in Chronic Liver Diseases. J. Physiol. Pharmacol. 2019, 70, 277–285. [Google Scholar] [CrossRef]
- Choi, E.S.; Kim, M.K.; Song, M.K.; Kim, J.M.; Kim, E.S.; Chung, W.J.; Park, K.S.; Cho, K.B.; Hwang, J.S.; Jang, B.K. Association between Serum Irisin Levels and Non-Alcoholic Fatty Liver Disease in Health Screen Examinees. PLoS ONE 2014, 9, e110680. [Google Scholar] [CrossRef] [PubMed]
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Abbate, M.; Montemayor, S.; Mascaró, C.M.; Casares, M.; Tejada, S.; Abete, I.; Zulet, M.A.; Tur, J.A.; et al. Oxidative Stress and Pro-Inflammatory Status in Patients with Non-Alcoholic Fatty Liver Disease. Antioxidants 2020, 9, 759. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Pérez, O.; Reyes-Garcia, R.; Muñoz-Torres, M.; Merino, E.; Boix, V.; Reus, S.; Giner, L.; Alfayate, R.; García-Fontana, B.; Sánchez-Payá, J.; et al. High Irisin Levels in Nondiabetic HIV-Infected Males Are Associated with Insulin Resistance, Nonalcoholic Fatty Liver Disease, and Subclinical Atherosclerosis. Clin. Endocrinol. 2018, 89, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Anastasilakis, A.D.; Margouta, A.; Mantzoros, C.S. Association Between Circulating Irisin and Homocysteine in Patients with Nonalcoholic Fatty Liver Disease. Endocrine 2014, 49, 560–562. [Google Scholar] [CrossRef]
- De La Torre-Saldaña, V.A.; Gómez-Sámano, M.Á.; Gómez-Pérez, F.J.; Rosas-Saucedo, J.; León-Suárez, A.; Grajales-Gómez, M.; Oseguera-Moguel, J.; Vega-Beyhart, A.; Cuevas-Ramos, D. Fasting Insulin and Alanine Amino Transferase, but not FGF21, Were Independent Parameters Related with Irisin Increment after Intensive Aerobic Exercising. Rev. Investig. Clin. 2019, 71, 133–140. [Google Scholar] [CrossRef]
- Wiecek, M.; Szymura, J.; Maciejczyk, M.; Kantorowicz, M.; Szygula, Z. Acute Anaerobic Exercise Affects the Secretion of Asprosin, Irisin, and Other Cytokines–A Comparison Between Sexes. Front. Physiol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Dong, H.N.; Park, S.Y.; Le, C.T.; Choi, D.-H.; Cho, E.-H. Irisin Regulates the Functions of Hepatic Stellate Cells. Endocrinol. Metab. 2020, 35, 647–655. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, Y.; Bi, J.; Wang, M.; Zhang, L.; Wang, T.; Wei, S.; Mou, X.; Lv, Y.; Wu, R. Involvement of Kindlin-2 in Irisin’s Protection Against Ischaemia Reperfusion-Induced Liver Injury in High-Fat Diet-Fed Mice. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Ge, Y.; Liu, N.; Jia, W.-D.; Li, J.-S.; Ma, J.-L.; Yu, J.-H.; Xu, G. Kindlin-2: A Novel Prognostic Biomarker for Patients with Hepatocellular Carcinoma. Pathol. Res. Pract. 2015, 211, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, Y.; Gao, Y.; Li, Q.; Zeng, Z.; Li, Y.; Chen, H. Kindlin-2 Regulates Hepatic Stellate Cells Activation and Liver Fibrogenesis. Cell Death Discov. 2018, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Hadinia, A.; Doustimotlagh, A.H.; Goodarzi, H.R.; Arya, A.; Jafarinia, M. Circulating Levels of Pro-inflammatory Cytokines in Patients with Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. Iran. J. Immunol. 2019, 16, 327–333. [Google Scholar]
- Pinto, A.P.; Da Rocha, A.L.; Cabrera, E.M.; Marafon, B.B.; Kohama, E.B.; Rovina, R.L.; Simabuco, F.M.; Junior, C.R.B.; De Moura, L.P.; Pauli, J.R.; et al. Role of Interleukin-6 in Inhibiting Hepatic Autophagy Markers in Exercised Mice. Cytokine 2020, 130, 155085. [Google Scholar] [CrossRef]
- Monteiro, P.A.; Prado, W.L.D.; Tenório, T.R.D.S.; Tomaz, L.M.; St-Pierre, D.H.; Lira, F. Immunometabolic Changes in Hepatocytes Arising from Obesity and the Practice of Physical Exercise. Curr. Pharm. Des. 2018, 24, 3200–3209. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Pedersen, B.K. Muscle-Derived Interleukin-6: Mechanisms for Activation and Possible Biological Roles. FASEB J. 2002, 16, 1335–1347. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Febbraio, M.; Saltin, B. Searching for the Exercise Factor: Is IL-6 a Candidate? J. Muscle Res. Cell Motil. 2003, 24, 113–119. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M. Muscle-Derived Interleukin-6—A Possible Link Between Skeletal Muscle, Adipose Tissue, Liver, and Brain. Brain Behav. Immun. 2005, 19, 371–376. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Shiba, N.; Maeda, T.; Matsugaki, T.; Takano, Y.; Itou, M.; Sakata, M.; Taniguchi, E.; Nagata, K.; Sata, M. Hybrid Training of Voluntary and Electrical Muscle Contractions Reduces Steatosis, Insulin Resistance, and IL-6 Levels in Patients with NAFLD: A Pilot Study. J. Gastroenterol. 2011, 46, 746–757. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carneros, D.; López-Lluch, G.; Bustos, M. Physiopathology of Lifestyle Interventions in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2020, 12, 3472. https://doi.org/10.3390/nu12113472
Carneros D, López-Lluch G, Bustos M. Physiopathology of Lifestyle Interventions in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients. 2020; 12(11):3472. https://doi.org/10.3390/nu12113472
Chicago/Turabian StyleCarneros, David, Guillermo López-Lluch, and Matilde Bustos. 2020. "Physiopathology of Lifestyle Interventions in Non-Alcoholic Fatty Liver Disease (NAFLD)" Nutrients 12, no. 11: 3472. https://doi.org/10.3390/nu12113472
APA StyleCarneros, D., López-Lluch, G., & Bustos, M. (2020). Physiopathology of Lifestyle Interventions in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients, 12(11), 3472. https://doi.org/10.3390/nu12113472