B Vitamins and Their Role in Immune Regulation and Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
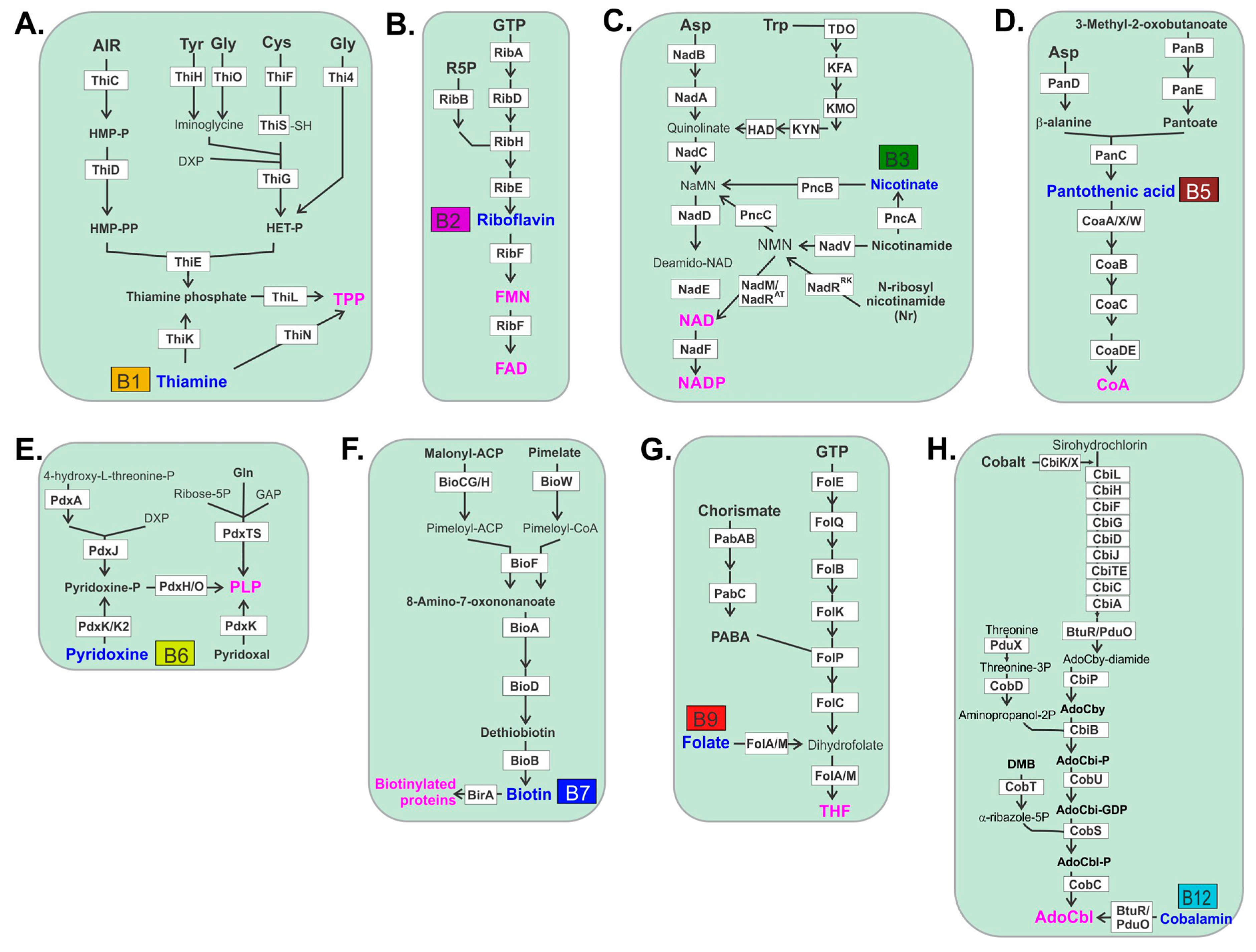
2. Gut Microbes Generate B Vitamins.
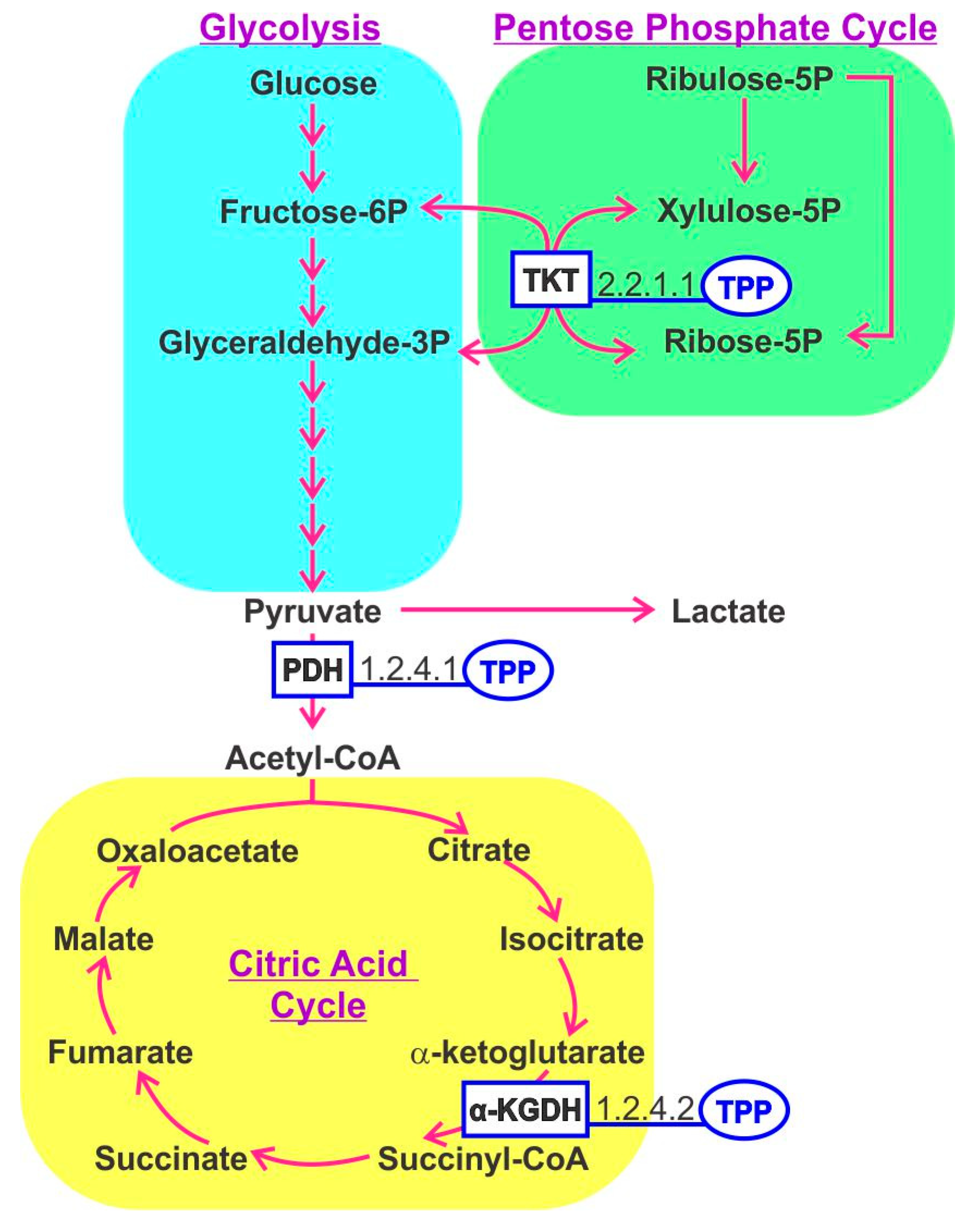
3. Thiamine B1
4. Riboflavin B2
5. Niacin B3
6. Pantothenic Acid B5
7. Pyridoxine B6
8. Biotin B7
9. Folate B9
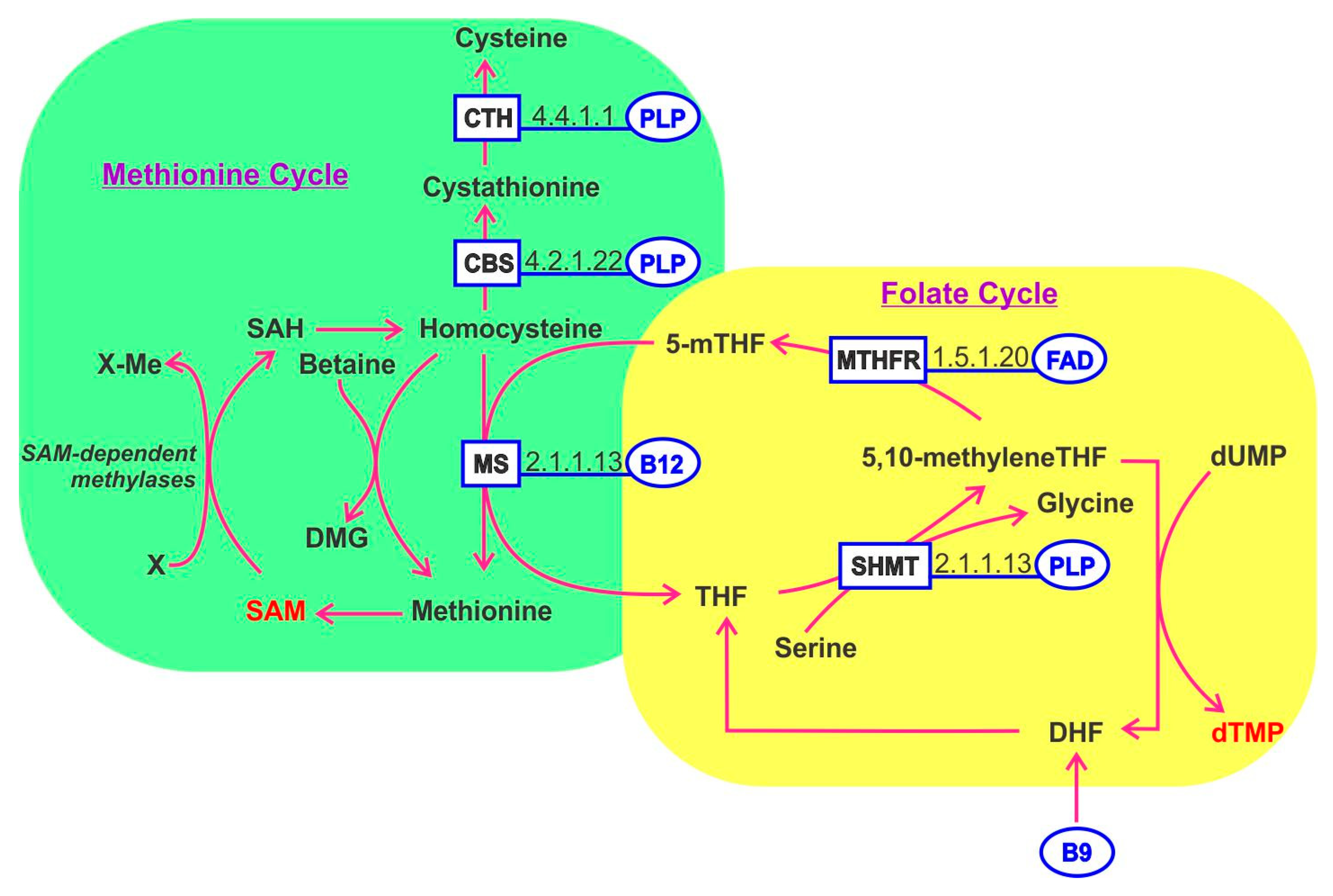
10. One-Carbon Metabolism
11. Cobalamin B12
12. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Whatham, A.; Bartlett, H.; Eperjesi, F.; Blumenthal, C.; Allen, J.; Suttle, C.; Gaskin, K. Vitamin and mineral deficiencies in the developed world and their effect on the eye and vision. Ophthalmic Physiol. Opt. 2008, 28, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Revuelta, J.L.; Buey, R.M.; Ledesma-Amaro, R.; Vandamme, E.J. Microbial biotechnology for the synthesis of (pro)vitamins, biopigments and antioxidants: Challenges and opportunities. Microb. Biotechnol. 2016, 9, 564–567. [Google Scholar] [CrossRef]
- Rodionov, D.A.; Arzamasov, A.A.; Khoroshkin, M.S.; Iablokov, S.N.; Leyn, S.A.; Peterson, S.N.; Novichkov, P.S.; Osterman, A.L. Micronutrient requirements and sharing capabilities of the human gut microbiome. Front. Microbiol. 2019, 10, 1316. [Google Scholar] [CrossRef]
- Sharma, V.; Rodionov, D.A.; Leyn, S.A.; Tran, D.; Iablokov, S.N.; Ding, H.; Peterson, D.A.; Osterman, A.L.; Peterson, S.N. B-vitamin sharing promotes stability of gut microbial communities. Front. Microbiol. 2019, 10, 1485. [Google Scholar] [CrossRef]
- Frank, R.A.; Leeper, F.J.; Luisi, B.F. Structure, mechanism and catalytic duality of thiamine-dependent enzymes. Cell Mol. Life Sci. 2007, 64, 892–905. [Google Scholar] [CrossRef]
- Mathis, D.; Shoelson, S.E. Immunometabolism: An emerging frontier. Nat. Rev. Immunol. 2011, 11, 81. [Google Scholar] [CrossRef]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic instruction of immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef]
- Shikina, T.; Hiroi, T.; Iwatani, K.; Jang, M.H.; Fukuyama, S.; Tamura, M.; Kubo, T.; Ishikawa, H.; Kiyono, H. IgA class switch occurs in the organized nasopharynx- and gut-associated lymphoid tissue, but not in the diffuse lamina propria of airways and gut. J. Immunol. 2004, 172, 6259–6264. [Google Scholar] [CrossRef]
- Kunisawa, J.; Sugiura, Y.; Wake, T.; Nagatake, T.; Suzuki, H.; Nagasawa, R.; Shikata, S.; Honda, K.; Hashimoto, E.; Suzuki, Y.; et al. Mode of bioenergetic metabolism during B cell differentiation in the intestine determines the distinct requirement for vitamin B1. Cell Rep. 2015, 13, 122–131. [Google Scholar] [CrossRef]
- Moallem, S.A.; Hosseinzadeh, H.; Farahi, S. A study of acute and chronic anti-nociceptive and anti-inflammatory effects of thiamine in mice. Iran. Biomed. J. 2008, 12, 173–178. [Google Scholar] [PubMed]
- Gibson, G.E.; Ksiezak-Reding, H.; Sheu, K.F.; Mykytyn, V.; Blass, J.P. Correlation of enzymatic, metabolic, and behavioral deficits in thiamin deficiency and its reversal. Neurochem. Res. 1984, 9, 803–814. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Chun, W.J.; Park, L.C.; Uchida, K.; Gibson, G.E. Oxidative stress is associated with region-specific neuronal death during thiamine deficiency. J. Neuropathol. Exp. Neurol. 1999, 58, 946–958. [Google Scholar] [CrossRef]
- Wegner, C.D.; Gundel, R.H.; Reilly, P.; Haynes, N.; Letts, L.G.; Rothlein, R. Intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of asthma. Science 1990, 247, 456–459. [Google Scholar] [CrossRef]
- Jones, P.T.; Anderson, R. Oxidative inhibition of polymorphonuclear leukocyte motility mediated by the peroxidase/H2O2/halide system: Studies on the reversible nature of the inhibition and mechanism of protection of migratory responsiveness by ascorbate, levamisole, thiamine and cysteine. Int. J. Immunopharmacol. 1983, 5, 377–389. [Google Scholar] [CrossRef]
- Yadav, U.C.; Kalariya, N.M.; Srivastava, S.K.; Ramana, K.V. Protective role of benfotiamine, a fat-soluble vitamin B1 analogue, in lipopolysaccharide-induced cytotoxic signals in murine macrophages. Free Radic. Biol. Med. 2010, 48, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E. Hydrocortisone, Ascorbic Acid and Thiamine (HAT Therapy) for the treatment of sepsis. Focus on ascorbic acid. Nutrients 2018, 10, 1762. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, A.M.; Telfer, A.B.; Shenkin, A. Thiamine deficiency in the critically ill. Intensive Care Med. 1988, 14, 384–387. [Google Scholar] [CrossRef]
- Liu, X.; Montissol, S.; Uber, A.; Ganley, S.; Grossestreuer, A.V.; Berg, K.; Heydrick, S.; Donnino, M.W. The effects of thiamine on breast cancer cells. Molecules 2018, 23, 1464. [Google Scholar] [CrossRef]
- Sweet, R.; Paul, A.; Zastre, J. Hypoxia induced upregulation and function of the thiamine transporter, SLC19A3 in a breast cancer cell line. Cancer Biol. Ther. 2010, 10, 1101–1111. [Google Scholar] [CrossRef]
- Jonus, H.C.; Hanberry, B.S.; Khatu, S.; Kim, J.; Luesch, H.; Dang, L.H.; Bartlett, M.G.; Zastre, J.A. The adaptive regulation of thiamine pyrophosphokinase-1 facilitates malignant growth during supplemental thiamine conditions. Oncotarget 2018, 9, 35422–35438. [Google Scholar] [CrossRef]
- Jonus, H.C.; Byrnes, C.C.; Kim, J.; Valle, M.L.; Bartlett, M.G.; Said, H.M.; Zastre, J.A. Thiamine mimetics sulbutiamine and benfotiamine as a nutraceutical approach to anticancer therapy. Biomed. Pharmacother. 2020, 121, 109648. [Google Scholar] [CrossRef]
- Comin-Anduix, B.; Boren, J.; Martinez, S.; Moro, C.; Centelles, J.J.; Trebukhina, R.; Petushok, N.; Lee, W.N.; Boros, L.G.; Cascante, M. The effect of thiamine supplementation on tumour proliferation. A metabolic control analysis study. Eur. J. Biochem. 2001, 268, 4177–4182. [Google Scholar] [CrossRef]
- Cancarini, I.; Krogh, V.; Agnoli, C.; Grioni, S.; Matullo, G.; Pala, V.; Pedraglio, S.; Contiero, P.; Riva, C.; Muti, P.; et al. Micronutrients involved in one-carbon metabolism and risk of breast cancer subtypes. PLoS ONE 2015, 10, e0138318. [Google Scholar] [CrossRef]
- Kabat, G.C.; Miller, A.B.; Jain, M.; Rohan, T.E. Dietary intake of selected B vitamins in relation to risk of major cancers in women. Br. J. Cancer 2008, 99, 816–821. [Google Scholar] [CrossRef]
- Bruce, W.R.; Furrer, R.; Shangari, N.; O’Brien, P.J.; Medline, A.; Wang, Y. Marginal dietary thiamin deficiency induces the formation of colonic aberrant crypt foci (ACF) in rats. Cancer Lett. 2003, 202, 125–129. [Google Scholar] [CrossRef]
- Powers, H.J. Current knowledge concerning optimum nutritional status of riboflavin, niacin and pyridoxine. Proc. Nutr. Soc. 1999, 58, 435–440. [Google Scholar] [CrossRef]
- Seekamp, A.; Hultquist, D.E.; Till, G.O. Protection by vitamin B2 against oxidant-mediated acute lung injury. Inflammation 1999, 23, 449–460. [Google Scholar] [CrossRef]
- Hashida, S.; Yuzawa, S.; Suzuki, N.N.; Fujioka, Y.; Takikawa, T.; Sumimoto, H.; Inagaki, F.; Fujii, H. Binding of FAD to cytochrome b558 is facilitated during activation of the phagocyte NADPH oxidase, leading to superoxide production. J. Biol. Chem. 2004, 279, 26378–26386. [Google Scholar] [CrossRef]
- Araki, S.; Suzuki, M.; Fujimoto, M.; Kimura, M. Enhancement of resistance to bacterial infection in mice by vitamin B2. J. Vet. Med. Sci. 1995, 57, 599–602. [Google Scholar] [CrossRef]
- Verdrengh, M.; Tarkowski, A. Riboflavin in innate and acquired immune responses. Inflamm. Res. 2005, 54, 390–393. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Tan, X.; Reis, J.C.; Badr, M.Z.; Papasian, C.J.; Morrison, D.C.; Qureshi, N. Suppression of nitric oxide induction and pro-inflammatory cytokines by novel proteasome inhibitors in various experimental models. Lipids Health Dis. 2011, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, K.; Prakash, M.D.; Kuol, N.; Nurgali, K.; Stojanovska, L.; Apostolopoulos, V. Anti-tumor effects of vitamin B2, B6 and B9 in promonocytic lymphoma cells. Int. J. Mol. Sci. 2019, 20, 3763. [Google Scholar] [CrossRef]
- Koay, H.F.; Gherardin, N.A.; Enders, A.; Loh, L.; Mackay, L.K.; Almeida, C.F.; Russ, B.E.; Nold-Petry, C.A.; Nold, M.F.; Bedoui, S.; et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat. Immunol. 2016, 17, 1300–1311. [Google Scholar] [CrossRef]
- Corbett, A.J.; Eckle, S.B.; Birkinshaw, R.W.; Liu, L.; Patel, O.; Mahony, J.; Chen, Z.; Reantragoon, R.; Meehan, B.; Cao, H.; et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 2014, 509, 361–365. [Google Scholar] [CrossRef]
- Hartmann, N.; McMurtrey, C.; Sorensen, M.L.; Huber, M.E.; Kurapova, R.; Coleman, F.T.; Mizgerd, J.P.; Hildebrand, W.; Kronenberg, M.; Lewinsohn, D.M.; et al. Riboflavin metabolism variation among clinical isolates of streptococcus pneumoniae results in differential activation of mucosal-associated invariant T cells. Am. J. Respir. Cell Mol. Biol. 2018, 58, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.; Kjer-Nielsen, L.; Le Nours, J.; Eckle, S.B.; Birkinshaw, R.; Beddoe, T.; Corbett, A.J.; Liu, L.; Miles, J.J.; Meehan, B.; et al. Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat. Commun. 2013, 4, 2142. [Google Scholar] [CrossRef]
- Toyosawa, T.; Suzuki, M.; Kodama, K.; Araki, S. Effects of intravenous infusion of highly purified vitamin B2 on lipopolysaccharide-induced shock and bacterial infection in mice. Eur. J. Pharmacol. 2004, 492, 273–280. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Imam, F.; Nadeem, A.; Al-Harbi, M.M.; Korashy, H.M.; Sayed-Ahmed, M.M.; Hafez, M.M.; Al-Shabanah, O.A.; Nagi, M.N.; Bahashwan, S. Riboflavin attenuates lipopolysaccharide-induced lung injury in rats. Toxicol. Mech. Methods 2015, 25, 417–423. [Google Scholar] [CrossRef]
- Schramm, M.; Wiegmann, K.; Schramm, S.; Gluschko, A.; Herb, M.; Utermohlen, O.; Kronke, M. Riboflavin (vitamin B2 ) deficiency impairs NADPH oxidase 2 (Nox2) priming and defense against Listeria monocytogenes. Eur. J. Immunol. 2014, 44, 728–741. [Google Scholar] [CrossRef]
- Gupta, T.K.; Vishnuvajjala, B.R.; Witiak, D.T.; Gerald, M.C. Antagonism of amphetamine stereotyped behavior by diastereoisomeric dihydrodibenzothiepin neuroleptics. Experientia 1977, 33, 65. [Google Scholar] [CrossRef]
- Pan, F.; Chen, Y.; He, J.Z.; Long, L.; Chen, Y.; Luo, H.J.; Xu, Y.W.; Pang, X.X.; Yang, Q.; Wang, J.J.; et al. Dietary riboflavin deficiency promotes N-nitrosomethylbenzylamine-induced esophageal tumorigenesis in rats by inducing chronic inflammation. Am. J. Cancer Res. 2019, 9, 2469–2481. [Google Scholar]
- Machado, D.; Shishido, S.M.; Queiroz, K.C.; Oliveira, D.N.; Faria, A.L.; Catharino, R.R.; Spek, C.A.; Ferreira, C.V. Irradiated riboflavin diminishes the aggressiveness of melanoma in vitro and in vivo. PLoS ONE 2013, 8, e54269. [Google Scholar] [CrossRef]
- Powers, H.J. Riboflavin (vitamin B-2) and health. Am. J. Clin. Nutr. 2003, 77, 1352–1360. [Google Scholar] [CrossRef]
- Zeng, J.; Gu, Y.; Fu, H.; Liu, C.; Zou, Y.; Chang, H. Association between one-carbon metabolism-related vitamins and risk of breast cancer: A systematic review and meta-analysis of prospective studies. Clin. Breast Cancer 2020, 20, e469–e480. [Google Scholar] [CrossRef] [PubMed]
- Clasen, J.L.; Heath, A.K.; Scelo, G.; Muller, D.C. Components of one-carbon metabolism and renal cell carcinoma: A systematic review and meta-analysis. Eur. J. Nutr. 2020. [Google Scholar] [CrossRef]
- Li, S.S.; Tan, H.Z.; Xu, Y.W.; Wu, Z.Y.; Wu, J.Y.; Zhao, X.K.; Wang, L.D.; Long, L.; Li, E.M.; Xu, L.Y.; et al. [The association between the whole blood riboflavin level and the occurrence, development and prognosis of esophageal squamous cell carcinoma]. Zhonghua Yu Fang Yi Xue Za Zhi 2019, 53, 1124–1129. [Google Scholar] [CrossRef]
- de Vogel, S.; Dindore, V.; van Engeland, M.; Goldbohm, R.A.; van den Brandt, P.A.; Weijenberg, M.P. Dietary folate, methionine, riboflavin, and vitamin B-6 and risk of sporadic colorectal cancer. J. Nutr. 2008, 138, 2372–2378. [Google Scholar] [CrossRef]
- Takata, Y.; Cai, Q.; Beeghly-Fadiel, A.; Li, H.; Shrubsole, M.J.; Ji, B.T.; Yang, G.; Chow, W.H.; Gao, Y.T.; Zheng, W.; et al. Dietary B vitamin and methionine intakes and lung cancer risk among female never smokers in China. Cancer Causes Control. 2012, 23, 1965–1975. [Google Scholar] [CrossRef]
- Kirkland, J.B.; Meyer-Ficca, M.L. Niacin. Adv. Food Nutr. Res. 2018, 83, 83–149. [Google Scholar] [CrossRef] [PubMed]
- Lohani, M.; Dhasmana, A.; Haque, S.; Dar, S.A.; Jawed, A.; Wahid, M.; Mandal, R.K.; Akhter, N.; Farasani, A.; Hobani, Y.H.; et al. Niacin deficiency modulates genes involved in cancer: Are smokers at higher risk? J. Cell Biochem. 2019, 120, 232–242. [Google Scholar] [CrossRef]
- Choi, H.J.; Jang, S.Y.; Hwang, E.S. High-dose nicotinamide suppresses ros generation and augments population expansion during CD8(+) T cell activation. Mol. Cells 2015, 38, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Wanders, D.; Graff, E.C.; White, B.D.; Judd, R.L. Niacin increases adiponectin and decreases adipose tissue inflammation in high fat diet-fed mice. PLoS ONE 2013, 8, e71285. [Google Scholar] [CrossRef]
- Karacaglar, E.; Atar, I.; Altin, C.; Yetis, B.; Cakmak, A.; Bayraktar, N.; Coner, A.; Ozin, B.; Muderrisoglu, H. The effects of niacin on inflammation in patients with non-ST elevated acute coronary syndrome. Acta Cardiol. Sin. 2015, 31, 120–126. [Google Scholar] [CrossRef]
- Su, G.; Sun, G.; Liu, H.; Shu, L.; Zhang, J.; Guo, L.; Huang, C.; Xu, J. Niacin suppresses progression of atherosclerosis by inhibiting vascular inflammation and apoptosis of vascular smooth muscle cells. Med. Sci. Monit. 2015, 21, 4081–4089. [Google Scholar] [CrossRef]
- Ganji, S.H.; Kukes, G.D.; Lambrecht, N.; Kashyap, M.L.; Kamanna, V.S. Therapeutic role of niacin in the prevention and regression of hepatic steatosis in rat model of nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G320–G327. [Google Scholar] [CrossRef]
- Kashyap, M.L.; Ganji, S.; Nakra, N.K.; Kamanna, V.S. Niacin for treatment of nonalcoholic fatty liver disease (NAFLD): Novel use for an old drug? J. Clin. Lipidol. 2019, 13, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Y.; van der Sluis, R.J.; van der Hoorn, J.W.; Princen, H.M.; Van Eck, M.; Van Berkel, T.J.; Rensen, P.C.; Hoekstra, M. Niacin reduces plasma CETP levels by diminishing liver macrophage content in CETP transgenic mice. Biochem. Pharmacol. 2012, 84, 821–829. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Matsui, T.C.; Gomides, L.F.; Godin, A.M.; Menezes, G.B.; de Matos Coelho, M.; Klein, A. Niacin inhibits carrageenan-induced neutrophil migration in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 533–540. [Google Scholar] [CrossRef]
- Digby, J.E.; Martinez, F.; Jefferson, A.; Ruparelia, N.; Chai, J.; Wamil, M.; Greaves, D.R.; Choudhury, R.P. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.Y.; Suh, G.J.; Kim, K.S.; Kwak, Y.H. Niacin attenuates lung inflammation and improves survival during sepsis by downregulating the nuclear factor-kappaB pathway. Crit. Care Med. 2011, 39, 328–334. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, H.J.; Rodriguez-Iturbe, B.; Vaziri, N.D. Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am. J. Physiol. Renal. Physiol. 2009, 297, F106–F113. [Google Scholar] [CrossRef] [PubMed]
- Takechi, R.; Pallebage-Gamarallage, M.M.; Lam, V.; Giles, C.; Mamo, J.C. Nutraceutical agents with anti-inflammatory properties prevent dietary saturated-fat induced disturbances in blood-brain barrier function in wild-type mice. J. Neuroinflamm. 2013, 10, 73. [Google Scholar] [CrossRef]
- Wakade, C.; Giri, B.; Malik, A.; Khodadadi, H.; Morgan, J.C.; Chong, R.K.; Baban, B. Niacin modulates macrophage polarization in Parkinson’s disease. J. NeuroImmunol. 2018, 320, 76–79. [Google Scholar] [CrossRef]
- Feingold, K.R.; Moser, A.; Shigenaga, J.K.; Grunfeld, C. Inflammation stimulates niacin receptor (GPR109A/HCA2) expression in adipose tissue and macrophages. J. Lipid Res. 2014, 55, 2501–2508. [Google Scholar] [CrossRef]
- Elangovan, S.; Pathania, R.; Ramachandran, S.; Ananth, S.; Padia, R.N.; Lan, L.; Singh, N.; Martin, P.M.; Hawthorn, L.; Prasad, P.D.; et al. The niacin/butyrate receptor GPR109A suppresses mammary tumorigenesis by inhibiting cell survival. Cancer Res. 2014, 74, 1166–1178. [Google Scholar] [CrossRef]
- Nikas, I.P.; Paschou, S.A.; Ryu, H.S. The role of nicotinamide in cancer chemoprevention and therapy. Biomolecules 2020, 10, 477. [Google Scholar] [CrossRef]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The role of SIRT1 on DNA damage response and epigenetic alterations in cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef]
- Surjana, D.; Halliday, G.M.; Damian, D.L. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J. Nucleic Acids 2010, 2010, 157591. [Google Scholar] [CrossRef]
- Fania, L.; Mazzanti, C.; Campione, E.; Candi, E.; Abeni, D.; Dellambra, E. Role of nicotinamide in genomic stability and skin cancer chemoprevention. Int. J. Mol. Sci. 2019, 20, 5946. [Google Scholar] [CrossRef]
- Antwi, S.O.; Petrick, J.L.; Campbell, P.T.; Norez, D.A.; Stevens, V.L.; Liao, L.M.; Roberts, L.R.; Patel, T.; McGlynn, K.A. One-carbon metabolism-related micronutrients intake and risk for hepatocellular carcinoma: A prospective cohort study. Int. J. Cancer 2020, 147, 2075–2090. [Google Scholar] [CrossRef]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent advances in NAMPT inhibitors: A novel immunotherapic strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef] [PubMed]
- Sabui, S.; Kapadia, R.; Ghosal, A.; Schneider, M.; Lambrecht, N.W.G.; Said, H.M. Biotin and pantothenic acid oversupplementation to conditional SLC5A6 KO mice prevents the development of intestinal mucosal abnormalities and growth defects. Am. J. Physiol. Cell Physiol. 2018, 315, C73–C79. [Google Scholar] [CrossRef]
- Ghosal, A.; Lambrecht, N.; Subramanya, S.B.; Kapadia, R.; Said, H.M. Conditional knockout of the Slc5a6 gene in mouse intestine impairs biotin absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G64–G71. [Google Scholar] [CrossRef]
- Nitto, T.; Onodera, K. Linkage between coenzyme a metabolism and inflammation: Roles of pantetheinase. J. Pharmacol. Sci. 2013, 123, 1–8. [Google Scholar] [CrossRef]
- Berruyer, C.; Martin, F.M.; Castellano, R.; Macone, A.; Malergue, F.; Garrido-Urbani, S.; Millet, V.; Imbert, J.; Duprè, S.; Pitari, G.; et al. Vanin-1-/- mice exhibit a glutathione-mediated tissue resistance to oxidative stress. Mol. Cell Biol. 2004, 24, 7214–7224. [Google Scholar] [CrossRef]
- Berruyer, C.; Pouyet, L.; Millet, V.; Martin, F.M.; LeGoffic, A.; Canonici, A.; Garcia, S.; Bagnis, C.; Naquet, P.; Galland, F. Vanin-1 licenses inflammatory mediator production by gut epithelial cells and controls colitis by antagonizing peroxisome proliferator-activated receptor gamma activity. J. Exp. Med. 2006, 203, 2817–2827. [Google Scholar] [CrossRef]
- Zhang, B.; Lo, C.; Shen, L.; Sood, R.; Jones, C.; Cusmano-Ozog, K.; Park-Snyder, S.; Wong, W.; Jeng, M.; Cowan, T.; et al. The role of vanin-1 and oxidative stress-related pathways in distinguishing acute and chronic pediatric ITP. Blood 2011, 117, 4569–4579. [Google Scholar] [CrossRef]
- Meghari, S.; Berruyer, C.; Lepidi, H.; Galland, F.; Naquet, P.; Mege, J.L. Vanin-1 controls granuloma formation and macrophage polarization in Coxiella burnetii infection. Eur. J. Immunol. 2007, 37, 24–32. [Google Scholar] [CrossRef]
- Pouyet, L.; Roisin-Bouffay, C.; Clément, A.; Millet, V.; Garcia, S.; Chasson, L.; Issaly, N.; Rostan, A.; Hofman, P.; Naquet, P.; et al. Epithelial vanin-1 controls inflammation-driven carcinogenesis in the colitis-associated colon cancer model. Inflamm. Bowel Dis. 2010, 16, 96–104. [Google Scholar] [CrossRef]
- Fedde, K.N.; Whyte, M.P. Alkaline phosphatase (tissue-nonspecific isoenzyme) is a phosphoethanolamine and pyridoxal-5’-phosphate ectophosphatase: Normal and hypophosphatasia fibroblast study. Am. J. Hum. Genet. 1990, 47, 767–775. [Google Scholar]
- DiSorbo, D.M.; Wagner, R., Jr.; Nathanson, L. In vivo and in vitro inhibition of B16 melanoma growth by vitamin B6. Nutr. Cancer 1985, 7, 43–52. [Google Scholar] [CrossRef]
- Matsuo, T.; Fujiwara, A.; Nakamura, K.; Sadzuka, Y. The effects of vitamin B(6) compounds on cell proliferation and melanogenesis in B16F10 melanoma cells. Oncol. Lett. 2018, 15, 5181–5184. [Google Scholar] [CrossRef]
- Zhang, P.; Suidasari, S.; Hasegawa, T.; Yanaka, N.; Kato, N. High concentrations of pyridoxal stimulate the expression of IGFBP1 in HepG2 cells through upregulation of the ERK/c-Jun pathway. Mol. Med. Rep. 2013, 8, 973–978. [Google Scholar] [CrossRef]
- Hartman, T.J.; Woodson, K.; Stolzenberg-Solomon, R.; Virtamo, J.; Selhub, J.; Barrett, M.J.; Albanes, D. Association of the B-vitamins pyridoxal 5’-phosphate (B(6)), B(12), and folate with lung cancer risk in older men. Am. J. Epidemiol. 2001, 153, 688–694. [Google Scholar] [CrossRef]
- Okazaki, Y.; Utama, Z.; Suidasari, S.; Zhang, P.; Yanaka, N.; Tomotake, H.; Sakaguchi, E.; Kato, N. Consumption of vitamin B(6) reduces fecal ratio of lithocholic acid to deoxycholic acid, a risk factor for colon cancer, in rats fed a high-fat diet. J. Nutr. Sci. Vitaminol. 2012, 58, 366–370. [Google Scholar] [CrossRef][Green Version]
- Kayashima, T.; Tanaka, K.; Okazaki, Y.; Matsubara, K.; Yanaka, N.; Kato, N. Consumption of vitamin B6 reduces colonic damage and protein expression of HSP70 and HO-1, the anti-tumor targets, in rats exposed to 1,2-dimethylhydrazine. Oncol. Lett. 2011, 2, 1243–1246. [Google Scholar] [CrossRef]
- Gebhard, K.J.; Gridley, D.S.; Stickney, D.R.; Shulz, T.D. Enhancement of immune status by high levels of dietary vitamin B-6 without growth inhibition of human malignant melanoma in athymic nude mice. Nutr. Cancer 1990, 14, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Marsili, S.; Vitale, I.; Senovilla, L.; Michels, J.; Garcia, P.; Vacchelli, E.; Chatelut, E.; Castedo, M.; Kroemer, G. Vitamin B6 metabolism influences the intracellular accumulation of cisplatin. Cell Cycle 2013, 12, 417–421. [Google Scholar] [CrossRef]
- Zhang, P.; Suidasari, S.; Hasegawa, T.; Yanaka, N.; Kato, N. Vitamin B6 activates p53 and elevates p21 gene expression in cancer cells and the mouse colon. Oncol. Rep. 2014, 31, 2371–2376. [Google Scholar] [CrossRef]
- Sujol, G.; Docquier, A.; Boulahtouf, A.; Castet-Nicolas, A.; Cavaillès, V. Vitamin B6 and cancer: From clinical data to molecularly mechanisms. Bull. Cancer 2011, 98, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vacchelli, E.; Michels, J.; Garcia, P.; Kepp, O.; Senovilla, L.; Vitale, I.; Kroemer, G. Effects of vitamin B6 metabolism on oncogenesis, tumor progression and therapeutic responses. Oncogene 2013, 32, 4995–5004. [Google Scholar] [CrossRef]
- Bourquin, F.; Capitani, G.; Grütter, M.G. PLP-dependent enzymes as entry and exit gates of sphingolipid metabolism. Protein Sci. 2011, 20, 1492–1508. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.; Li, Q.; Xin, Y.; Fang, X.; Tian, Y.; Ma, J.; Wang, J.; Wang, Q.; Zhang, R.; Wang, J.; et al. Intake of dietary one-carbon metabolism-related B vitamins and the risk of esophageal cancer: A dose-response meta-analysis. Nutrients 2018, 10, 835. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, L.; Wang, Q.; Shen, J. Pyridoxine for prevention of hand-foot syndrome caused by chemotherapy: A systematic review. PLoS ONE 2013, 8, e72245. [Google Scholar] [CrossRef]
- Hymes, J.; Wolf, B. Biotinidase and its roles in biotin metabolism. Clin. Chim. Acta 1996, 255, 1–11. [Google Scholar] [CrossRef]
- Chauhan, J.; Dakshinamurti, K. Transcriptional regulation of the glucokinase gene by biotin in starved rats. J. Biol. Chem. 1991, 266, 10035–10038. [Google Scholar]
- Spence, J.T.; Koudelka, A.P. Effects of biotin upon the intracellular level of cGMP and the activity of glucokinase in cultured rat hepatocytes. J. Biol. Chem. 1984, 259, 6393–6396. [Google Scholar]
- Agrawal, S.; Agrawal, A.; Said, H.M. Biotin deficiency enhances the inflammatory response of human dendritic cells. Am. J. Physiol. Cell Physiol. 2016, 311, C386–C391. [Google Scholar] [CrossRef] [PubMed]
- Sghaier, R.; Zarrouk, A.; Nury, T.; Badreddine, I.; O’Brien, N.; Mackrill, J.J.; Vejux, A.; Samadi, M.; Nasser, B.; Caccia, C.; et al. Biotin attenuation of oxidative stress, mitochondrial dysfunction, lipid metabolism alteration and 7beta-hydroxycholesterol-induced cell death in 158N murine oligodendrocytes. Free Radic. Res. 2019, 53, 535–561. [Google Scholar] [CrossRef]
- Järvinen, E.; Ismail, K.; Muniandy, M.; Bogl, L.H.; Heinonen, S.; Tummers, M.; Miettinen, S.; Kaprio, J.; Rissanen, A.; Ollikainen, M.; et al. Biotin-dependent functions in adiposity: A study of monozygotic twin pairs. Int. J. Obes. 2016, 40, 788–795. [Google Scholar] [CrossRef]
- Kuroishi, T. Regulation of Immunological and inflammatory functions by biotin. Can. J. Physiol. Pharmacol. 2015, 93, 1091–1096. [Google Scholar] [CrossRef]
- Honein, M.A.; Paulozzi, L.J.; Mathews, T.J.; Erickson, J.D.; Wong, L.Y. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA 2001, 285, 2981–2986. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Waly, M.I.; Taranikanti, V.; Guizani, N.; Ali, A.; Rahman, M.S.; Al-Attabi, Z.; Al-Malky, R.N.; Al-Maskari, S.N.M.; Al-Ruqaishi, B.R.S.; et al. Folate/Vitamin B12 supplementation combats oxidative stress-associated carcinogenesis in a rat model of colon cancer. Nutr. Cancer 2019, 71, 100–110. [Google Scholar] [CrossRef]
- Courtemanche, C.; Elson-Schwab, I.; Mashiyama, S.T.; Kerry, N.; Ames, B.N. Folate deficiency inhibits the proliferation of primary human CD8+ T lymphocytes in vitro. J. Immunol. 2004, 173, 3186–3192. [Google Scholar] [CrossRef]
- Courtemanche, C.; Huang, A.C.; Elson-Schwab, I.; Kerry, N.; Ng, B.Y.; Ames, B.N. Folate deficiency and ionizing radiation cause DNA breaks in primary human lymphocytes: A comparison. FASEB J. 2004, 18, 209–211. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hirota, K.; Nagahama, K.; Ohkawa, K.; Takahashi, T.; Nomura, T.; Sakaguchi, S. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity 2007, 27, 145–159. [Google Scholar] [CrossRef]
- Wu, C.H.; Huang, T.C.; Lin, B.F. Folate deficiency affects dendritic cell function and subsequent T helper cell differentiation. J. Nutr. Biochem. 2017, 41, 65–72. [Google Scholar] [CrossRef]
- Makino, E.; Fukuyama, T.; Watanabe, Y.; Tajiki-Nishino, R.; Tajima, H.; Ohnuma-Koyama, A.; Takahashi, N.; Ohtsuka, R.; Okazaki, Y. Subacute oral administration of folic acid elicits anti-inflammatory response in a mouse model of allergic dermatitis. J. Nutr. Biochem. 2019, 67, 14–19. [Google Scholar] [CrossRef]
- Kok, D.E.; O’Flanagan, C.H.; Coleman, M.F.; Ashkavand, Z.; Hursting, S.D.; Krupenko, S.A. Effects of folic acid withdrawal on transcriptomic profiles in murine triple-negative breast cancer cell lines. Biochimie 2020, 173, 114–122. [Google Scholar] [CrossRef]
- Jhaveri, M.S.; Wagner, C.; Trepel, J.B. Impact of extracellular folate levels on global gene expression. Mol. Pharmacol. 2001, 60, 1288–1295. [Google Scholar] [CrossRef]
- Hansen, M.F.; Jensen, S.; Füchtbauer, E.M.; Martensen, P.M. High folic acid diet enhances tumour growth in PyMT-induced breast cancer. Br. J. Cancer 2017, 116, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Crott, J.W.; Choi, S.W.; Ordovas, J.M.; Ditelberg, J.S.; Mason, J.B. Effects of dietary folate and aging on gene expression in the colonic mucosa of rats: Implications for carcinogenesis. Carcinogenesis 2004, 25, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Crott, J.W.; Liu, Z.; Keyes, M.K.; Choi, S.W.; Jang, H.; Moyer, M.P.; Mason, J.B. Moderate folate depletion modulates the expression of selected genes involved in cell cycle, intracellular signaling and folate uptake in human colonic epithelial cell lines. J. Nutr. Biochem. 2008, 19, 328–335. [Google Scholar] [CrossRef]
- Terzis, A.J.; Fiskerstrand, T.; Refsum, H.; Ueland, P.M.; Arnold, H.; Bjerkvig, R. Proliferation, migration and invasion of human glioma cells exposed to antifolate drugs. Int. J. Cancer 1993, 54, 112–118. [Google Scholar] [CrossRef]
- Kim, S.J.; Zuchniak, A.; Sohn, K.J.; Lubinski, J.; Demsky, R.; Eisen, A.; Akbari, M.R.; Kim, Y.I.; Narod, S.A.; Kotsopoulos, J. Plasma folate, vitamin B-6, and vitamin B-12 and breast cancer risk in BRCA1- and BRCA2-mutation carriers: A prospective study. Am. J. Clin. Nutr. 2016, 104, 671–677. [Google Scholar] [CrossRef]
- Kim, S.J.; Zhang, C.X.W.; Demsky, R.; Armel, S.; Kim, Y.I.; Narod, S.A.; Kotsopoulos, J. Folic acid supplement use and breast cancer risk in BRCA1 and BRCA2 mutation carriers: A case-control study. Breast Cancer Res. Treat. 2019, 174, 741–748. [Google Scholar] [CrossRef]
- Kim, Y.I. Folate, colorectal carcinogenesis, and DNA methylation: Lessons from animal studies. Environ. Mol. Mutagen. 2004, 44, 10–25. [Google Scholar] [CrossRef]
- Tomaszewski, J.J.; Cummings, J.L.; Parwani, A.V.; Dhir, R.; Mason, J.B.; Nelson, J.B.; Bacich, D.J.; O’Keefe, D.S. Increased cancer cell proliferation in prostate cancer patients with high levels of serum folate. Prostate 2011, 71, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Dinney, C.P.; Ye, Y.; Grossman, H.B.; Lerner, S.P.; Wu, X. Is folic acid safe for non-muscle-invasive bladder cancer patients? An evidence-based cohort study. Am. J. Clin. Nutr. 2018, 107, 208–216. [Google Scholar] [CrossRef]
- Kuo, C.S.; Lin, C.Y.; Wu, M.Y.; Lu, C.L.; Huang, R.F. Relationship between folate status and tumour progression in patients with hepatocellular carcinoma. Br. J. Nutr. 2008, 100, 596–602. [Google Scholar] [CrossRef]
- Su, Y.H.; Huang, W.C.; Huang, T.H.; Huang, Y.J.; Sue, Y.K.; Huynh, T.T.; Hsiao, M.; Liu, T.Z.; Wu, A.T.; Lin, C.M. Folate deficient tumor microenvironment promotes epithelial-to-mesenchymal transition and cancer stem-like phenotypes. Oncotarget 2016, 7, 33246–33256. [Google Scholar] [CrossRef]
- Wang, T.P.; Hsu, S.H.; Feng, H.C.; Huang, R.F. Folate deprivation enhances invasiveness of human colon cancer cells mediated by activation of sonic hedgehog signaling through promoter hypomethylation and cross action with transcription nuclear factor-kappa B pathway. Carcinogenesis 2012, 33, 1158–1168. [Google Scholar] [CrossRef]
- Larsson, S.C.; Giovannucci, E.; Wolk, A. Dietary folate intake and incidence of ovarian cancer: The Swedish Mammography Cohort. J. Natl. Cancer Inst. 2004, 96, 396–402. [Google Scholar] [CrossRef][Green Version]
- Dugué, P.A.; Chamberlain, J.A.; Bassett, J.K.; Hodge, A.M.; Brinkman, M.T.; Joo, J.E.; Jung, C.H.; Wong, E.M.; Makalic, E.; Schmidt, D.F.; et al. Overall lack of replication of associations between dietary intake of folate and vitamin B-12 and DNA methylation in peripheral blood. Am. J. Clin. Nutr. 2020, 111, 228–230. [Google Scholar] [CrossRef]
- Wu, X.Y.; Lu, L. Vitamin B6 deficiency, genome instability and cancer. Asian Pac. J. Cancer Prev. 2012, 13, 5333–5338. [Google Scholar] [CrossRef]
- Paul, L.; Selhub, J. Interaction between excess folate and low vitamin B12 status. Mol. Asp. Med. 2017, 53, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Guéant, J.L.; Oussalah, A.; Zgheib, R.; Siblini, Y.; Hsu, S.B.; Namour, F. Genetic, epigenetic and genomic mechanisms of methionine dependency of cancer and tumor-initiating cells: What could we learn from folate and methionine cycles. Biochimie 2020, 173, 123–128. [Google Scholar] [CrossRef]
- Guéant, J.L.; Caillerez-Fofou, M.; Battaglia-Hsu, S.; Alberto, J.M.; Freund, J.N.; Dulluc, I.; Adjalla, C.; Maury, F.; Merle, C.; Nicolas, J.P.; et al. Molecular and cellular effects of vitamin B12 in brain, myocardium and liver through its role as co-factor of methionine synthase. Biochimie 2013, 95, 1033–1040. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Lalla, E.; Lu, Y.; Gleason, M.R.; Wolf, B.M.; Tanji, N.; Ferran, L.J., Jr.; Kohl, B.; Rao, V.; Kisiel, W.; et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J. Clin. Investig. 2001, 107, 675–683. [Google Scholar] [CrossRef]
- Dusitanond, P.; Eikelboom, J.W.; Hankey, G.J.; Thom, J.; Gilmore, G.; Loh, K.; Yi, Q.; Klijn, C.J.; Langton, P.; van Bockxmeer, F.M.; et al. Homocysteine-lowering treatment with folic acid, cobalamin, and pyridoxine does not reduce blood markers of inflammation, endothelial dysfunction, or hypercoagulability in patients with previous transient ischemic attack or stroke: A randomized substudy of the VITATOPS trial. Stroke 2005, 36, 144–146. [Google Scholar] [CrossRef]
- Chen, X.; Ahamada, H.; Zhang, T.; Bai, Z.; Wang, C. Association of intake folate and related gene polymorphisms with breast cancer. J. Nutr. Sci. Vitaminol. 2019, 65, 459–469. [Google Scholar] [CrossRef]
- Cheng, W.W.; Wang, Z.K.; Shangguan, H.F.; Zhu, Q.; Zhang, H.Y. Are vitamins relevant to cancer risks? A Mendelian randomization investigation. Nutrition 2020, 78, 110870. [Google Scholar] [CrossRef]
- Brasky, T.M.; Ray, R.M.; Navarro, S.L.; Schenk, J.M.; Newton, A.M.; Neuhouser, M.L. Supplemental one-carbon metabolism related B vitamins and lung cancer risk in the Women’s Health Initiative. Int. J. Cancer 2020, 147, 1374–1384. [Google Scholar] [CrossRef]
- Arendt, J.F.H.; Sørensen, H.T.; Horsfall, L.J.; Petersen, I. Elevated vitamin B12 levels and cancer risk in UK primary care: A THIN database cohort study. Cancer Epidemiol. Biomark. Prev. 2019, 28, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Collin, S.M. Folate and B12 in prostate cancer. Adv. Clin. Chem. 2013, 60, 1–63. [Google Scholar] [CrossRef] [PubMed]
- Tamura, J.; Kubota, K.; Murakami, H.; Sawamura, M.; Matsushima, T.; Tamura, T.; Saitoh, T.; Kurabayshi, H.; Naruse, T. Immunomodulation by vitamin B12: Augmentation of CD8+ T lymphocytes and natural killer (NK) cell activity in vitamin B12-deficient patients by methyl-B12 treatment. Clin. Exp. Immunol. 1999, 116, 28–32. [Google Scholar] [CrossRef]
- Partearroyo, T.; Úbeda, N.; Montero, A.; Achón, M.; Varela-Moreiras, G. Vitamin B(12) and folic acid imbalance modifies NK cytotoxicity, lymphocytes B and lymphoprolipheration in aged rats. Nutrients 2013, 5, 4836–4848. [Google Scholar] [CrossRef]
- Lin, C.Y.; Kuo, C.S.; Lu, C.L.; Wu, M.Y.; Huang, R.F. Elevated serum vitamin B(12) levels in association with tumor markers as the prognostic factors predictive for poor survival in patients with hepatocellular carcinoma. Nutr. Cancer 2010, 62, 190–197. [Google Scholar] [CrossRef]
- Lo-Bisgaard, T.; Espelund, U.; Frystyk, J.; Rasmussen, T.R.; Nexo, E.; Arendt, J.F.H. Vitamin B12 and its binding proteins in patients with non-small cell lung cancer referred to fast-track diagnostic work-up for lung cancer. Scand. J. Clin. Lab. Investig. 2020, 80, 14–19. [Google Scholar] [CrossRef]
- Collin, S.M.; Metcalfe, C.; Refsum, H.; Lewis, S.J.; Zuccolo, L.; Smith, G.D.; Chen, L.; Harris, R.; Davis, M.; Marsden, G.; et al. Circulating folate, vitamin B12, homocysteine, vitamin B12 transport proteins, and risk of prostate cancer: A case-control study, systematic review, and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1632–1642. [Google Scholar] [CrossRef]
- Fanidi, A.; Carreras-Torres, R.; Larose, T.L.; Yuan, J.M.; Stevens, V.L.; Weinstein, S.J.; Albanes, D.; Prentice, R.; Pettinger, M.; Cai, Q.; et al. Is high vitamin B12 status a cause of lung cancer? Int. J. Cancer 2019, 145, 1499–1503. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, C.T.; Rodionov, D.A.; Osterman, A.L.; Peterson, S.N. B Vitamins and Their Role in Immune Regulation and Cancer. Nutrients 2020, 12, 3380. https://doi.org/10.3390/nu12113380
Peterson CT, Rodionov DA, Osterman AL, Peterson SN. B Vitamins and Their Role in Immune Regulation and Cancer. Nutrients. 2020; 12(11):3380. https://doi.org/10.3390/nu12113380
Chicago/Turabian StylePeterson, Christine Tara, Dmitry A. Rodionov, Andrei L. Osterman, and Scott N. Peterson. 2020. "B Vitamins and Their Role in Immune Regulation and Cancer" Nutrients 12, no. 11: 3380. https://doi.org/10.3390/nu12113380
APA StylePeterson, C. T., Rodionov, D. A., Osterman, A. L., & Peterson, S. N. (2020). B Vitamins and Their Role in Immune Regulation and Cancer. Nutrients, 12(11), 3380. https://doi.org/10.3390/nu12113380