B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease


Abstract
1. Introduction
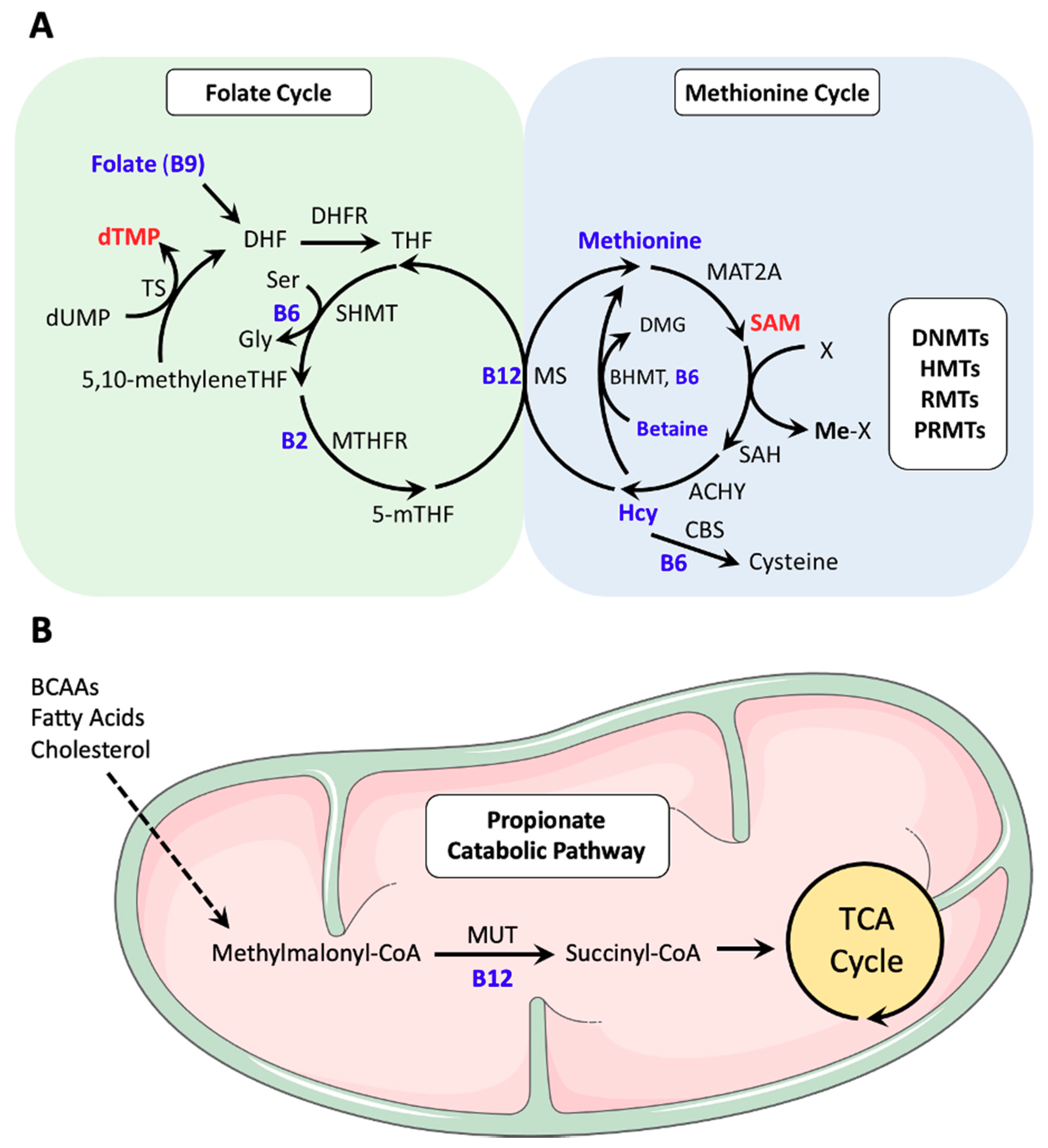
2. Key Regulators of One-Carbon Metabolism
2.1. Methionine
2.2. Folate
2.3. Vitamin B12
2.4. Co-Dependence of the Folate and Methionine Cycles
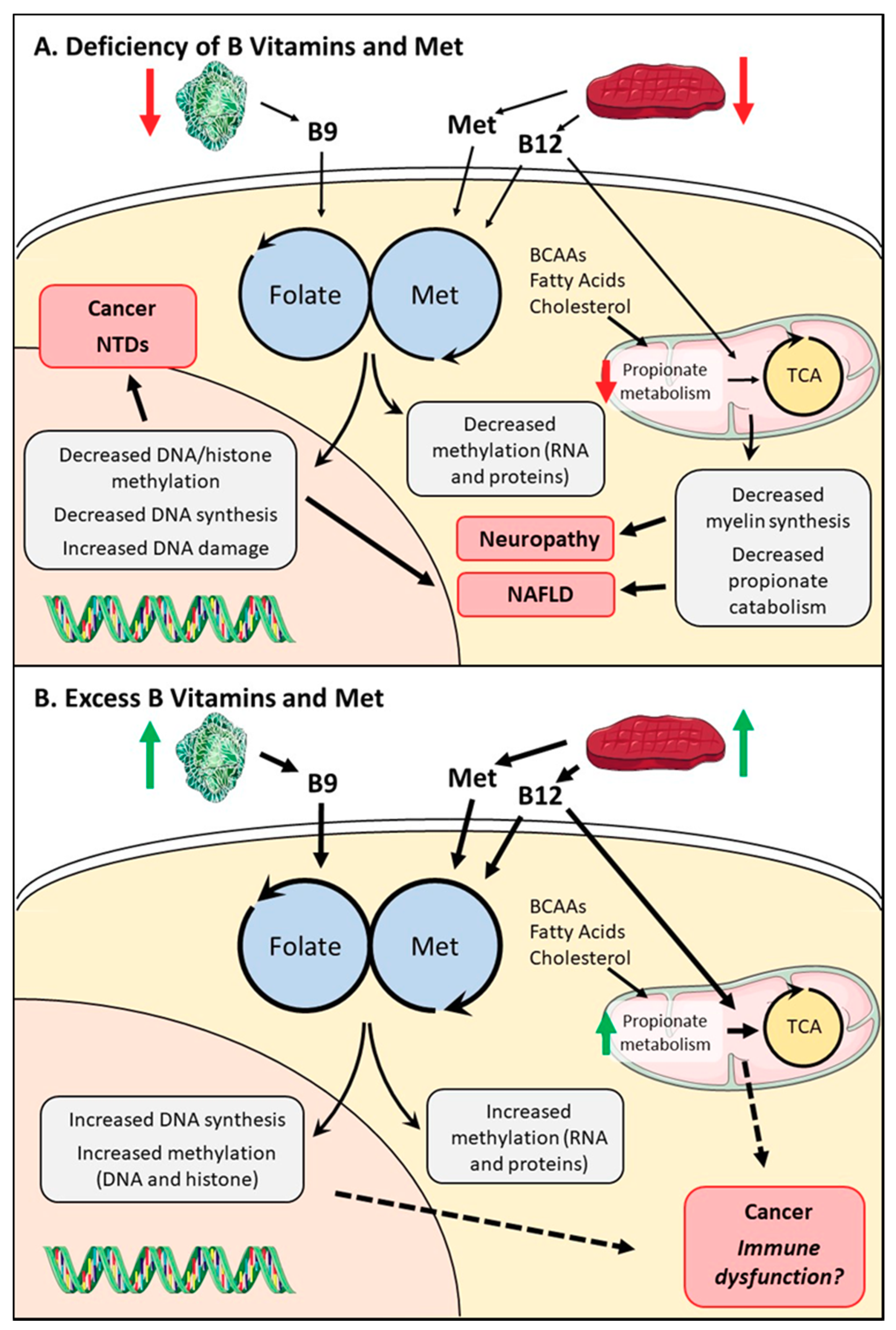
3. Methionine, Folate, and B12 Deficiency on One-Carbon Metabolism: Causes and Consequences
3.1. Causes of Deficiency
3.2. Developmental and Neurological Consequences of B9/B12 Deficiency
3.3. Hematological Consequences of B9/B12 Deficiency
3.4. One-Carbon Metabolism Defiencies and Colorectal Cancer
3.5. Non-Alcoholic Fatty Liver Diseases and One-Carbon Metabolism
3.6. Fortification and Supplementation for the Treatment of B9/B12 Deficiency
4. The Role of Methionine, Folate, and B12 Excess in Disease Progression
4.1. High Serum B9/B12 Levels and Cancer
4.2. Excess One-Carbon Metabolites in Immunity and Organ Function
5. Targeting One-Carbon Metabolism for the Treatment of Disease
5.1. Folate and Methionine-Cycle Targeted Therapies
5.2. Dietary Interventions of One-Carbon Metabolism
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, C.; Nott, T.J.; Jin, J.; Pawson, T. Deciphering arginine methylation: Tudor tells the tale. Nat. Rev. Mol. Cell Biol. 2011, 12, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, B.J.; McCarthy, E.L.; Booker, S.J. Radical S-Adenosylmethionine Enzymes in Human Health and Disease. Annu. Rev. Biochem. 2016, 85, 485–514. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Schoonjans, K.; Auwerx, J. Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metab. 2010, 12, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Lanouette, S.; Mongeon, V.; Figeys, D.; Couture, J.-F. The functional diversity of protein lysine methylation. Mol. Syst. Biol. 2014, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Chiuve, S.E.; Giovannucci, E.L.; Hankinson, S.E.; Zeisel, S.H.; Dougherty, L.W.; Willett, W.C.; Rimm, E.B. The association between betaine and choline intakes and the plasma concentrations of homocysteine in women. Am. J. Clin. Nutr. 2007, 86, 1073–1081. [Google Scholar] [CrossRef]
- Guéant, J.-L.; Oussalah, A.; Zgheib, R.; Siblini, Y.; Hsu, S.B.; Namour, F. Genetic, epigenetic and genomic mechanisms of methionine dependency of cancer and tumor-initiating cells: What could we learn from folate and methionine cycles. Biochimie 2020, 173, 123–128. [Google Scholar] [CrossRef]
- Guillamot, M.; Cimmino, L.; Aifantis, I. The Impact of DNA Methylation in Hematopoietic Malignancies. Trends Cancer 2016, 2, 70–83. [Google Scholar] [CrossRef]
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.-L.; Brito, A.; Guéant, J.-L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.-H.; et al. Vitamin B12 deficiency. Nat. Rev. Dis. Primers 2017, 3, 17040. [Google Scholar] [CrossRef]
- Kerns, J.C.; Gutierrez, J.L. Thiamin. Adv. Nutr. 2017, 8, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline. The National Academies Collection: Reports funded by National Institutes of Health. In Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; National Academies Press (US): Washington, DC, USA, 1998. [Google Scholar] [CrossRef]
- Sauve, A.A. NAD+ and vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Plesofsky-Vig, N.; Brambl, R. Pantothenic acid and coenzyme A in cellular modification of proteins. Annu. Rev. Nutr. 1988, 8, 461–482. [Google Scholar] [CrossRef] [PubMed]
- Ueland, P.M.; Ulvik, A.; Rios-Avila, L.; Midttun, Ø.; Gregory, J.F. Direct and Functional Biomarkers of Vitamin B6 Status. Annu. Rev. Nutr. 2015, 35, 33–70. [Google Scholar] [CrossRef]
- McMahon, R.J. Biotin in metabolism and molecular biology. Annu. Rev. Nutr. 2002, 22, 221–239. [Google Scholar] [CrossRef]
- Tong, X.; Zhao, F.; Thompson, C.B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37. [Google Scholar] [CrossRef]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Lunt, S.Y.; Dayton, T.L.; Fiske, B.P.; Israelsen, W.J.; Mattaini, K.R.; Vokes, N.I.; Stephanopoulos, G.; Cantley, L.C.; Metallo, C.M.; et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 325–334. [Google Scholar] [CrossRef]
- Methionine in National Center for Biotechnology Information. PubChem Database. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/6137 (accessed on 15 September 2020).
- Mastrototaro, L.; Sponder, G.; Saremi, B.; Aschenbach, J.R. Gastrointestinal methionine shuttle: Priority handling of precious goods. IUBMB Life 2016, 68, 924–934. [Google Scholar] [CrossRef]
- Zhao, R.; Matherly, L.H.; Goldman, I.D. Membrane transporters and folate homeostasis: Intestinal absorption and transport into systemic compartments and tissues. Expert Rev. Mol. Med. 2009, 11, e4. [Google Scholar] [CrossRef]
- Antony, A.C. Folate receptors. Annu. Rev. Nutr. 1996, 16, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, F.; Bito, T. Vitamin B12 sources and microbial interaction. Exp. Biol. Med. 2018, 243, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, F. Vitamin B12 sources and bioavailability. Exp. Biol. Med. 2007, 232, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Coda, R.; Chamlagain, B.; Varmanen, P.; Piironen, V.; Katina, K. Co-fermentation of Propionibacterium freudenreichii and Lactobacillus brevis in Wheat Bran for in situ Production of Vitamin B12. Front. Microbiol. 2019, 10, 1541. [Google Scholar] [CrossRef] [PubMed]
- Kozyraki, R.; Cases, O. Vitamin B12 absorption: Mammalian physiology and acquired and inherited disorders. Biochimie 2013, 95, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Gailus, S.; Höhne, W.; Gasnier, B.; Nürnberg, P.; Fowler, B.; Rutsch, F. Insights into lysosomal cobalamin trafficking: Lessons learned from cblF disease. J. Mol. Med. 2010, 88, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Coelho, D.; Kim, J.C.; Miousse, I.R.; Fung, S.; du Moulin, M.; Buers, I.; Suormala, T.; Burda, P.; Frapolli, M.; Stucki, M.; et al. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 2012, 44, 1152–1155. [Google Scholar] [CrossRef]
- Alam, A.; Woo, J.-S.; Schmitz, J.; Prinz, B.; Root, K.; Chen, F.; Bloch, J.S.; Zenobi, R.; Locher, K.P. Structural basis of transcobalamin recognition by human CD320 receptor. Nat. Commun. 2016, 7, 12100. [Google Scholar] [CrossRef]
- Cai, X.C.; Kapilashrami, K.; Luo, M. Synthesis and Assays of Inhibitors of Methyltransferases. Methods Enzymol. 2016, 574, 245–308. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Hoffman, R.M.; Bertino, J.R. Exploiting methionine restriction for cancer treatment. Biochem. Pharmacol. 2018, 154, 170–173. [Google Scholar] [CrossRef]
- Koutmos, M.; Datta, S.; Pattridge, K.A.; Smith, J.L.; Matthews, R.G. Insights into the reactivation of cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2009, 106, 18527–18532. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Wongkittichote, P.; Cunningham, G.; Summar, M.L.; Pumbo, E.; Forny, P.; Baumgartner, M.R.; Chapman, K.A. Tricarboxylic acid cycle enzyme activities in a mouse model of methylmalonic aciduria. Mol. Genet. Metab. 2019, 128, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Caterino, M.; Cevenini, A.; Jung, V.; Chhuon, C.; Lipecka, J.; Fedele, R.; Guerrera, I.C.; Ruoppolo, M. Proteomics Reveals that Methylmalonyl-CoA Mutase Modulates Cell Architecture and Increases Susceptibility to Stress. Int. J. Mol. Sci. 2020, 21, 4998. [Google Scholar] [CrossRef]
- Costanzo, M.; Cevenini, A.; Marchese, E.; Imperlini, E.; Raia, M.; Del Vecchio, L.; Caterino, M.; Ruoppolo, M. Label-Free Quantitative Proteomics in a Methylmalonyl-CoA Mutase-Silenced Neuroblastoma Cell Line. Int. J. Mol. Sci. 2018, 19, 3580. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-F.S.; Ho, Y.-H.; Lin, H.-L.; Wei, J.-S.; Liu, T.-Z. Folate Deficiency Induces a Cell Cycle-Specific Apoptosis in HepG2 Cells. J. Nutr. 1999, 129, 25–31. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Sun, Q.; He, B.; Jia, Y.; Cai, D.; Zhao, R. Folate deprivation induces cell cycle arrest at G0/G1 phase and apoptosis in hippocampal neuron cells through down-regulation of IGF-1 signaling pathway. Int. J. Biochem. Cell Biol. 2016, 79, 222–230. [Google Scholar] [CrossRef]
- Battaglia-Hsu, S.F.; Akchiche, N.; Noel, N.; Alberto, J.M.; Jeannesson, E.; Orozco-Barrios, C.E.; Martinez-Fong, D.; Daval, J.L.; Gueant, J.L. Vitamin B12 deficiency reduces proliferation and promotes differentiation of neuroblastoma cells and up-regulates PP2A, proNGF, and TACE. Proc. Natl. Acad. Sci. USA 2009, 106, 21930–21935. [Google Scholar] [CrossRef]
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine Metabolism Regulates Maintenance and Differentiation of Human Pluripotent Stem Cells. Cell Metab. 2014, 19, 780–794. [Google Scholar] [CrossRef]
- Choi, S.W.; Friso, S.; Ghandour, H.; Bagley, P.J.; Selhub, J.; Mason, J.B. Vitamin B-12 deficiency induces anomalies of base substitution and methylation in the DNA of rat colonic epithelium. J. Nutr. 2004, 134, 750–755. [Google Scholar] [CrossRef]
- Fernandez-Roig, S.; Lai, S.C.; Murphy, M.M.; Fernandez-Ballart, J.; Quadros, E.V. Vitamin B12 deficiency in the brain leads to DNA hypomethylation in the TCblR/CD320 knockout mouse. Nutr. Metab. 2012, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.A.; Gretz, D.M.; Taylor, P.C.; James, S.J.; Pogribny, I.P.; Miller, B.J.; Henning, S.M.; Swendseid, M.E. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J. Nutr. 1998, 128, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gomez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Battaglia-Hsu, S.F.; Ghemrawi, R.; Coelho, D.; Dreumont, N.; Mosca, P.; Hergalant, S.; Gauchotte, G.; Sequeira, J.M.; Ndiongue, M.; Houlgatte, R.; et al. Inherited disorders of cobalamin metabolism disrupt nucleocytoplasmic transport of mRNA through impaired methylation/phosphorylation of ELAVL1/HuR. Nucleic Acids Res. 2018, 46, 7844–7857. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Arnold, C.; Battaglia-Hsu, S.F.; Pourié, G.; Trinh, I.; Bassila, C.; Rashka, C.; Wiedemann, A.; Flayac, J.; Robert, A.; et al. SIRT1 activation rescues the mislocalization of RNA-binding proteins and cognitive defects induced by inherited cobalamin disorders. Metabolism 2019, 101, 153992. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P., Jr.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Molloy, A.M.; Kirke, P.N.; Brody, L.C.; Scott, J.M.; Mills, J.L. Effects of folate and vitamin B12 deficiencies during pregnancy on fetal, infant, and child development. Food Nutr. Bull. 2008, 29, S101–S111, discussion S112–S115. [Google Scholar] [CrossRef]
- Bailey, R.L.; Carmel, R.; Green, R.; Pfeiffer, C.M.; Cogswell, M.E.; Osterloh, J.D.; Sempos, C.T.; Yetley, E.A. Monitoring of vitamin B-12 nutritional status in the United States by using plasma methylmalonic acid and serum vitamin B-12. Am. J. Clin. Nutr. 2011, 94, 552–561. [Google Scholar] [CrossRef]
- Rickes, E.L.; Brink, N.G.; Koniuszy, F.R.; Wood, T.R.; Folkers, K. Crystalline Vitamin B12. Science 1948, 107, 396. [Google Scholar] [CrossRef]
- Smith, E.L. Purification of Anti-pernicious Anæmia Factors from Liver. Nature 1948, 161, 638–639. [Google Scholar] [CrossRef]
- Lahner, E.; Annibale, B. Pernicious anemia: New insights from a gastroenterological point of view. World J. Gastroenterol. 2009, 15, 5121–5128. [Google Scholar] [CrossRef] [PubMed]
- Vuylsteke, P.; Bertrand, C.; Verhoef, G.E.; Vandenberghe, P. Case of megaloblastic anemia caused by intestinal taeniasis. Ann. Hematol. 2004, 83, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Cordingley, F.T.; Crawford, G.P. Giardia infection causes vitamin B12 deficiency. Aust. N. Z. J. Med. 1986, 16, 78–79. [Google Scholar] [CrossRef]
- Quigley, E.M.; Murray, J.A.; Pimentel, M. AGA Clinical Practice Update on Small Intestinal Bacterial Overgrowth: Expert Review. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, M.; Gupta, N.; Singh, T.; Velpandian, T. Cobalamin and folic Acid status in relation to the etiopathogenesis of pancytopenia in adults at a tertiary care centre in north India. Anemia 2012, 2012, 707402. [Google Scholar] [CrossRef] [PubMed]
- Aroda, V.R.; Edelstein, S.L.; Goldberg, R.B.; Knowler, W.C.; Marcovina, S.M.; Orchard, T.J.; Bray, G.A.; Schade, D.S.; Temprosa, M.G.; White, N.H.; et al. Long-term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J. Clin. Endocrinol. Metab. 2016, 101, 1754–1761. [Google Scholar] [CrossRef]
- Bauman, W.A.; Shaw, S.; Jayatilleke, E.; Spungen, A.M.; Herbert, V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care 2000, 23, 1227–1231. [Google Scholar] [CrossRef]
- Lam, J.R.; Schneider, J.L.; Zhao, W.; Corley, D.A. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA 2013, 310, 2435–2442. [Google Scholar] [CrossRef]
- Andersen, K.J.; Schjønsby, H. Intrinsic factor-mediated binding of cyanocobalamin to cholestyramine. J. Pharm. Sci. 1978, 67, 1626–1627. [Google Scholar] [CrossRef]
- Amess, J.A.; Burman, J.F.; Rees, G.M.; Nancekievill, D.G.; Mollin, D.L. Megaloblastic haemopoiesis in patients receiving nitrous oxide. Lancet 1978, 2, 339–342. [Google Scholar] [CrossRef]
- Danishpajooh, I.O.; Gudi, T.; Chen, Y.; Kharitonov, V.G.; Sharma, V.S.; Boss, G.R. Nitric oxide inhibits methionine synthase activity in vivo and disrupts carbon flow through the folate pathway. J. Biol. Chem. 2001, 276, 27296–27303. [Google Scholar] [CrossRef] [PubMed]
- Drummond, J.T.; Matthews, R.G. Nitrous oxide degradation by cobalamin-dependent methionine synthase: Characterization of the reactants and products in the inactivation reaction. Biochemistry 1994, 33, 3732–3741. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.; Lumb, M.; Perry, J.; Chanarin, I.; Minty, B.; Halsey, M.J.; Nunn, J.F. Selective inactivation of vitamin B12 in rats by nitrous oxide. Lancet 1978, 2, 1023–1024. [Google Scholar] [CrossRef]
- Hu, Y.; Raffield, L.M.; Polfus, L.M.; Moscati, A.; Nadkarni, G.; Preuss, M.H.; Zhong, X.; Wei, Q.; Rich, S.S.; Li, Y.; et al. A common TCN1 loss-of-function variant is associated with lower vitamin B12 concentration in African Americans. Blood 2018, 131, 2859–2863. [Google Scholar] [CrossRef] [PubMed]
- Namour, F.; Olivier, J.-L.; Abdelmouttaleb, I.; Adjalla, C.; Debard, R.; Salvat, C.; Guéant, J.-L. Transcobalamin codon 259 polymorphism in HT-29 and Caco-2 cells and in Caucasians: Relation to transcobalamin and homocysteine concentration in blood. Blood J. Am. Soc. Hematol. 2001, 97, 1092–1098. [Google Scholar] [CrossRef]
- Guéant, J.-L.; Chabi, N.W.; Guéant-Rodriguez, R.-M.; Mutchinick, O.M.; Debard, R.; Payet, C.; Lu, X.; Villaume, C.; Bronowicki, J.-P.; Quadros, E.V.; et al. Environmental influence on the worldwide prevalence of a 776C→G variant in the transcobalamin gene (TCN2). J. Med Genet. 2007, 44, 363. [Google Scholar] [CrossRef]
- Serrano, M.; Pérez-Dueñas, B.; Montoya, J.; Ormazabal, A.; Artuch, R. Genetic causes of cerebral folate deficiency: Clinical, biochemical and therapeutic aspects. Drug Discov. Today 2012, 17, 1299–1306. [Google Scholar] [CrossRef]
- Steele, J.W.; Kim, S.-E.; Finnell, R.H. One-carbon metabolism and folate transporter genes: Do they factor prominently in the genetic etiology of neural tube defects? Biochimie 2020, 173, 27–32. [Google Scholar] [CrossRef]
- Froese, D.S.; Gravel, R.A. Genetic disorders of vitamin B12 metabolism: Eight complementation groups—Eight genes. Expert Rev. Mol. Med. 2010, 12, e37. [Google Scholar] [CrossRef]
- Sarafoglou, K.; Rodgers, J.; Hietala, A.; Matern, D.; Bentler, K. Expanded Newborn Screening for Detection of Vitamin B12 Deficiency. JAMA 2011, 305, 1198–1200. [Google Scholar] [CrossRef]
- Gramer, G.; Fang-Hoffmann, J.; Feyh, P.; Klinke, G.; Monostori, P.; Mütze, U.; Posset, R.; Weiss, K.H.; Hoffmann, G.F.; Okun, J.G. Newborn Screening for Vitamin B12 Deficiency in Germany Strategies, Results, and Public Health Implications. J. Pediatr. 2020, 216, 165–172.e4. [Google Scholar] [CrossRef]
- Smithells, R.W.; Sheppard, S.; Schorah, C.J. Vitamin deficiencies and neural tube defects. Arch. Dis. Child. 1976, 51, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Bailey, L.B.; Berry, R.J. Folic acid food fortification-its history, effect, concerns, and future directions. Nutrients 2011, 3, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Smithells, R.W.; Sheppard, S.; Schorah, C.J.; Seller, M.J.; Nevin, N.C.; Harris, R.; Read, A.P.; Fielding, D.W. Possible prevention of neural-tube defects by periconceptional vitamin supplementation. Lancet 1980, 1, 339–340. [Google Scholar] [CrossRef]
- Laurence, K.M.; James, N.; Miller, M.H.; Tennant, G.B.; Campbell, H. Double-blind randomised controlled trial of folate treatment before conception to prevent recurrence of neural-tube defects. Br. Med. J. (Clin. Res. Ed.) 1981, 282, 1509–1511. [Google Scholar] [CrossRef] [PubMed]
- Czeizel, A.E.; Dudas, I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N. Engl. J. Med. 1992, 327, 1832–1835. [Google Scholar] [CrossRef]
- MRC Vitamin Study Research Group. Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. Lancet 1991, 338, 131–137. [Google Scholar] [CrossRef]
- Centers for Disease Control. Use of folic acid for prevention of spina bifida and other neural tube defects—1983–1991. MMWR Morb. Mortal. Wkly. Rep. 1991, 40, 513–516. [Google Scholar]
- Centers for Disease Control. Recommendations for the Use of Folic Acid to Reduce the Number of Cases of Spina Bifida and Other Neural Tube Defects. MMWR Recomm. Rep. 1992, 41, 1–7. [Google Scholar]
- Greene, N.D.; Stanier, P.; Moore, G.E. The emerging role of epigenetic mechanisms in the etiology of neural tube defects. Epigenetics 2011, 6, 875–883. [Google Scholar] [CrossRef]
- Alata Jimenez, N.; Torres Pérez, S.A.; Sánchez-Vásquez, E.; Fernandino, J.I.; Strobl-Mazzulla, P.H. Folate deficiency prevents neural crest fate by disturbing the epigenetic Sox2 repression on the dorsal neural tube. Dev. Biol. 2018, 444, S193–S201. [Google Scholar] [CrossRef] [PubMed]
- Botto, L.D.; Yang, Q. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: A HuGE review. Am. J. Epidemiol. 2000, 151, 862–877. [Google Scholar] [CrossRef]
- Imbard, A.; Benoist, J.-F.; Blom, H.J. Neural tube defects, folic acid and methylation. Int. J. Environ. Res. Public Health 2013, 10, 4352–4389. [Google Scholar] [CrossRef]
- Kim, D.J.; Venkataraman, A.; Jain, P.C.; Wiesler, E.P.; DeBlasio, M.; Klein, J.; Tu, S.S.; Lee, S.; Medzhitov, R.; Iwasaki, A. Vitamin B12 and folic acid alleviate symptoms of nutritional deficiency by antagonizing aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 15837–15845. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Dalla Torre, C.; Citton, V.; Manara, R.; Pompanin, S.; Binotto, G.; Adami, F. Cobalamin deficiency: Clinical picture and radiological findings. Nutrients 2013, 5, 4521–4539. [Google Scholar] [CrossRef] [PubMed]
- Whyte, E.M.; Mulsant, B.H.; Butters, M.A.; Qayyum, M.; Towers, A.; Sweet, R.A.; Klunk, W.; Wisniewski, S.; DeKosky, S.T. Cognitive and behavioral correlates of low vitamin B12 levels in elderly patients with progressive dementia. Am. J. Geriatr. Psychiatry 2002, 10, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Green, R. Vitamin B12 deficiency from the perspective of a practicing hematologist. Blood 2017, 129, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Konda, M.; Godbole, A.; Pandey, S.; Sasapu, A. Vitamin B12 deficiency mimicking acute leukemia. Proceedings (Bayl. Univ. Med. Cent.) 2019, 32, 589–592. [Google Scholar] [CrossRef]
- Singh, N.; Qayyum, S.; Wasik, M.A.; Luger, S.M. Combined B12 and folate deficiency presenting as an aggressive hematologic malignancy. Am. J. Hematol. 2015, 90, 964–965. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Gehrke, C.W.; Kuo, K.C.; Ehrlich, M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988, 48, 1159–1161. [Google Scholar]
- Friso, S.; Choi, S.W. The potential cocarcinogenic effect of vitamin B12 deficiency. Clin. Chem. Lab. Med. 2005, 43, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J. Folate and cancer: How DNA damage, repair and methylation impact on colon carcinogenesis. J. Inherit. Metab. Dis. 2011, 34, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I. Folate and colorectal cancer: An evidence-based critical review. Mol. Nutr. Food Res. 2007, 51, 267–292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-Y.; Wan, X.-Y.; Cao, J.-W. Dietary Methionine Intake and Risk of Incident Colorectal Cancer: A Meta-Analysis of 8 Prospective Studies Involving 431,029 Participants. PLoS ONE 2013, 8, e83588. [Google Scholar] [CrossRef]
- Pufulete, M.; Al-Ghnaniem, R.; Leather, A.J.; Appleby, P.; Gout, S.; Terry, C.; Emery, P.W.; Sanders, T.A. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: A case control study. Gastroenterology 2003, 124, 1240–1248. [Google Scholar] [CrossRef]
- Lim, U.; Flood, A.; Choi, S.W.; Albanes, D.; Cross, A.J.; Schatzkin, A.; Sinha, R.; Katki, H.A.; Cash, B.; Schoenfeld, P.; et al. Genomic methylation of leukocyte DNA in relation to colorectal adenoma among asymptomatic women. Gastroenterology 2008, 134, 47–55. [Google Scholar] [CrossRef] [PubMed]
- 99. Sharma, A.; Vatapalli, R.; Abdelfatah, E.; Wyatt McMahon, K.; Kerner, Z.; Guzzetta, A.; Singh, J.; Zahnow, C.; Baylin, S.B.; Yerram, S.; et al. Hypomethylating agents synergize with irinotecan to improve response to chemotherapy in colorectal cancer cells. PLoS ONE 2017, 12, e0176139. [Google Scholar] [CrossRef]
- Radziejewska, A.; Muzsik, A.; Milagro, F.I.; Martínez, J.A.; Chmurzynska, A. One-Carbon Metabolism and Nonalcoholic Fatty Liver Disease: The Crosstalk between Nutrients, Microbiota, and Genetics. Lifestyle Genom. 2020, 13, 53–63. [Google Scholar] [CrossRef]
- Stahl, E.C.; Haschak, M.J.; Popovic, B.; Brown, B.N. Macrophages in the Aging Liver and Age-Related Liver Disease. Front. Immunol. 2018, 9, 2795. [Google Scholar] [CrossRef]
- Eudy, B.J.; McDermott, C.E.; Fernandez, G.; Mathews, C.E.; Lai, J.; da Silva, R.P. Disruption of hepatic one-carbon metabolism impairs mitochondrial function and enhances macrophage activity in methionine-choline-deficient mice. J. Nutr. Biochem. 2020, 81, 108381. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, Y.; Park, J.B.; Kwak, J.-H.; Kim, K.-J.; Kim, J.-H.; Song, H.; Cho, J.-Y.; Hwang, D.Y.; Kim, K.S.; et al. Comparative study of fatty liver induced by methionine and choline-deficiency in C57BL/6N mice originating from three different sources. Lab. Anim. Res. 2017, 33, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Aissa, A.F.; Tryndyak, V.; de Conti, A.; Melnyk, S.; Gomes, T.D.; Bianchi, M.L.; James, S.J.; Beland, F.A.; Antunes, L.M.; Pogribny, I.P. Effect of methionine-deficient and methionine-supplemented diets on the hepatic one-carbon and lipid metabolism in mice. Mol. Nutr. Food Res. 2014, 58, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Mahamid, M.; Mahroum, N.; Bragazzi, N.L.; Shalaata, K.; Yavne, Y.; Adawi, M.; Amital, H.; Watad, A. Folate and B12 Levels Correlate with Histological Severity in NASH Patients. Nutrients 2018, 10, 440. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Uña, M.; Varela-Rey, M.; Cano, A.; Fernández-Ares, L.; Beraza, N.; Aurrekoetxea, I.; Martínez-Arranz, I.; García-Rodríguez, J.L.; Buqué, X.; Mestre, D.; et al. Excess S-adenosylmethionine reroutes phosphatidylethanolamine towards phosphatidylcholine and triglyceride synthesis. Hepatology 2013, 58, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Da Costa, K.-A.; Franklin, P.D.; Alexander, E.A.; Lamont, J.T.; Sheard, N.F.; Beiser, A. Choline, an essential nutrient for humans. FASEB J. 1991, 5, 2093–2098. [Google Scholar] [CrossRef]
- Guerrerio, A.L.; Colvin, R.M.; Schwartz, A.K.; Molleston, J.P.; Murray, K.F.; Diehl, A.; Mohan, P.; Schwimmer, J.B.; Lavine, J.E.; Torbenson, M.S.; et al. Choline intake in a large cohort of patients with nonalcoholic fatty liver disease. Am. J. Clin. Nutr. 2012, 95, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Dahlhoff, C.; Worsch, S.; Sailer, M.; Hummel, B.A.; Fiamoncini, J.; Uebel, K.; Obeid, R.; Scherling, C.; Geisel, J.; Bader, B.L.; et al. Methyl-donor supplementation in obese mice prevents the progression of NAFLD, activates AMPK and decreases acyl-carnitine levels. Mol. Metab. 2014, 3, 565–580. [Google Scholar] [CrossRef]
- Crown, S.B.; Marze, N.; Antoniewicz, M.R. Catabolism of Branched Chain Amino Acids Contributes Significantly to Synthesis of Odd-Chain and Even-Chain Fatty Acids in 3T3-L1 Adipocytes. PLoS ONE 2015, 10, e0145850. [Google Scholar] [CrossRef]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21. [Google Scholar] [CrossRef]
- Ghosh, S.; Sinha, J.K.; Putcha, U.K.; Raghunath, M. Severe but Not Moderate Vitamin B12 Deficiency Impairs Lipid Profile, Induces Adiposity, and Leads to Adverse Gestational Outcome in Female C57BL/6 Mice. Front. Nutr. 2016, 3, 1. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Food Standards: Amendment of Standards of Identity for Enriched Grain Products to Require Addition of Folic Acid; Correction; Department of Health and Human Services: Washington, DC, USA, 1996; Volume 61. [Google Scholar]
- Centers for Disease Control and Prevention. CDC Grand Rounds: Additional opportunities to prevent neural tube defects with folic acid fortification. MMWR Morb. Mortal. Wkly. Rep. 2010, 59, 980–984. [Google Scholar]
- Liu, J.J.; Ward, R.L. Folate and one-carbon metabolism and its impact on aberrant DNA methylation in cancer. Adv. Genet. 2010, 71, 79–121. [Google Scholar] [CrossRef] [PubMed]
- Honein, M.A.; Paulozzi, L.J.; Mathews, T.J.; Erickson, J.D.; Wong, L.Y. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA 2001, 285, 2981–2986. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.J.; Rasmussen, S.A.; Flores, A.; Kirby, R.S.; Edmonds, L.D. Decline in the prevalence of spina bifida and anencephaly by race/ethnicity: 1995–2002. Pediatrics 2005, 116, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Glass, G.B.J.; Skeggs, H.R.; Lee, D.H.; Jones, E.L.; Hardy, W.W. Hydroxocobalamin. I. Blood Levels and Urinary Excretion of Vitamin B12 in Man After a Single Parenteral Dose of Aqueous Hydroxocobalamin, Aqueous Cyanocobalamin and Cyanocobalamin Zinc-Tannate Complex. Blood 1961, 18, 511–521. [Google Scholar] [CrossRef]
- Beck, W.S. Diagnosis of megaloblastic anemia. Annu. Rev. Med. 1991, 42, 311–322. [Google Scholar] [CrossRef]
- Rader, J.I.; Weaver, C.M.; Angyal, G. Total folate in enriched cereal-grain products in the United States following fortification. Food Chem. 2000, 70, 275–289. [Google Scholar] [CrossRef]
- Quinlivan, E.P.; Gregory, J.F., 3rd. Effect of food fortification on folic acid intake in the United States. Am. J. Clin. Nutr. 2003, 77, 221–225. [Google Scholar] [CrossRef]
- Arendt, J.F.B.; Nexo, E. Cobalamin related parameters and disease patterns in patients with increased serum cobalamin levels. PLoS ONE 2012, 7, e45979. [Google Scholar] [CrossRef]
- Flores-Guerrero, J.L.; Minović, I.; Groothof, D.; Gruppen, E.G.; Riphagen, I.J.; Kootstra-Ros, J.; Muller Kobold, A.; Hak, E.; Navis, G.; Gansevoort, R.T.; et al. Association of Plasma Concentration of Vitamin B12 With All-Cause Mortality in the General Population in the Netherlands. JAMA Netw. Open 2020, 3, e1919274. [Google Scholar] [CrossRef]
- Yang, J.; Li, H.; Deng, H.; Wang, Z. Association of One-Carbon Metabolism-Related Vitamins (Folate, B6, B12), Homocysteine and Methionine With the Risk of Lung Cancer: Systematic Review and Meta-Analysis. Front. Oncol. 2018, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Essén, A.; Santaolalla, A.; Garmo, H.; Hammar, N.; Walldius, G.; Jungner, I.; Malmström, H.; Holmberg, L.; Van Hemelrijck, M. Baseline serum folate, vitamin B12 and the risk of prostate and breast cancer using data from the Swedish AMORIS cohort. Cancer Causes Control. 2019, 30, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.B.; Dickstein, A.; Jacques, P.F.; Haggarty, P.; Selhub, J.; Dallal, G.; Rosenberg, I.H. A Temporal Association between Folic Acid Fortification and an Increase in Colorectal Cancer Rates May Be Illuminating Important Biological Principles: A Hypothesis. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1325. [Google Scholar] [CrossRef] [PubMed]
- Arendt, J.F.; Pedersen, L.; Nexo, E.; Sorensen, H.T. Elevated plasma vitamin B12 levels as a marker for cancer: A population-based cohort study. J. Natl. Cancer Inst. 2013, 105, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Arendt, J.F.H.; Sorensen, H.T.; Horsfall, L.J.; Petersen, I. Elevated Vitamin B12 Levels and Cancer Risk in UK Primary Care: A THIN Database Cohort Study. Cancer Epidemiol. Biomark. Prev. 2019, 28, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Andrès, E.; Serraj, K.; Zhu, J.; Vermorken, A.J.M. The pathophysiology of elevated vitamin B12 in clinical practice. QJM Int. J. Med. 2013, 106, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Collin, S.M.; Metcalfe, C.; Refsum, H.; Lewis, S.J.; Zuccolo, L.; Smith, G.D.; Chen, L.; Harris, R.; Davis, M.; Marsden, G.; et al. Circulating Folate, Vitamin B12, Homocysteine, Vitamin B12, Transport Proteins, and Risk of Prostate Cancer: A Case-Control Study, Systematic Review, and Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1632. [Google Scholar] [CrossRef] [PubMed]
- Chiche, L.; Jean, R.; Romain, F.; Roux, F.; Thomas, G.; Canavese, S.; Branger, S.; Harlé, J.R.; Durand, J.M. Implications cliniques de la découverte d’une hypervitaminémie B12 en médecine interne. Rev. Med. Interne 2008, 29, 187–194. [Google Scholar] [CrossRef]
- Beard, M.F.; Pitney, W.R.; Sanneman, E.H. Serum concentrations of vitamin B12 in patients suffering from leukemia. Blood 1954, 9, 789–794. [Google Scholar] [CrossRef]
- Mollin, D.; Ross, G. Serum vitamin B12 concentrations in leukaemia and in some other haematological conditions. Br. J. Haematol. 1955, 1, 155–172. [Google Scholar] [CrossRef]
- Gilbert, H.S.; Krauss, S.; Pasternack, B.; Herbert, V.; Wasserman, L.R. Serum vitamin B12 content and unsaturated vitamin B12-binding capacity in myeloproliferative disease: Value in differential diagnosis and as indicators of disease activity. Ann. Intern. Med. 1969, 71, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Gimsing, P. Cobalamin forms and analogues in plasma and myeloid cells during chronic myelogenous leukaemia related to clinical condition. Br. J. Haematol. 1995, 89, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.A.; Finkler, A.E. Abnormal Transport of Vitamin B12 in Plasma in Chronic Myelogenous Leukemia. Nature 1964, 204, 1207. [Google Scholar] [CrossRef] [PubMed]
- Gimsing, P.; Overballe-Petersen, C.; Hippe, E. Cobalamin and cobalamin-binding proteins in plasma related to the clinical condition in chronic myelogenous leukemia. Leukemia 1995, 9, 1604–1609. [Google Scholar] [PubMed]
- Deneuville, T.; Mario, N.; Tiev, K.P.; Toledano, C.; Josselin-Mahr, L.; Gain, M.; Pateron, D.; Guidet, B.; Retbi, A.; Taright, N.; et al. Concentration plasmatique élevée de la vitamine B12: Un indicateur des maladies hépatiques ou tumorales? Rev. Med. Interne 2009, 30. [Google Scholar] [CrossRef]
- Carmel, R. Extreme elevation of serum transcobalamin I in patients with metastatic cancer. N. Engl. J. Med. 1975, 292, 282–284. [Google Scholar] [CrossRef]
- Frémont, S.; Champigneulle, B.; Gérard, P.; Felden, F.; Lambert, D.; Guéant, J.L.; Nicolas, J.P. Blood transcobalamin levels in malignant hepatoma. Tumour Biol. 1991, 12, 353–359. [Google Scholar] [CrossRef]
- Liu, G.; Wang, Y.; Yue, M.; Zhao, L.; Guo, Y.-D.; Liu, Y.; Yang, H.; Liu, F.; Zhang, X.; Zhi, L.; et al. High expression of TCN1 is a negative prognostic biomarker and can predict neoadjuvant chemosensitivity of colon cancer. Sci. Rep. (Nat. Publ. Group) 2020, 10. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Wei, Y.C.; Tian, Y.F.; Sun, D.P.; Sheu, M.J.; Yang, C.C.; Lin, L.C.; Lin, C.Y.; Hsing, C.H.; Li, W.S.; et al. Overexpression of Transcobalamin 1 is an Independent Negative Prognosticator in Rectal Cancers Receiving Concurrent Chemoradiotherapy. J. Cancer 2017, 8, 1330–1337. [Google Scholar] [CrossRef]
- Brasky, T.M.; White, E.; Chen, C.-L. Long-Term, Supplemental, One-Carbon Metabolism-Related Vitamin B Use in Relation to Lung Cancer Risk in the Vitamins and Lifestyle (VITAL) Cohort. J. Clin. Oncol. 2017, 35, 3440–3448. [Google Scholar] [CrossRef]
- Fanidi, A.; Carreras-Torres, R.; Larose, T.L.; Yuan, J.-M.; Stevens, V.L.; Weinstein, S.J.; Albanes, D.; Prentice, R.; Pettinger, M.; Cai, Q.; et al. Is high vitamin B12 status a cause of lung cancer? Int. J. Cancer 2019, 145, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.C.; Grau, M.V.; Haile, R.W.; Sandler, R.S.; Summers, R.W.; Bresalier, R.S.; Burke, C.A.; McKeown-Eyssen, G.E.; Baron, J.A. Folic acid and risk of prostate cancer: Results from a randomized clinical trial. J. Natl. Cancer Inst. 2009, 101, 432–435. [Google Scholar] [CrossRef]
- Oliai Araghi, S.; Kiefte-de Jong, J.C.; van Dijk, S.C.; Swart, K.M.A.; van Laarhoven, H.W.; van Schoor, N.M.; de Groot, L.; Lemmens, V.; Stricker, B.H.; Uitterlinden, A.G.; et al. Folic Acid and Vitamin B12 Supplementation and the Risk of Cancer: Long-term Follow-up of the B Vitamins for the Prevention of Osteoporotic Fractures (B-PROOF) Trial. Cancer Epidemiol. Biomark. Prev. 2019, 28, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Naghshi, S.; Sadeghi, O.; Willett, W.C.; Esmaillzadeh, A. Dietary intake of total, animal, and plant proteins and risk of all cause, cardiovascular, and cancer mortality: Systematic review and dose-response meta-analysis of prospective cohort studies. BMJ 2020, 370, m2412. [Google Scholar] [CrossRef] [PubMed]
- Troen, A.M.; Mitchell, B.; Sorensen, B.; Wener, M.H.; Johnston, A.; Wood, B.; Selhub, J.; McTiernan, A.; Yasui, Y.; Oral, E.; et al. Unmetabolized folic acid in plasma is associated with reduced natural killer cell cytotoxicity among postmenopausal women. J. Nutr. 2006, 136, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Nemkov, T.; Casas-Selves, M.; Bilousova, G.; Zaberezhnyy, V.; Higa, K.C.; Serkova, N.J.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Folate dietary insufficiency and folic acid supplementation similarly impair metabolism and compromise hematopoiesis. Haematologica 2017, 102, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.-Y.; Lee, T.-H.; Wang, H.-H.; Yang, T.-H.; Chang, I.J.; Huang, Y.-C. Serum Homocysteine, Folate, and Vitamin B12 Levels in Patients with Systemic Lupus Erythematosus: A Meta-Analysis and Meta-Regression. J. Am. Coll. Nutr. 2020, 1–11. [Google Scholar] [CrossRef]
- Ermens, A.A.; Vlasveld, L.T.; Lindemans, J. Significance of elevated cobalamin (vitamin B12) levels in blood. Clin. Biochem. 2003, 36, 585–590. [Google Scholar] [CrossRef]
- Baker, H.; Leevy, C.B.; DeAngelis, B.; Frank, O.; Baker, E.R. Cobalamin (Vitamin B12) and Holotranscobalamin Changes in Plasma and Liver Tissue in Alcoholics with Liver Disease. J. Am. Coll. Nutr. 1998, 17, 235–238. [Google Scholar] [CrossRef]
- McMahon, G.M.; Hwang, S.-J.; Tanner, R.M.; Jacques, P.F.; Selhub, J.; Muntner, P.; Fox, C.S. The association between vitamin B12, albuminuria and reduced kidney function: An observational cohort study. BMC Nephrol. 2015, 16, 7. [Google Scholar] [CrossRef]
- Carmel, R.; Vasireddy, H.; Aurangzeb, I.; George, K. High serum cobalamin levels in the clinical setting--clinical associations and holo-transcobalamin changes. Clin. Lab. Haematol. 2001, 23, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.M.; Beckett, A.G. Serum vitamin B12 in renal failure. J. Clin. Pathol. 1962, 15, 456–458. [Google Scholar] [CrossRef]
- Julian, T.; Syeed, R.; Glascow, N.; Angelopoulou, E.; Zis, P. B12 as a Treatment for Peripheral Neuropathic Pain: A Systematic Review. Nutrients 2020, 12, 2221. [Google Scholar] [CrossRef] [PubMed]
- Xǔ, G.; Xu, S.; Tang, W.-Z.; Xú, G.; Cheng, C.; Xu, J. Local Injection of Methylcobalamin Combined with Lidocaine for Acute Herpetic Neuralgia. Pain Med. 2015, 17, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Lv, Z.-W.; Feng, Y.; Tang, W.-Z.; Xu, G. A Single-Center Randomized Controlled Trial of Local Methylcobalamin Injection for Subacute Herpetic Neuralgia. Pain Med. 2013, 14, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Bowen, R.A.; Drake, S.K.; Vanjani, R.; Huey, E.D.; Grafman, J.; Horne, M.K., 3rd. Markedly increased vitamin B12 concentrations attributable to IgG-IgM-vitamin B12 immune complexes. Clin. Chem. 2006, 52, 2107–2114. [Google Scholar] [CrossRef]
- Yadav, D.K.; Shrestha, S.; Lillycrop, K.A.; Joglekar, C.V.; Pan, H.; Holbrook, J.D.; Fall, C.H.D.; Yajnik, C.S.; Chandak, G.R. Vitamin B12 supplementation influences methylation of genes associated with Type 2 diabetes and its intermediate traits. Epigenomics 2017, 10, 71–90. [Google Scholar] [CrossRef]
- Trautmann, C.; Bock, A.; Urbach, A.; Hübner, C.A.; Engmann, O. Acute vitamin B12 supplementation evokes antidepressant response and alters Ntrk-2. Neuropharmacology 2020, 171, 108112. [Google Scholar] [CrossRef]
- de Queiroz, K.B.; Cavalcante-Silva, V.; Lopes, F.L.; Rocha, G.A.; D’Almeida, V.; Coimbra, R.S. Vitamin B12 is neuroprotective in experimental pneumococcal meningitis through modulation of hippocampal DNA methylation. J. Neuroinflammation 2020, 17, 96. [Google Scholar] [CrossRef]
- Chang, S.E.; Littlefield, J.W. Elevated dihydrofolate reductase messenger RNA levels in methotrexate-resistant BHK cells. Cell 1976, 7, 391–396. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.C.; Prigot, A.; Wright, B.; Weintraub, S.; Wright, L.T. An evaluation of folic acid antagonists in adults with neoplastic diseases: A study of 93 patients with incurable neoplasms. J. Natl. Med. Assoc. 1951, 43, 211–240. [Google Scholar] [PubMed]
- Evans, W.E.; Crom, W.R.; Abromowitch, M.; Dodge, R.; Look, A.T.; Bowman, W.P.; George, S.L.; Pui, C.H. Clinical pharmacodynamics of high-dose methotrexate in acute lymphocytic leukemia. Identification of a relation between concentration and effect. N. Engl. J. Med. 1986, 314, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Vogler, W.R.; Huguley, C.M., Jr.; Kerr, W. Toxicity and Antitumor Effect of Divided Doses of Methotrexate. Arch. Intern. Med. 1965, 115, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Hersh, E.M.; Wong, V.G.; Henderson, E.S.; Freireich, E.J. Hepatotoxic effects of methotrexate. Cancer 1966, 19, 600–606. [Google Scholar] [CrossRef]
- Levitt, M.; Mosher, M.B.; DeConti, R.C.; Farber, L.R.; Skeel, R.T.; Marsh, J.C.; Mitchell, M.S.; Papac, R.J.; Thomas, E.D.; Bertino, J.R. Improved therapeutic index of methotrexate with “leucovorin rescue”. Cancer Res. 1973, 33, 1729–1734. [Google Scholar]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Vogler, W.R.; Jacobs, J. Toxic and therapeutic effects of methotrexate-folinic acid (Leucovorin) in advanced cancer and leukemia. Cancer 1971, 28, 894–901. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Coblyn, J.S.; Fox, D.A.; Fraser, P.A.; Holdsworth, D.E.; Glass, D.N.; Trentham, D.E. Efficacy of low-dose methotrexate in rheumatoid arthritis. N. Engl. J. Med. 1985, 312, 818–822. [Google Scholar] [CrossRef]
- Cronstein, B.N. Low-dose methotrexate: A mainstay in the treatment of rheumatoid arthritis. Pharmacol. Rev. 2005, 57, 163–172. [Google Scholar] [CrossRef]
- Rossetti, M.; Spreafico, R.; Saidin, S.; Chua, C.; Moshref, M.; Leong, J.Y.; Tan, Y.K.; Thumboo, J.; van Loosdregt, J.; Albani, S. Ex vivo-expanded but not in vitro-induced human regulatory T cells are candidates for cell therapy in autoimmune diseases thanks to stable demethylation of the FOXP3 regulatory T cell-specific demethylated region. J. Immunol. 2015, 194, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Amjadi, P.; Green, P.; Read, J.E.; Brennan, F.; Gregory, B.; Williams, R.O. Methotrexate Restores Regulatory T Cell Function Through Demethylation of the FoxP3 Upstream Enhancer in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 1182–1192. [Google Scholar] [CrossRef]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 2012, 30, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Linehan, W.M.; Helman, L.J. Targeting cancer metabolism. Clin. Cancer Res. 2012, 18, 5537–5545. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Q.; Yin, F.-Q.; Zhang, W.; Yan, L.-H.; Li, L. MTRR silencing inhibits growth and cisplatin resistance of ovarian carcinoma via inducing apoptosis and reducing autophagy. Am. J. Transl. Res. 2015, 7, 1510–1527. [Google Scholar]
- Richie, J.P., Jr.; Leutzinger, Y.; Parthasarathy, S.; Malloy, V.; Orentreich, N.; Zimmerman, J.A. Methionine restriction increases blood glutathione and longevity in F344 rats. FASEB J. 1994, 8, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Buehner, G.; Chang, Y.; Harper, J.M.; Sigler, R.; Smith-Wheelock, M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 2005, 4, 119–125. [Google Scholar] [CrossRef]
- Orentreich, N.; Matias, J.R.; DeFelice, A.; Zimmerman, J.A. Low methionine ingestion by rats extends life span. J. Nutr. 1993, 123, 269–274. [Google Scholar] [CrossRef]
- Zimmerman, J.A.; Malloy, V.; Krajcik, R.; Orentreich, N. Nutritional control of aging. Exp. Gerontol. 2003, 38, 47–52. [Google Scholar] [CrossRef]
- Ables, G.P.; Perrone, C.E.; Orentreich, D.; Orentreich, N. Methionine-restricted C57BL/6J mice are resistant to diet-induced obesity and insulin resistance but have low bone density. PLoS ONE 2012, 7, e51357. [Google Scholar] [CrossRef]
- Malloy, V.L.; Krajcik, R.A.; Bailey, S.J.; Hristopoulos, G.; Plummer, J.D.; Orentreich, N. Methionine restriction decreases visceral fat mass and preserves insulin action in aging male Fischer 344 rats independent of energy restriction. Aging Cell 2006, 5, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Barcena, C.; Quiros, P.M.; Durand, S.; Mayoral, P.; Rodriguez, F.; Caravia, X.M.; Marino, G.; Garabaya, C.; Fernandez-Garcia, M.T.; Kroemer, G.; et al. Methionine Restriction Extends Lifespan in Progeroid Mice and Alters Lipid and Bile Acid Metabolism. Cell Rep. 2018, 24, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Grandison, R.C.; Piper, M.D.; Partridge, L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 2009, 462, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yu, L.; Fang, J.; Hu, C.A.; Yin, J.; Ni, H.; Ren, W.; Duraipandiyan, V.; Chen, S.; Al-Dhabi, N.A.; et al. Methionine restriction on oxidative stress and immune response in dss-induced colitis mice. Oncotarget 2017, 8, 44511–44520. [Google Scholar] [CrossRef] [PubMed]
- Martinez, Y.; Li, X.; Liu, G.; Bin, P.; Yan, W.; Mas, D.; Valdivie, M.; Hu, C.A.; Ren, W.; Yin, Y. The role of methionine on metabolism, oxidative stress, and diseases. Amino Acids 2017, 49, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Dixon, T.; Jung, S.; Graff, E.C.; Forney, L.A.; Gettys, T.W.; Wanders, D. Dietary Methionine Restriction Reduces Inflammation Independent of FGF21 Action. Obesity 2019, 27, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Sadighi Akha, A.A.; Miller, R.A.; Harper, J.M. Life-span extension in mice by preweaning food restriction and by methionine restriction in middle age. J. Gerontol. Biol. Sci. Med. Sci. 2009, 64, 711–722. [Google Scholar] [CrossRef]
- Wanders, D.; Ghosh, S.; Stone, K.P.; Van, N.T.; Gettys, T.W. Transcriptional impact of dietary methionine restriction on systemic inflammation: Relevance to biomarkers of metabolic disease during aging. Biofactors 2014, 40, 13–26. [Google Scholar] [CrossRef]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.F.; Fraga, M.F.; Avila, S.; Guo, M.; Pollan, M.; Herman, J.G.; Esteller, M. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003, 63, 1114–1121. [Google Scholar] [PubMed]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih Ie, M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Cavuoto, P.; Fenech, M.F. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat. Rev. 2012, 38, 726–736. [Google Scholar] [CrossRef]
- Cellarier, E.; Durando, X.; Vasson, M.P.; Farges, M.C.; Demiden, A.; Maurizis, J.C.; Madelmont, J.C.; Chollet, P. Methionine dependency and cancer treatment. Cancer Treat. Rev. 2003, 29, 489–499. [Google Scholar] [CrossRef]
- Sugimura, T.; Birnbaum, S.M.; Winitz, M.; Greenstein, J.P. Quantitative nutritional studies with water-soluble, chemically defined diets. VIII. The forced feeding of diets each lacking in one essential amino acid. Arch. Biochem. Biophys. 1959, 81, 448–455. [Google Scholar] [CrossRef]
- Guo, H.Y.; Herrera, H.; Groce, A.; Hoffman, R.M. Expression of the biochemical defect of methionine dependence in fresh patient tumors in primary histoculture. Cancer Res. 1993, 53, 2479–2483. [Google Scholar] [PubMed]
- Halpern, B.C.; Clark, B.R.; Hardy, D.N.; Halpern, R.M.; Smith, R.A. The effect of replacement of methionine by homocystine on survival of malignant and normal adult mammalian cells in culture. Proc. Natl. Acad. Sci. USA 1974, 71, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Mecham, J.O.; Rowitch, D.; Wallace, C.D.; Stern, P.H.; Hoffman, R.M. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem. Biophys. Res. Commun. 1983, 117, 429–434. [Google Scholar] [CrossRef]
- Hoshiya, Y.; Kubota, T.; Inada, T.; Kitajima, M.; Hoffman, R.M. Methionine-depletion modulates the efficacy of 5-fluorouracil in human gastric cancer in nude mice. Anticancer Res. 1997, 17, 4371–4375. [Google Scholar] [PubMed]
- Sinha, R.; Cooper, T.K.; Rogers, C.J.; Sinha, I.; Turbitt, W.J.; Calcagnotto, A.; Perrone, C.E.; Richie, J.P., Jr. Dietary methionine restriction inhibits prostatic intraepithelial neoplasia in TRAMP mice. Prostate 2014, 74, 1663–1673. [Google Scholar] [CrossRef]
- Hens, J.R.; Sinha, I.; Perodin, F.; Cooper, T.; Sinha, R.; Plummer, J.; Perrone, C.E.; Orentreich, D. Methionine-restricted diet inhibits growth of MCF10AT1-derived mammary tumors by increasing cell cycle inhibitors in athymic nude mice. BMC Cancer 2016, 16, 349. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, J.H.; Lee, E.; Jang, Y.J.; Son, J.E.; Kwon, J.Y.; Lim, T.-G.; Kim, S.; Park, J.H.Y.; Kim, J.-E.; et al. Methionine deprivation suppresses triple-negative breast cancer metastasis in vitro and in vivo. Oncotarget 2016, 7, 67223–67234. [Google Scholar] [CrossRef]
- Miousse, I.R.; Tobacyk, J.; Quick, C.M.; Jamshidi-Parsian, A.; Skinner, C.M.; Kore, R.; Melnyk, S.B.; Kutanzi, K.R.; Xia, F.; Griffin, R.J.; et al. Modulation of dietary methionine intake elicits potent, yet distinct, anticancer effects on primary versus metastatic tumors. Carcinogenesis 2018, 39, 1117–1126. [Google Scholar] [CrossRef]
- Goseki, N.; Yamazaki, S.; Shimojyu, K.; Kando, F.; Maruyama, M.; Endo, M.; Koike, M.; Takahashi, H. Synergistic effect of methionine-depleting total parenteral nutrition with 5-fluorouracil on human gastric cancer: A randomized, prospective clinical trial. Jpn. J. Cancer Res. 1995, 86, 484–489. [Google Scholar] [CrossRef]
- Molina-Montes, E.; Salamanca-Fernández, E.; Garcia-Villanova, B.; Sánchez, M.J. The Impact of Plant-Based Dietary Patterns on Cancer-Related Outcomes: A Rapid Review and Meta-Analysis. Nutrients 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Olteanu, H.; Munson, T.; Banerjee, R. Differences in the efficiency of reductive activation of methionine synthase and exogenous electron acceptors between the common polymorphic variants of human methionine synthase reductase. Biochemistry 2002, 41, 13378–13385. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, D.J.; Kluijtmans, L.A.; Barbaux, S.; McMaster, D.; Young, I.S.; Yarnell, J.W.; Evans, A.; Whitehead, A.S. The methionine synthase reductase (MTRR) A66G polymorphism is a novel genetic determinant of plasma homocysteine concentrations. Atherosclerosis 2001, 157, 451–456. [Google Scholar] [CrossRef]
- Fang, D.H.; Ji, Q.; Fan, C.H.; An, Q.; Li, J. Methionine synthase reductase A66G polymorphism and leukemia risk: Evidence from published studies. Leuk. Lymphoma 2014, 55, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, S.; Wang, M.; He, J.; Xi, S. Association of MTRR A66G polymorphism with cancer susceptibility: Evidence from 85 studies. J. Cancer 2017, 8, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Allen, A.E.; Locasale, J.W. The Molecular Link from Diet to Cancer Cell Metabolism. Mol. Cell 2020, 78, 1034–1044. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Vitamin | Biological Function |
|---|---|
| B1 (thiamine) | cofactor for enzymes in glucose metabolism, amino acid catabolism, nucleotide synthesis, and fatty acid synthesis [11] |
| B2 (riboflavin) | precursor for flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) for cellular respiration [12] |
| B3 (nicotinamide) | precursor for nicotinamide adenine dinucleotide (NAD) utilized in biosynthetic pathways, energy metabolism, and protection from reactive oxygen species [13] |
| B5 (pantothenic acid) | precursor for coenzyme A (coA), an acyl-carrier required for the activity of many enzymes [14] |
| B6 (pyridoxine) | cofactor for over 150 enzymes involved mainly in amino acid synthesis and degradation [15] |
| B7 (biotin) | plays an essential role in carboxylation reactions [16] and also has many applications in laboratory research |
| B9 (folate) | substrate for nucleotide synthesis and methyl-donors in the one-carbon metabolism pathway [12] |
| B12 (cobalamin) | cofactor for enzymes in one-carbon metabolism and the propionate catabolic pathway [12] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyon, P.; Strippoli, V.; Fang, B.; Cimmino, L. B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease. Nutrients 2020, 12, 2867. https://doi.org/10.3390/nu12092867
Lyon P, Strippoli V, Fang B, Cimmino L. B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease. Nutrients. 2020; 12(9):2867. https://doi.org/10.3390/nu12092867
Chicago/Turabian StyleLyon, Peter, Victoria Strippoli, Byron Fang, and Luisa Cimmino. 2020. "B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease" Nutrients 12, no. 9: 2867. https://doi.org/10.3390/nu12092867
APA StyleLyon, P., Strippoli, V., Fang, B., & Cimmino, L. (2020). B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease. Nutrients, 12(9), 2867. https://doi.org/10.3390/nu12092867