Intracellular pH Regulation of Skeletal Muscle in the Milieu of Insulin Signaling


{kind=link}
{kind=link}
Abstract
1. Introduction
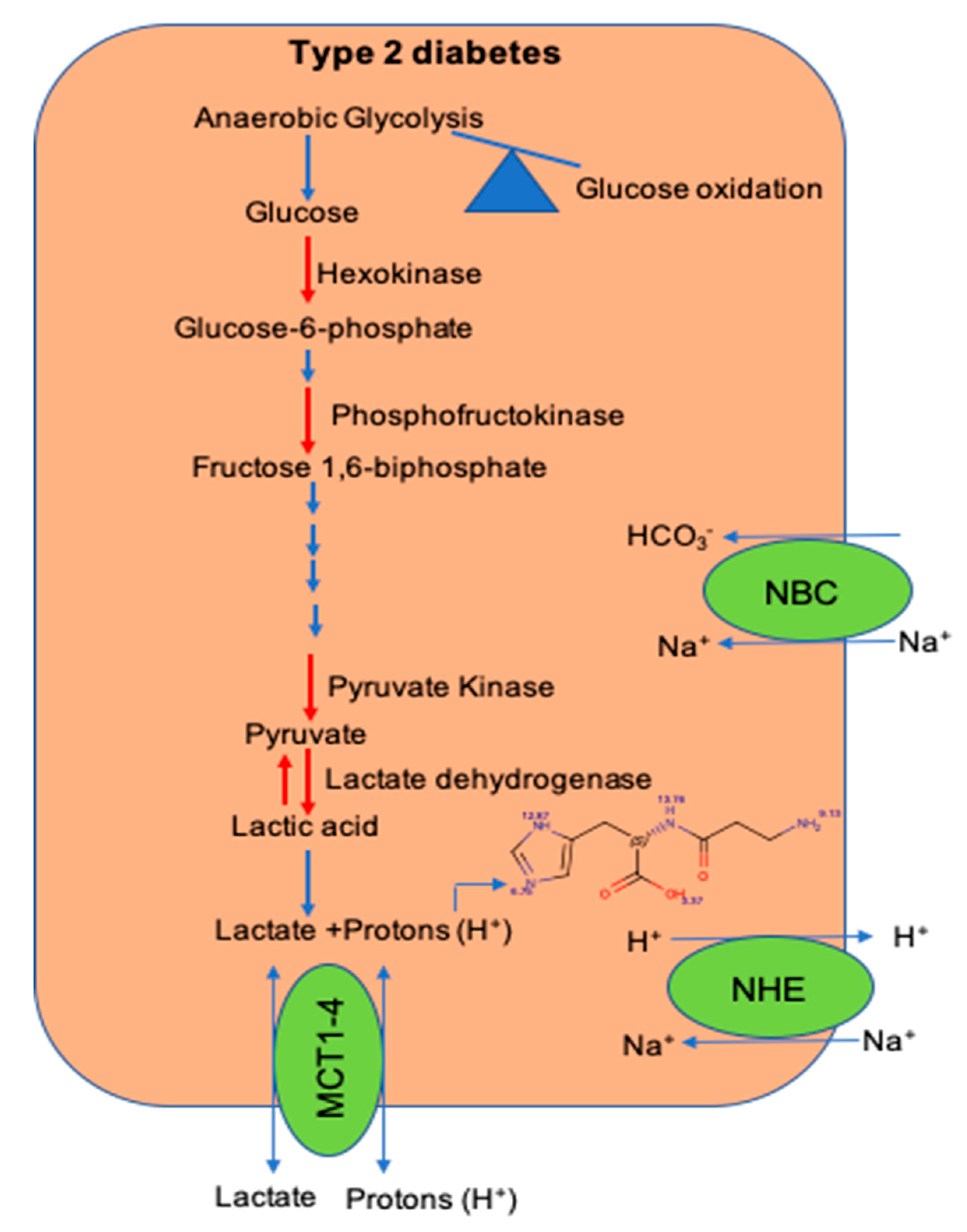
2. Defects in Glycolytic Pathway During Type 2 Diabetes
3. Acidosis and Insulin Resistance
4. Maintenance of Intracellular pH ([pH]i) in the Muscle
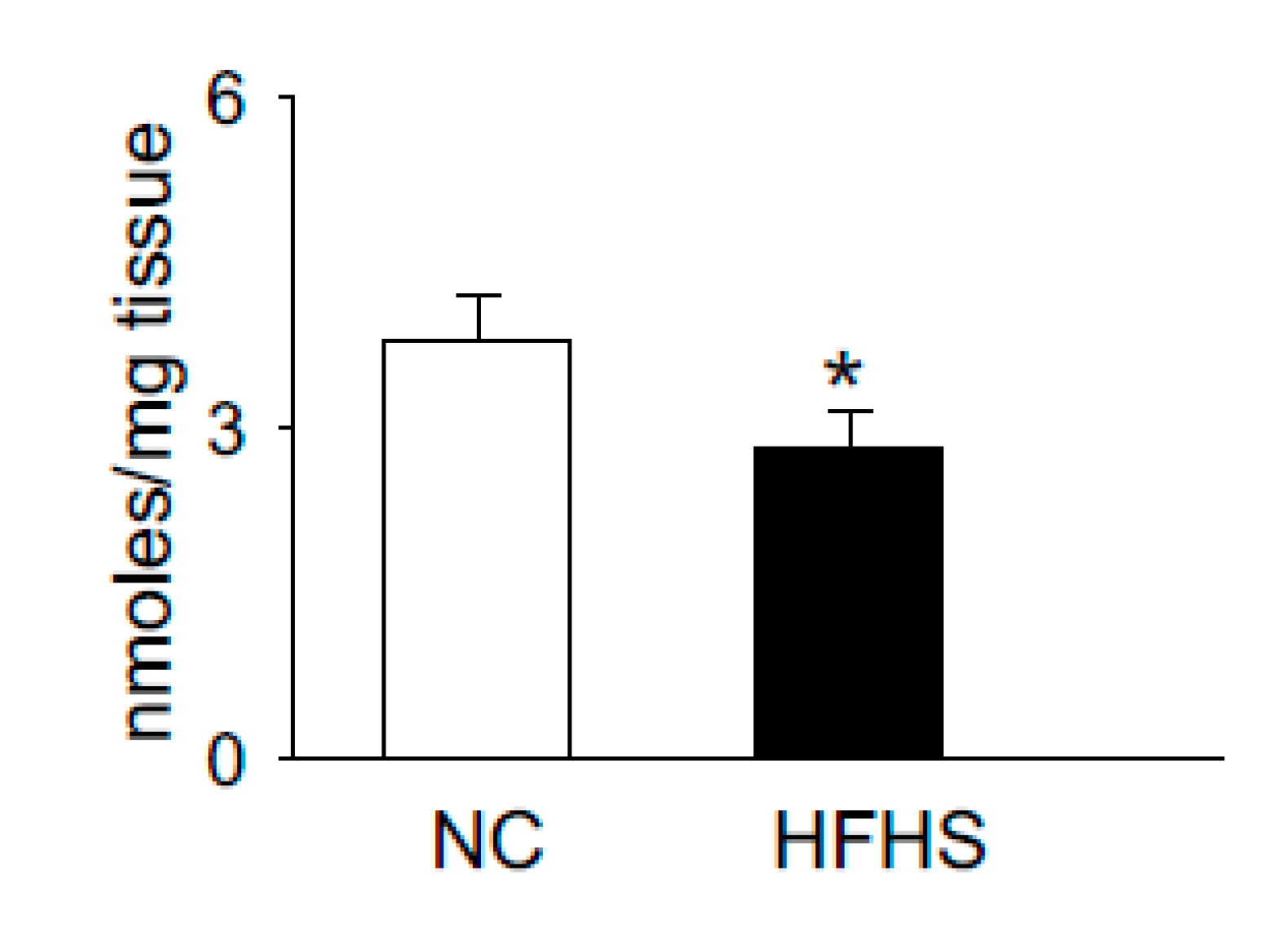
5. Histidyl Dipeptides and Intracellular pH [pH]i Regulation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lillioja, S.; Mott, D.M.; Howard, B.V.; Bennett, P.H.; Yki-Jarvinen, H.; Freymond, D.; Nyomba, B.L.; Zurlo, F.; Swinburn, B.; Bogardus, C. Impaired Glucose Tolerance as a Disorder of Insulin Action. Longitudinal and cross-sectional studies in Pima Indians. N. Eng. J. Med. 1988, 318, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Warram, J.H.; Martin, B.C.; Krolewski, A.S.; Soeldner, J.S.; Kahn, C.R. Slow Glucose Removal Rate and Hyperinsulinemia Precede the Development of Type II Diabetes in the Offspring of Diabetic Parents. Ann. Intern. Med. 1990, 113, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The metabolic syndrome: Role of skeletal muscle metabolism. Ann. Med. 2006, 38, 389–402. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I. Ectopic Fat in Insulin Resistance, Dyslipidemia, and Cardiometabolic Disease. N. Eng. J. Med. 2014, 371, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Krook, A.; Björnholm, M.; Galuska, D.; Jiang, X.J.; Fahlman, R.; Myers, M.G.; Wallberg-Henriksson, H.; Zierath, J.R. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 2000, 49, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Bouzakri, K.; Koistinen, H.; Zierath, J.R. Molecular Mechanisms of Skeletal Muscle Insulin Resistance in Type 2 Diabetes. Curr. Diabetes Rev. 2005, 1, 167–174. [Google Scholar] [CrossRef]
- Bailey, J.L.; Zheng, B.; Hu, Z.; Price, S.R.; Mitch, W.E. Chronic Kidney Disease Causes Defects in Signaling through the Insulin Receptor Substrate/Phosphatidylinositol 3-Kinase/Akt Pathway: Implications for Muscle Atrophy. J. Am. Soc. Nephrol. 2006, 17, 1388–1394. [Google Scholar] [CrossRef]
- Deivanayagam, S.; Mohammed, B.S.; E Vitola, B.; Naguib, G.H.; Keshen, T.H.; Kirk, E.P.; Klein, S. Nonalcoholic fatty liver disease is associated with hepatic and skeletal muscle insulin resistance in overweight adolescents. Am. J. Clin. Nutr. 2008, 88, 257–262. [Google Scholar] [CrossRef]
- Kobayashi, S.; Maesato, K.; Moriya, H.; Ohtake, T.; Ikeda, T. Insulin resistance in patients with chronic kidney disease. Am. J. Kidney Dis. 2005, 45, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Nerpin, E.; Risérus, U.; Ingelsson, E.; Sundström, J.; Jobs, M.; Larsson, A.; Basu, S.; Ärnlöv, J. Insulin Sensitivity Measured With Euglycemic Clamp Is Independently Associated With Glomerular Filtration Rate in a Community-Based Cohort. Diabetes Care 2008, 31, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Perreault, M.; Marette, A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med. 2001, 7, 1138–1143. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Steensberg, A.; Schjerling, P. Muscle-derived interleukin-6: Possible biological effects. J. Physiol. 2001, 536, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Bruunsgaard, H. Physical activity and modulation of systemic low-level inflammation. J. Leukoc. Biol. 2005, 78, 819–835. [Google Scholar] [CrossRef]
- Anderson, E.J.; Vistoli, G.; Katunga, L.A.; Funai, K.; Regazzoni, L.; Monroe, T.B.; Gilardoni, E.; Cannizzaro, L.; Colzani, M.; De Maddis, D.; et al. A carnosine analog mitigates metabolic disorders of obesity by reducing carbonyl stress. J. Clin. Investig. 2018, 128, 5280–5293. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Urakawa, H.; Katsuki, A.; Sumida, Y.; Gabazza, E.C.; Murashima, S.; Morioka, K.; Maruyama, N.; Kitagawa, N.; Tanaka, T.; Hori, Y.; et al. Oxidative Stress Is Associated with Adiposity and Insulin Resistance in Men. J. Clin. Endocrinol. Metab. 2003, 88, 4673–4676. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Maddux, B.A.; See, W.; Lawrence, J.C., Jr.; Goldfine, A.L.; Goldfine, I.D.; Evans, J.L. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes 2001, 50, 404–410. [Google Scholar] [CrossRef]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Hemi, R.; Kanety, H.; Bashan, N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Khamaisi, M.; Bashan, N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia 1999, 42, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Franch, H.A.; Raissi, S.; Wang, X.; Zheng, B.; Bailey, J.L.; Price, S.R. Acidosis impairs insulin receptor substrate-1-associated phosphoinositide 3-kinase signaling in muscle cells: Consequences on proteolysis. Am. J. Physiol. Physiol. 2004, 287, F700–F706. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.-H.; Cho, J.M.; Kim, Y.-N.; Kim, S.S.; Kim, H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2012, 19, 83–92. [Google Scholar] [CrossRef]
- Chang, P.Y.; Jensen, J.; Printz, R.L.; Granner, D.K.; Ivy, J.L.; E Moller, D. Overexpression of hexokinase II in transgenic mice. Evidence that increased phosphorylation augments muscle glucose uptake. J. Biol. Chem. 1996, 271, 14834–14839. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Bertoldo, A.; Ng, J.M.; Azuma, K.; Pencek, R.R.; Kelley, C.; Price, J.C.; Cobelli, C.; Kelley, D.E. Interactions Among Glucose Delivery, Transport, and Phosphorylation That Underlie Skeletal Muscle Insulin Resistance in Obesity and Type 2 Diabetes: Studies With Dynamic PET Imaging. Diabetes 2013, 63, 1058–1068. [Google Scholar] [CrossRef]
- Vestergaard, H.; Bjørbaek, C.; Hansen, T.; Larsen, F.S.; Granner, D.K.; Pedersen, O. Impaired activity and gene expression of hexokinase II in muscle from non-insulin-dependent diabetes mellitus patients. J. Clin. Investig. 1995, 96, 2639–2645. [Google Scholar] [CrossRef]
- Ducluzeau, P.-H.; Perretti, N.; Laville, M.; Andreelli, F.; Vega, N.; Riou, J.-P.; Vidal, H. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue. Evidence for specific defects in type 2 diabetes. Diabetes 2001, 50, 1134–1142. [Google Scholar] [CrossRef]
- Cusi, K.J.; Pratipanawatr, T.; Koval, J.; Printz, R.; Ardehali, H.; Granner, D.K.; DeFronzo, R.A.; Mandarino, L.J. Exercise increases hexokinase II mRNA, but not activity in obesity and type 2 diabetes. Metabolism 2001, 50, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, S.; Pietilä, M.; Halmekytö, M.; Suppola, S.; Pirinen, E.; Deeb, S.S.; Jänne, J.; Laakso, M. Hexokinase II-deficient mice. Prenatal death of homozygotes without disturbances in glucose tolerance in heterozygotes. J. Biol. Chem. 1999, 274, 22517–22523. [Google Scholar] [CrossRef] [PubMed]
- Fueger, P.T.; Bracy, D.P.; Malabanan, C.M.; Pencek, R.R.; Granner, D.K.; Wasserman, D.H. Hexokinase II overexpression improves exercise-stimulated but not insulin-stimulated muscle glucose uptake in high-fat-fed C57BL/6J mice. Diabetes 2004, 53, 306–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dunaway, G.A.; Kasten, T.P.; Sebo, T.; Trapp, R. Analysis of the phosphofructokinase subunits and isoenzymes in human tissues. Biochem. J. 1988, 251, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.C.; Rudolphi, O.; Ek, B.; Exelbert, R.; Plotz, P.H.; Raben, N. Glycogenosis type VII (Tarui disease) in a Swedish family: Two novel mutations in muscle phosphofructokinase gene (PFK-M) resulting in intron retentions. Am. J. Hum. Genet. 1996, 59, 59–65. [Google Scholar]
- Ristow, M.; Vorgerd, M.; Möhlig, M.; Schatz, H.; Pfeiffer, A. Deficiency of phosphofructo-1-kinase/muscle subtype in humans impairs insulin secretion and causes insulin resistance. J. Clin. Investig. 1997, 100, 2833–2841. [Google Scholar] [CrossRef]
- Keildson, S.; Fadista, J.; Ladenvall, C.; Hedman, A.K.; Elgzyri, T.; Small, K.S.; Grundberg, E.; Nica, A.C.; Glass, D.; Richards, J.B.; et al. Expression of Phosphofructokinase in Skeletal Muscle Is Influenced by Genetic Variation and Associated With Insulin Sensitivity. Diabetes 2013, 63, 1154–1165. [Google Scholar] [CrossRef]
- Trivedi, B.; Danforth, W.H. Effect of pH on the kinetics of frog muscle phosphofructokinase. J. Biol. Chem. 1966, 241, 4110–4112. [Google Scholar]
- Scheuermann-Freestone, M.; Madsen, P.L.; Manners, D.; Blamire, A.M.; Buckingham, R.E.; Styles, P.; Radda, G.K.; Neubauer, S.; Clarke, K. Abnormal Cardiac and Skeletal Muscle Energy Metabolism in Patients With Type 2 Diabetes. Circulation 2003, 107, 3040–3046. [Google Scholar] [CrossRef]
- Bouché, C.; Serdy, S.; Kahn, C.R.; Goldfine, A.B. The Cellular Fate of Glucose and Its Relevance in Type 2 Diabetes. Endocr. Rev. 2004, 25, 807–830. [Google Scholar] [CrossRef]
- Wu, P.; Inskeep, K.; Bowker-Kinley, M.M.; Popov, K.M.; Harris, R.A. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 1999, 48, 1593–1599. [Google Scholar] [CrossRef]
- Rahimi, Y.; Camporez, J.P.G.; Petersen, M.C.; Pesta, D.; Perry, R.J.; Jurczak, M.J.; Cline, G.W.; Shulman, G.I. Genetic activation of pyruvate dehydrogenase alters oxidative substrate selection to induce skeletal muscle insulin resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 16508–16513. [Google Scholar] [CrossRef]
- Avogaro, A.; Toffolo, G.; Miola, M.; Valerio, A.; Tiengo, A.; Cobelli, C.; Del Prato, S. Intracellular lactate- and pyruvate-interconversion rates are increased in muscle tissue of non-insulin-dependent diabetic individuals. J. Clin. Investig. 1996, 98, 108–115. [Google Scholar] [CrossRef]
- Reaven, G.M.; Hollenbeck, C.; Jeng, C.Y.; Wu, M.S.; Chen, Y.D. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 1988, 37, 1020–1024. [Google Scholar] [CrossRef]
- Crawford, S.O.; Hoogeveen, R.C.; Brancati, F.L.; Astor, B.C.; Ballantyne, C.M.; Schmidt, M.I.; Young, J.H. Association of blood lactate with type 2 diabetes: The Atherosclerosis Risk in Communities Carotid MRI Study. Int. J. Epidemiol. 2010, 39, 1647–1655. [Google Scholar] [CrossRef]
- Juraschek, S.P.; Selvin, E.; Miller, E.R.; Brancati, F.L.; Young, J.H. Plasma lactate and diabetes risk in 8045 participants of the atherosclerosis risk in communities study. Ann. Epidemiol. 2013, 23, 791–796. [Google Scholar] [CrossRef]
- Juel, C. Lactate-proton cotransport in skeletal muscle. Physiol. Rev. 1997, 77, 321–358. [Google Scholar] [CrossRef]
- Juel, C. Muscle pH regulation: Role of training. Acta Physiol. Scand. 1998, 162, 359–366. [Google Scholar] [CrossRef]
- Choi, C.S.; Kim, Y.B.; Lee, F.N.; Zabolotny, J.M.; Kahn, B.B.; Youn, J.H. Lactate induces insulin resistance in skeletal muscle by suppressing glycolysis and impairing insulin signaling. Am. J. Physiol. Metab. 2002, 283, E233–E240. [Google Scholar] [CrossRef]
- Xu, H.; Jia, T.; Huang, X.; Riserus, U.; Cederholm, T.; Ärnlöv, J.; Sjögren, P.; Lindholm, B.; Carrero, J.J. Dietary acid load, insulin sensitivity and risk of type 2 diabetes in community-dwelling older men. Diabetologia 2014, 57, 1561–1568. [Google Scholar] [CrossRef]
- Jong, J.C.K.D.; Li, Y.; Chen, M.; Curhan, G.C.; Mattei, J.; Malik, V.S.; Forman, J.P.; Franco, O.H.; Hu, F.B. Diet-dependent acid load and type 2 diabetes: Pooled results from three prospective cohort studies. Diabetologia 2016, 60, 270–279. [Google Scholar] [CrossRef]
- Fagherazzi, G.; Vilier, A.; Bonnet, F.; Lajous, M.; Balkau, B.; Boutron-Ruault, M.C.; Clavel-Chapelon, F. Dietary acid load and risk of type 2 diabetes: The E3N-EPIC cohort study. Diabetologia 2013, 57, 313–320. [Google Scholar] [CrossRef]
- Maalouf, N.M.; Cameron, M.A.; Moe, O.W.; Adams-Huet, B.; Sakhaee, K. Low Urine pH: A Novel Feature of the Metabolic Syndrome. Clin. J. Am. Soc. Nephrol. 2007, 2, 883–888. [Google Scholar] [CrossRef]
- Maalouf, N.M.; Cameron, M.A.; Moe, O.W.; Sakhaee, K. Metabolic basis for low urine pH in type 2 diabetes. Clin. J. Am. Soc. Nephrol. 2010, 5, 1277–1281. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Beckles, A.D. Glucose intolerance following chronic metabolic acidosis in man. Am. J. Physiol. Metab. 1979, 236, E328–E334. [Google Scholar] [CrossRef]
- Yamada, T.; Zhang, S.J.; Westerblad, H.; Katz, A. β-Hydroxybutyrate inhibits insulin-mediated glucose transport in mouse oxidative muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E364–E373. [Google Scholar] [CrossRef]
- Souto, G.; Donapetry, C.; Calviño, J.; Adeva, M.M. Metabolic Acidosis-Induced Insulin Resistance and Cardiovascular Risk. Metab. Syndr. Relat. Disord. 2011, 9, 247–253. [Google Scholar] [CrossRef]
- Farwell, W.R.; Taylor, E.N. Serum bicarbonate, anion gap and insulin resistance in the National Health and Nutrition Examination Survey. Diabet. Med. J. Br. Diabet. Assoc. 2008, 25, 798–804. [Google Scholar] [CrossRef]
- May, R.C.; Kelly, R.A.; Mitch, W.E. Mechanisms for defects in muscle protein metabolism in rats with chronic uremia. Influence of metabolic acidosis. J. Clin. Investig. 1987, 79, 1099–1103. [Google Scholar] [CrossRef]
- Garibotto, G.; Sofia, A.; Russo, R.; Paoletti, E.; Bonanni, A.; Parodi, E.L.; Viazzi, F.; Verzola, D. Insulin sensitivity of muscle protein metabolism is altered in patients with chronic kidney disease and metabolic acidosis. Kidney Int. 2015, 88, 1419–1426. [Google Scholar] [CrossRef]
- Cuthbert, C.; Alberti, K. Acidemia and Insulin Resistance in the Diabetic Ketoacidotic Rat. Metabolism 1978, 27, 1903–1916. [Google Scholar] [CrossRef]
- Mak, R.H. Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int. 1998, 54, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Hayata, H.; Miyazaki, H.; Niisato, N.; Yokoyama, N.; Marunaka, Y. Lowered extracellular pH is involved in the pathogenesis of skeletal muscle insulin resistance. Biochem. Biophys. Res. Commun. 2014, 445, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Berezhnov, A.V.; Soutar, M.P.M.; Fedotova, E.I.; Frolova, M.S.; Plun-Favreau, H.; Zinchenko, V.P.; Abramov, A.Y. Intracellular pH Modulates Autophagy and Mitophagy. J. Biol. Chem. 2016, 291, 8701–8708. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Møller, A.B.; Kampmann, U.; Hedegaard, J.; Thorsen, K.; Nordentoft, I.K.; Vendelbo, M.H.; Møller, N.; Jessen, N. Altered gene expression and repressed markers of autophagy in skeletal muscle of insulin resistant patients with type 2 diabetes. Sci. Rep. 2017, 7, 43775. [Google Scholar] [CrossRef]
- Yang, L.; Lin, H.; Lin, W.; Xu, X. Exercise Ameliorates Insulin Resistance of Type 2 Diabetes through Motivating Short-Chain Fatty Acid-Mediated Skeletal Muscle Cell Autophagy. Biol. Basel 2020, 9, 203. [Google Scholar] [CrossRef]
- Cappellari, G.G.; Zanetti, M.; Semolic, A.; Vinci, P.; Ruozi, G.; Falcione, A.; Filigheddu, N.; Guarnieri, G.; Graziani, A.; Giacca, M.; et al. Unacylated Ghrelin Reduces Skeletal Muscle Reactive Oxygen Species Generation and Inflammation and Prevents High-Fat Diet Induced Hyperglycemia and Whole-Body Insulin Resistance in Rodents. Diabetes 2016, 65, 874–886. [Google Scholar] [CrossRef]
- Liu, Y.; Palanivel, R.; Rai, E.; Park, M.; Gabor, T.V.; Scheid, M.P.; Xu, A.; Sweeney, G. Adiponectin Stimulates Autophagy and Reduces Oxidative Stress to Enhance Insulin Sensitivity During High-Fat Diet Feeding in Mice. Diabetes 2014, 64, 36–48. [Google Scholar] [CrossRef]
- Juel, C. Regulation of pH in human skeletal muscle: Adaptations to physical activity. Acta Physiol. Oxf. 2008, 193, 17–24. [Google Scholar] [CrossRef]
- Chatel, B.; Bendahan, D.; Hourdé, C.; Pellerin, L.; Lengacher, S.; Magistretti, P.J.; Le Fur, Y.; Vilmen, C.; Bernard, M.; Messonnier, L.A. Role of MCT1 and CAII in skeletal muscle pH homeostasis, energetics, and function: In vivo insights from MCT1 haploinsufficient mice. FASEB J. 2017, 31, 2562–2575. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family?from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflügers Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, N.U.; Roughton, F.J.W. Carbonic anhydrase. Its preparation and properties. J. Physiol. 1933, 80, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Monazzami, A.; Rajabi, H.; Ghrakhanlou, R.; Yari, K.; Rahimi, Z. Modulation of oxidative and glycolytic skeletal muscle fibers Na+/H+ exchanger1 (NHE1) and Na+/HCO3- co-transporter1 (NBC1) genes and proteins expression in type 2 diabetic rat (Streptozotocin + high fat diet) following long term endurance training. Cell Mol. Biol. Noisy Grand 2017, 63, 11–18. [Google Scholar] [CrossRef]
- Kristensen, J.M.; Kristensen, M.; Juel, C. Expression of Na+/HCO3- co-transporter proteins (NBCs) in rat and human skeletal muscle. Acta Physiol. Scand. 2004, 182, 69–76. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Pette, D.; Staron, R.S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 2000, 50, 500–509. [Google Scholar] [CrossRef]
- Bonen, A.; Mccullagh, K.J.A.; Putman, C.T.; Hultman, E.; Jones, N.L.; Heigenhauser, G.J.F. Short-term training increases human muscle MCT1 and femoral venous lactate in relation to muscle lactate. Am. J. Physiol. Content 1998, 274, E102–E107. [Google Scholar] [CrossRef]
- Pilegaard, H.; Mohr, T.; Kjaer, M.; Juel, C. Lactate/H+ transport in skeletal muscle from spinal-cord-injured patients. Scand. J. Med. Sci. Sports 1998, 8, 98–101. [Google Scholar] [CrossRef]
- Dubouchaud, H.; Granier, P.; Mercier, J.; Le Peuch, C.; Préfaut, C. Lactate uptake by skeletal muscle sarcolemmal vesicles decreases after 4 wk of hindlimb unweighting in rats. J. Appl. Physiol. 1996, 80, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Dubouchaud, H.; Butterfield, G.E.; Wolfel, E.E.; Bergman, B.C.; Brooks, G.A. Endurance training, expression, and physiology of LDH, MCT1, and MCT4 in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E571–E579. [Google Scholar] [CrossRef]
- Bonen, A. Lactate transporters (MCT proteins) in heart and skeletal muscles. Med. Sci. Sports Exerc. 2000, 32, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Juel, C.; Halestrap, A.P. Lactate transport in skeletal muscle - role and regulation of the monocarboxylate transporter. J. Physiol. 1999, 517, 633–642. [Google Scholar] [CrossRef]
- Pilegaard, H.; Domino, K.; Noland, T.; Juel, C.; Hellsten, Y.; Halestrap, A.P.; Bangsbo, J. Effect of high-intensity exercise training on lactate/H+ transport capacity in human skeletal muscle. Am. J. Physiol. Content 1999, 276, E255–E261. [Google Scholar] [CrossRef] [PubMed]
- Pilegaard, H.; Terzis, G.; Halestrap, A.; Juel, C. Distribution of the lactate/H+ transporter isoforms MCT1 and MCT4 in human skeletal muscle. Am. J. Physiol. Content 1999, 276, E843–E848. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Jackson, V.N.; Heddle, C.; Price, N.T.; Pilegaard, H.; Juel, C.; Bonen, A.; Montgomery, I.; Hutter, O.F.; Halestrap, A.P. Lactic Acid Efflux from White Skeletal Muscle Is Catalyzed by the Monocarboxylate Transporter Isoform MCT3. J. Biol. Chem. 1998, 273, 15920–15926. [Google Scholar] [CrossRef] [PubMed]
- Juel, C.; Holten, M.K.; Dela, F. Effects of strength training on muscle lactate release and MCT1 and MCT4 content in healthy and type 2 diabetic humans. J. Physiol. 2004, 556, 297–304. [Google Scholar] [CrossRef]
- Lengacher, S.; Nehiri-Sitayeb, T.; Steiner, N.; Carneiro, L.; Favrod, C.; Preitner, F.; Thorens, B.; Stehle, J.-C.; Dix, L.; Pralong, F.; et al. Resistance to Diet-Induced Obesity and Associated Metabolic Perturbations in Haploinsufficient Monocarboxylate Transporter 1 Mice. PLoS ONE 2013, 8, e82505. [Google Scholar] [CrossRef]
- Wetzel, P.; Gros, G. Carbonic Anhydrases in Striated Muscle. In Experientia Supplementum; The Carbonic Anhydrases: New Horizons; Chegwidden, W.R., Carter, N.D., Edwards, Y.H., Eds.; Birkhäuser: Basel, Switzerland; pp. 375–399.
- Mullen, E.; Ohlendieck, K. Proteomic profiling of non-obese type 2 diabetic skeletal muscle. Int. J. Mol. Med. 2010, 25, 445–458. [Google Scholar] [CrossRef]
- Liu, M.; Walter, G.A.; Pathare, N.C.; Forster, R.E.; Vandenborne, K. A quantitative study of bioenergetics in skeletal muscle lacking carbonic anhydrase III using 31P magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA 2006, 104, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Christ, C.Y.; Hunt, D.; Hancock, J.; García-Macedo, R.; Mandarino, L.J.; Ivy, J.L. Exercise training improves muscle insulin resistance but not insulin receptor signaling in obese Zucker rats. J. Appl. Physiol. 1985 2002, 92, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; Reichart, D.; Olefsky, J. Exercise and thiazolidinedione therapy normalize insulin action in the obese Zucker fatty rat. Diabetes 2000, 49, 2154–2159. [Google Scholar] [CrossRef][Green Version]
- Becker-Zimmermann, K.; Berger, M.; Berchtold, P.; Gries, F.A.; Herberg, L.; Schwenen, M. Treadmill training improves intravenous glucose tolerance and insulin sensitivity in fatty zucker rats. Diabetologia 1982, 22, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Metz, L.; Vermaelen, M.; Lambert-Cordillac, K.; Broca, C.; Sirvent, P.; Raynaud, E.; Mercier, J. Endurance training increases lactate transport in male Zucker fa/fa rats. Biochem. Biophys. Res. Commun. 2005, 331, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Nikooie, R.; Rajabi, H.; Gharakhanlu, R.; Atabi, F.; Omidfar, K.; Aveseh, M.; Larijani, B. Exercise-induced changes of MCT1 in cardiac and skeletal muscles of diabetic rats induced by high-fat diet and STZ. J. Physiol. Biochem. 2013, 69, 865–877. [Google Scholar] [CrossRef]
- Abe, H. Role of histidine-related compounds as intracellular proton buffering constituents in vertebrate muscle. Biochem. Moscow 2000, 65, 757–765. [Google Scholar]
- Culbertson, J.Y.; Kreider, R.B.; Greenwood, M.; Cooke, M.B. Effects of Beta-Alanine on Muscle Carnosine and Exercise Performance:A Review of the Current Literature. Nutrients 2010, 2, 75–98. [Google Scholar] [CrossRef]
- Tanokura, M.; Tasumi, M.; Miyazawa, T. 1H Nuclear magnetic resonance studies of histidine-containing di- and tripeptides. Estimation of the effects of charged groups on the pKa value of the imidazole ring. Biopolymers 1976, 15, 393–401. [Google Scholar] [CrossRef]
- Davey, C. The significance of carnosine and anserine in striated skeletal muscle. Arch. Biochem. Biophys. 1960, 89, 303–308. [Google Scholar] [CrossRef]
- Mannion, A.F.; Jakeman, P.M.; Dunnett, M.; Harris, R.C.; Willan, P.L.T. Carnosine and anserine concentrations in the quadriceps femoris muscle of healthy humans. Eur. J. Appl Physiol. Occup. Physiol. 1992, 64, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and Pathophysiology of Carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef] [PubMed]
- Hoetker, D.; Chung, W.; Zhang, D.; Zhao, J.; Schmidtke, V.K.; Riggs, D.W.; Derave, W.; Bhatnagar, A.; Bishop, D.J.; Baba, S.P. Exercise alters and beta-alanine combined with exercise augments histidyl dipeptide levels and scavenges lipid peroxidation products in human skeletal muscle. J. Appl. Physiol. 1985 2018, 1767–1778, 125. [Google Scholar] [CrossRef]
- Drozak, J.; Veiga-Da-Cunha, M.; Vertommen, D.; Stroobant, V.; Van Schaftingen, E. Molecular Identification of Carnosine Synthase as ATP-grasp Domain-containing Protein 1 (ATPGD1). J. Biol. Chem. 2010, 285, 9346–9356. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.W.; O’Fallon, J.V. The subcellular distribution of carnosine, carnosine synthetase, and carnosinase in mouse olfactory tissues. Brain Res. 1979, 173, 99–108. [Google Scholar] [CrossRef]
- Ng, R.H.; Marshall, F.D.; Henn, F.A.; Sellström, Å. Metabolism of carnosine and homocarnosine in subcellular fractions and neuronal and glial cell-enriched fractions of rabbit brain. J. Neurochem. 1977, 28, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, A.; Eggleton, P. The titration constants of anserine, carnosine and some related compounds. Biochem. J. 1938, 32, 209–211. [Google Scholar] [CrossRef]
- Aldini, G.; Orioli, M.; Rossoni, G.; Savi, F.; Braidotti, P.; Vistoli, G.; Yeum, K.-J.; Negrisoli, G.; Carini, M. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J. Cell. Mol. Med. 2010, 15, 1339–1354. [Google Scholar] [CrossRef]
- Baba, S.P.; Hoetker, J.D.; Merchant, M.; Klein, J.B.; Cai, J.; Barski, O.A.; Conklin, D.J.; Bhatnagar, A. Role of Aldose Reductase in the Metabolism and Detoxification of Carnosine-Acrolein Conjugates. J. Biol. Chem. 2013, 288, 28163–28179. [Google Scholar] [CrossRef]
- Ihara, H.; Kakihana, Y.; Yamakage, A.; Kai, K.; Shibata, T.; Nishida, M.; Yamada, K.-I.; Uchida, K.; Yamda, K.-I. 2-Oxo-histidine–containing dipeptides are functional oxidation products. J. Biol. Chem. 2018, 294, 1279–1289. [Google Scholar] [CrossRef]
- Baran, E.J. Metal complexes of carnosine. Biochem. Mosc. 2000, 65, 789–797. [Google Scholar]
- Gualano, B.; Everaert, I.; Stegen, S.; Artioli, G.G.; Taes, Y.; Roschel, H.; Achten, E.; Otaduy, M.C.; Júnior, A.H.L.; Harris, R.; et al. Reduced muscle carnosine content in type 2, but not in type 1 diabetic patients. Amino Acids 2011, 43, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Mong, M.C.; Chao, C.Y.; Yin, M.C. Histidine and carnosine alleviated hepatic steatosis in mice consumed high saturated fat diet. Eur. J. Pharmacol. 2011, 653, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Sauerhöfer, S.; Yuan, G.; Braun, G.S.; Deinzer, M.; Neumaier, M.; Gretz, N.; Floege, J.; Kriz, W.; Van Der Woude, F.; Moeller, M.J. L-Carnosine, a Substrate of Carnosinase-1, Influences Glucose Metabolism. Diabetes 2007, 56, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- De Courten, B.; Jakubova, M.; De Courten, M.P.; Kukurova, I.J.; Vallova, S.; Krumpolec, P.; Valkovic, L.; Kurdiova, T.; Garzon, D.; Barbaresi, S.; et al. Effects of carnosine supplementation on glucose metabolism: Pilot clinical trial. Obesity Silver Spring 2016, 24, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Regazzoni, L.; De Courten, B.; Garzon, D.; Altomare, A.; Marinello, C.; Jakubova, M.; Vallova, S.; Krumpolec, P.; Carini, M.; Ukropec, J.; et al. A carnosine intervention study in overweight human volunteers: Bioavailability and reactive carbonyl species sequestering effect. Sci. Rep. 2016, 6, 27224. [Google Scholar] [CrossRef]
- Teufel, M.; Saudek, V.; Ledig, J.-P.; Bernhardt, A.; Boularand, S.; Carreau, A.; Cairns, N.J.; Carter, C.J.; Cowley, D.J.; Duverger, D.; et al. Sequence Identification and Characterization of Human Carnosinase and a Closely Related Non-specific Dipeptidase. J. Biol. Chem. 2002, 278, 6521–6531. [Google Scholar] [CrossRef]
- Orioli, M.; Vistoli, G.; Regazzoni, L.; Pedretti, A.; Lapolla, A.; Rossoni, G.; Canevotti, R.; Gamberoni, L.; Previtali, M.; Carini, M.; et al. Design, synthesis, ADME properties, and pharmacological activities of beta-alanyl-D-histidine (D-carnosine) prodrugs with improved bioavailability. Chem. Med. Chem. 2011, 6, 1269–1282. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Ricci, C.; Scipioni, A.; Fantauzzi, C.B.; Giaccari, A.; Salomone, E.; Canevotti, R.; Lapolla, A.; Orioli, M.; et al. D-carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a Western diet through reduction of carbonyl stress and inflammation. Br. J. Pharmacol. 2012, 166, 1344–1356. [Google Scholar] [CrossRef]
- Everaert, I.; De Naeyer, H.; Taes, Y.; Derave, W. Gene expression of carnosine-related enzymes and transporters in skeletal muscle. Eur. J. Appl. Physiol. 2013, 113, 1169–1179. [Google Scholar] [CrossRef]
- Ingram, K.H.; Hill, H.; Moellering, D.; Hill, B.G.; Lara-Castro, C.; Newcomer, B.; Brandon, L.J.; Ingalls, C.P.; Penumetcha, M.; Rupp, J.C.; et al. Skeletal muscle lipid peroxidation and insulin resistance in humans. J. Clin. Endocrinol. Metab. 2012, 97, E1182–E1186. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posa, D.K.; Baba, S.P. Intracellular pH Regulation of Skeletal Muscle in the Milieu of Insulin Signaling. Nutrients 2020, 12, 2910. https://doi.org/10.3390/nu12102910
Posa DK, Baba SP. Intracellular pH Regulation of Skeletal Muscle in the Milieu of Insulin Signaling. Nutrients. 2020; 12(10):2910. https://doi.org/10.3390/nu12102910
Chicago/Turabian StylePosa, Dheeraj Kumar, and Shahid P. Baba. 2020. "Intracellular pH Regulation of Skeletal Muscle in the Milieu of Insulin Signaling" Nutrients 12, no. 10: 2910. https://doi.org/10.3390/nu12102910
APA StylePosa, D. K., & Baba, S. P. (2020). Intracellular pH Regulation of Skeletal Muscle in the Milieu of Insulin Signaling. Nutrients, 12(10), 2910. https://doi.org/10.3390/nu12102910