Melon GliSODin® Prevents Diet-Induced NASH Onset by Reducing Fat Synthesis and Improving Liver Function


 and
and
Abstract
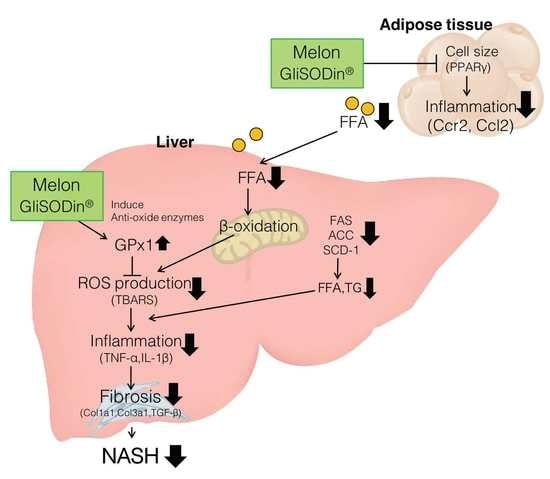
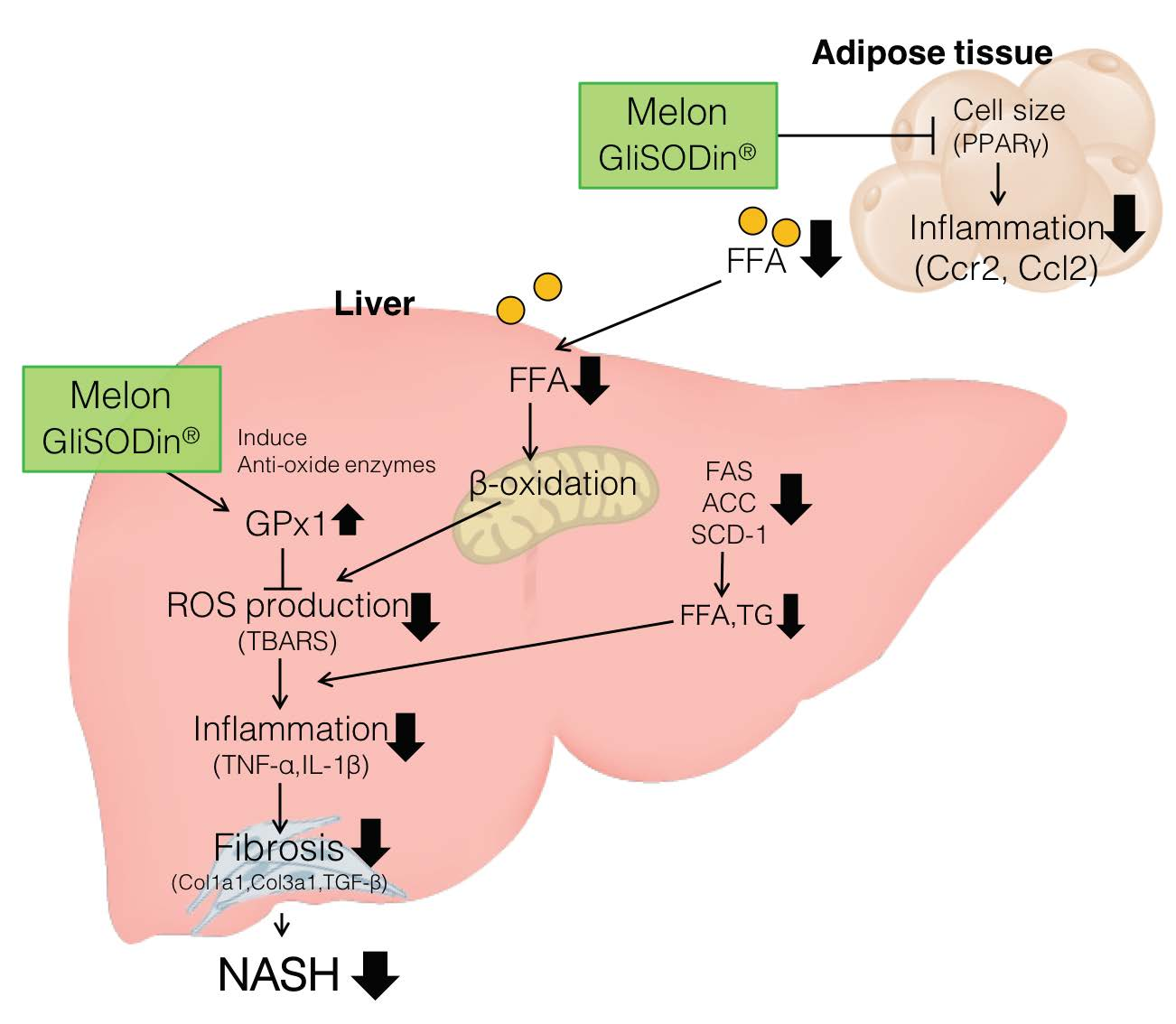
1. Introduction
2. Materials and Methods
2.1. Melon Extract
2.2. Histology and Staining Analysis
2.3. Lipid Parameters: Blood Chemistry and Liver Tissue Analysis
2.4. mRNA Expression Analysis via Quantitative RT-PCR
2.5. 2-Thiobarbituric Acid Reactive Substances (TBARS) Measurement
2.6. Statistical Analysis
3. Results
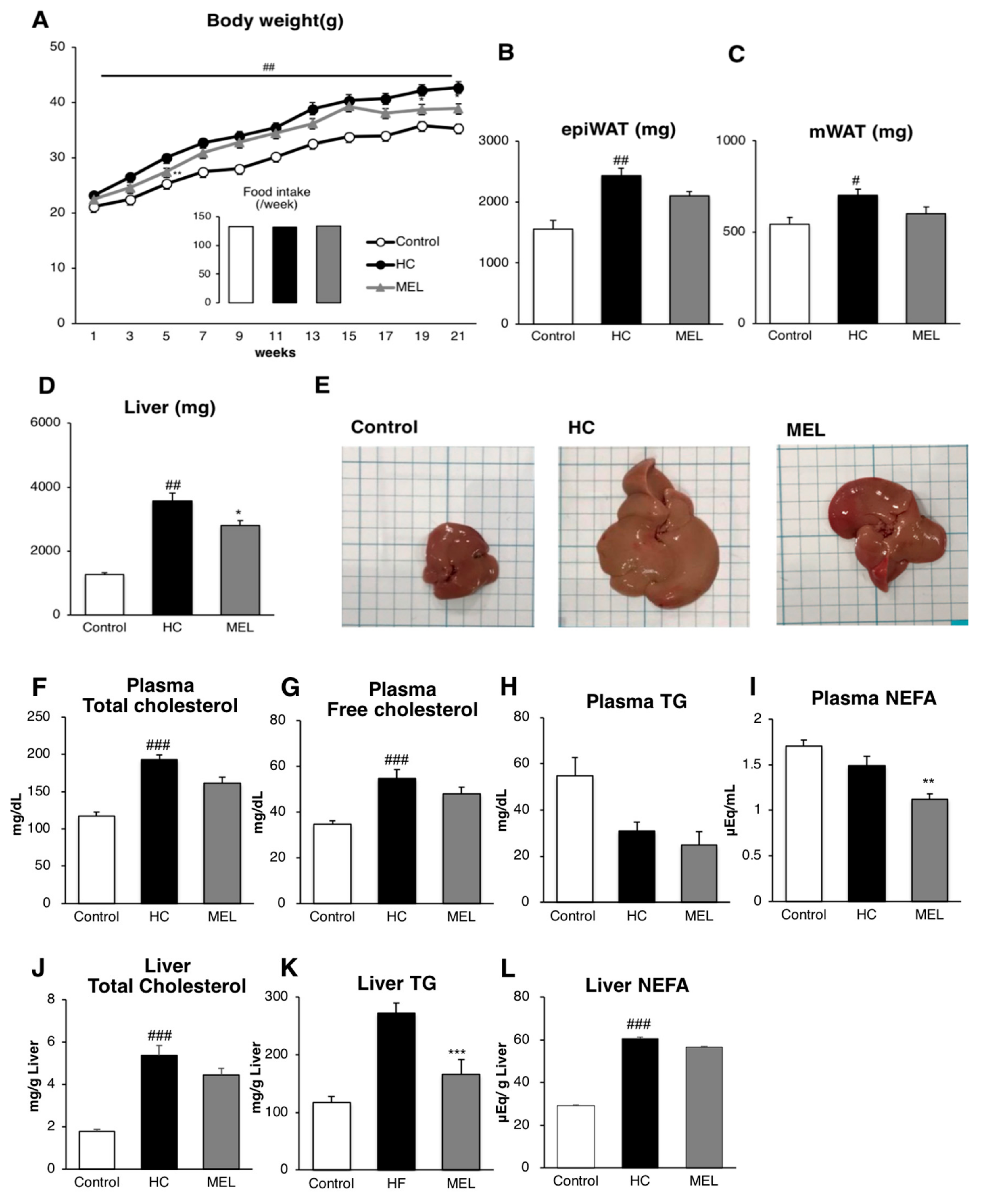
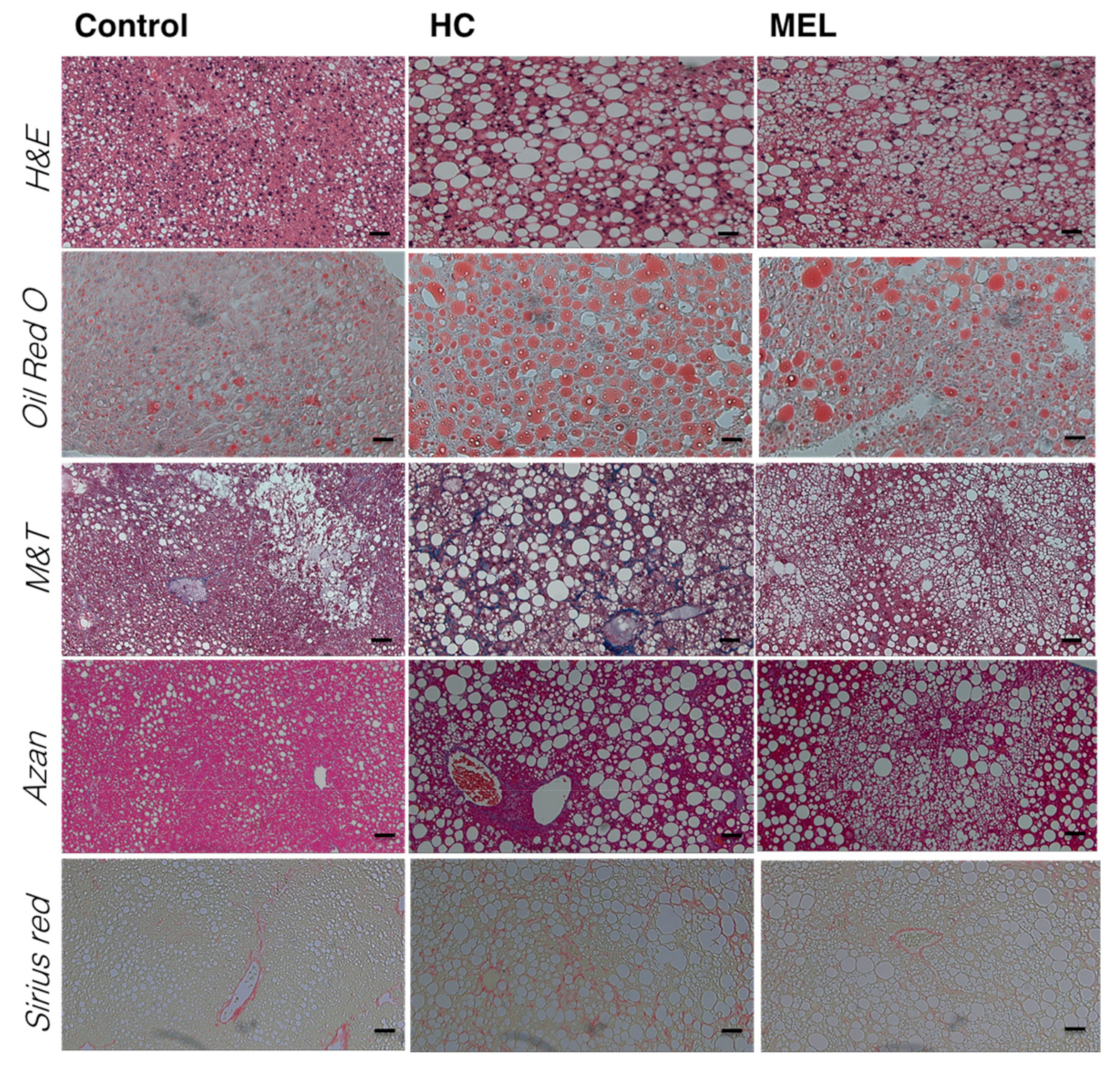
3.1. Melon GliSODin® Prevents Lipid Accumulation in the Mouse Model of Diet-Induced NASH
3.2. Melon GliSODin® Prevents Liver Fibrosis in A Mouse Model Of Hc-Induced NASH
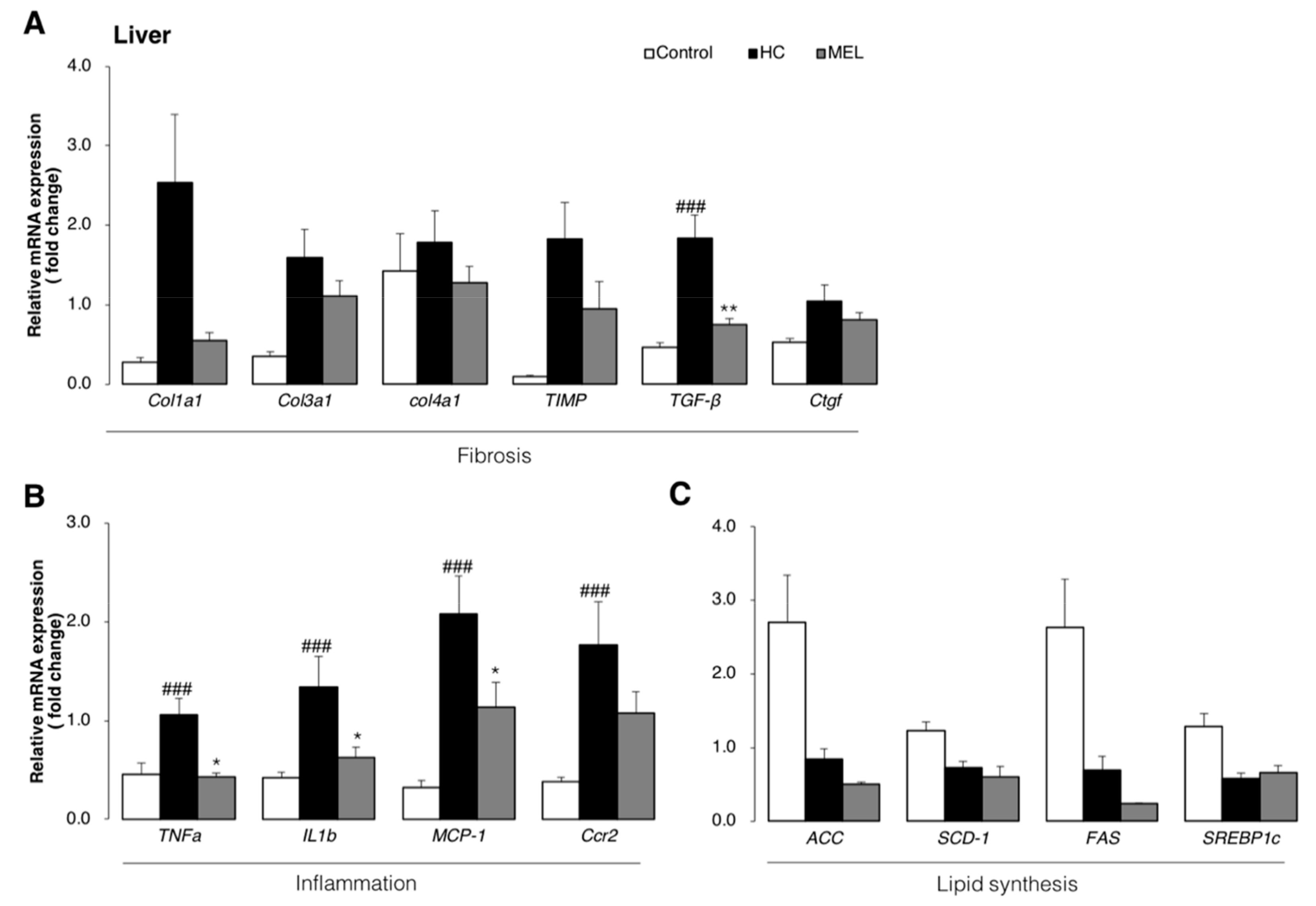
3.3. Melon GliSODin® Attenuated Liver Inflammation and Fibrosis at the Molecular Level
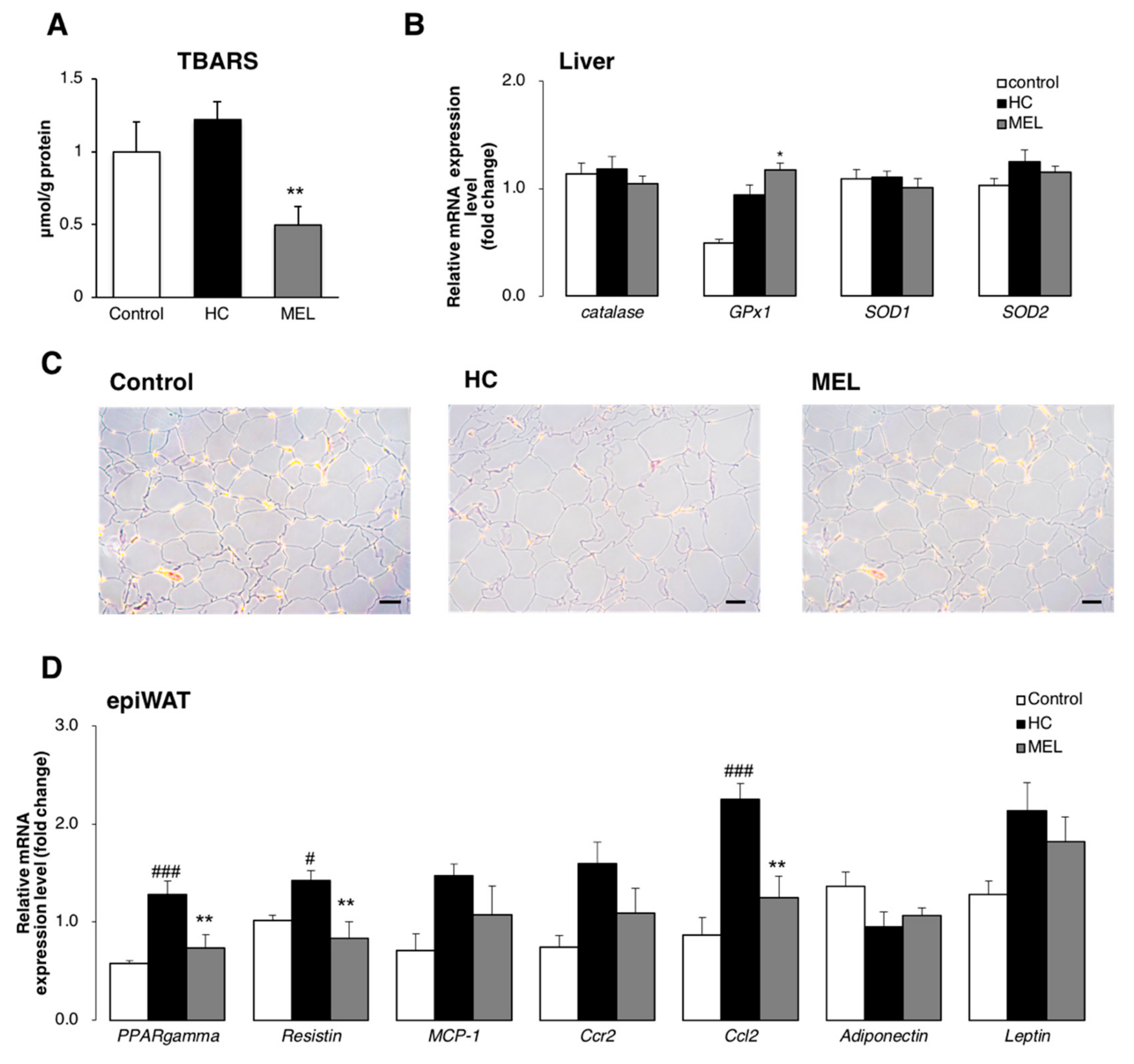
3.4. Melon GliSODin® Boosts Antioxidation Defence Systems in the Hc Diet Loaded Liver.
3.5. Melon GliSODin® Reduces the Size of Adipocytes and Suppresses the Flux of Free Fatty Acids (FFA) in the Liver
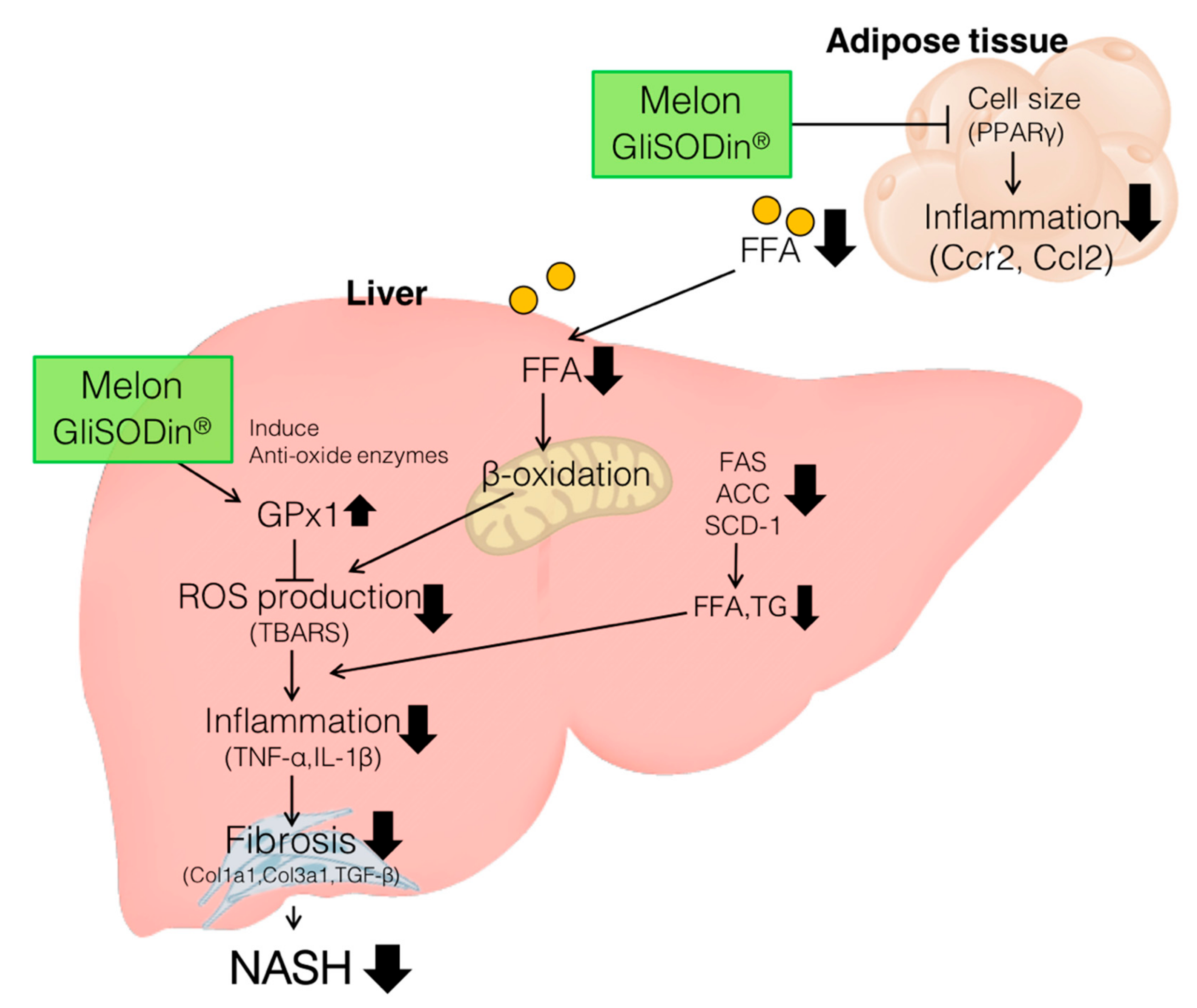
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Forni, C.; Facchiano, F.; Bartoli, M.; Pieretti, S.; Facchiano, A.; D’Arcangelo, D.; Norelli, S.; Valle, G.; Nisini, R.; Beninati, S.; et al. Beneficial Role of Phytochemicals on Oxidative Stress and Age-Related Diseases. Biomed. Res. Int. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar] [PubMed]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin Resistance in Nonalcoholic Fatty Liver Disease. Curr. Pharm. Des. 2010, 16, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Joanny Menvielle-Bourg, F. Superoxide Dismutase (SOD), a Powerful Antioxidant, is now available Orally. Phytothérapie 2005, 3, 1–4. [Google Scholar]
- Ore, A.; Akinloye, O.A. Oxidative Stress and Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina (Kaunas). 2019, 55, 26. [Google Scholar] [CrossRef]
- Vouldoukis, I.; Conti, M.; Krauss, P.; Kamaté, C.; Blazquez, S.; Tefit, M.; Mazier, D.; Calenda, A.; Dugas, B. Supplementation with gliadin-combined plant superoxide dismutase extract promotes antioxidant defences and protects against oxidative stress. Phyther. Res. 2004, 18, 957–962. [Google Scholar] [CrossRef]
- Vouldoukis, I.; Lacan, D.; Kamate, C.; Coste, P.; Calenda, A.; Mazier, D.; Conti, M.; Dugas, B. Antioxidant and anti-inflammatory properties of a Cucumis melo LC. extract rich in superoxide dismutase activity. J. Ethnopharmacol. 2004, 94, 67–75. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta and fibrosis. World J. Gastroenterol. 2007, 13, 3056–3062. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Karimi Galougahi, K.; Liu, C.-C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Kaser, S. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 2005, 54, 117–121. [Google Scholar] [CrossRef]
- Matsunami, T.; Sato, Y.; Ariga, S.; Sato, T.; Shimomura, T.; Kashimura, H.; Hasegawa, Y.; Yukawa, M. Regulation of synthesis and oxidation of fatty acids by adiponectin receptors (AdipoR1/R2) and insulin receptor substrate isoforms (IRS-1/-2) of the liver in a nonalcoholic steatohepatitis animal model. Metabolism. 2011, 60, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal β-oxidation—A metabolic pathway with multiple functions. Biochim. Biophys. Acta - Mol. Cell Res. 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Wojtczak, L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic. Biol. Med. 2008, 45, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Invest. 2005, 115, 1111–1119. [Google Scholar] [CrossRef]
- Berg, A.H.; Scherer, P.E. Adipose Tissue, Inflammation, and Cardiovascular Disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate Induces Insulin Resistance in H4IIEC3 Hepatocytes through Reactive Oxygen Species Produced by Mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| 18S | TTCTGGCCAACGGTCTAGACAAC | CCAGTGGTCTTGGTGTGCTGA |
| Acc | ACCCACTCCACTGTTTGTGA | CCTTGGAATTCAGGAGAGGA |
| Catalase | CCAGCGACCAGATGAAGCAG | CCACTCTCTCAGGAATCCGC |
| Ccl2 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| Ccr2 | AGCACATGTGGTGAATCCAA | TGCCATCATAAAGGAGCCA |
| Ccr2 | AGCACATGTGGTGAATCCAA | TGCCATCATAAAGGAGCCA |
| Col1a1 | CCTCAGGGTATTGCTGGACAAC | TTGATCCAGAAGGACCTTGTTTG |
| Col3a1 | TTGATGTGCAGCTGGCATTC | GCCACTGGCCTGATCCATAT |
| Col4a1 | CACATTTTCCACAGCCAGAG | GTCTGGCTTCTGCTGCTCTT |
| Ctgf | ACCCGAGTTACCAATGACAATACC | CCGCAGAACTTAGCCCTGTATG |
| FAS | TCTGCCAGTGAGTTGAGGAC | CTGCAGAGAAGCGAGCATAC |
| GPx1 | AGTCCACCGTGTATGCCTTCT | GAGACGCGACATTCTCAATGA |
| IL1b | CTGAACTCAACTGTGAAATGCCA | AAAGGTTTGGAAGCAGCCCT |
| MCP-1 | CCACTCACCTGCTGCTACTCAT | TGGTGATCCTCTTGTAGCTCTCC |
| PPARgamma | TGGCCACCTCTTTGCTCTGCTC | AGGCCGAGAAGGAGAAGCTGTTG |
| Resistin | CCCTCCTTTTCCTTTTCTTCCTTG | AGACTGCTGTGCCTTCTGGG |
| Scd1 | CTCCTGCTGATGTGCTTCAT | AAGGTGCTAACGAACAGGCT |
| SOD1 | TGAGGTCCTGCACTGGTAC | CAAGCGGTGAACCAGTTGTG |
| SOD2 | TTAACGCGCAGATCATGCA | GGTGGCGTTGAGATTGTTCA |
| Srebp1c | CGTGAGCTACCTGGACTGAA | CGGGACAGCTTAGCCTCTAC |
| TGF-β | TTGCTTCAGCTCCACAGAGA | TGGTTGTAGAGGGCAAGGAC |
| TIMP | AGGTGGTCTCGTTGATTTCT | GTAAGGCCTGTAGCTGTGCC |
| TNFa | CTGGGACAGTGACCTGGACT | GCACCTCAGGGAAGAGTCTG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, A.; Kitamura, N.; Yokoyama, Y.; Uchida, S.; Kumadaki, K.; Tsubota, K.; Watanabe, M. Melon GliSODin® Prevents Diet-Induced NASH Onset by Reducing Fat Synthesis and Improving Liver Function. Nutrients 2019, 11, 1779. https://doi.org/10.3390/nu11081779
Nakamura A, Kitamura N, Yokoyama Y, Uchida S, Kumadaki K, Tsubota K, Watanabe M. Melon GliSODin® Prevents Diet-Induced NASH Onset by Reducing Fat Synthesis and Improving Liver Function. Nutrients. 2019; 11(8):1779. https://doi.org/10.3390/nu11081779
Chicago/Turabian StyleNakamura, Anna, Naho Kitamura, Yoko Yokoyama, Sena Uchida, Kayo Kumadaki, Kazuo Tsubota, and Mitsuhiro Watanabe. 2019. "Melon GliSODin® Prevents Diet-Induced NASH Onset by Reducing Fat Synthesis and Improving Liver Function" Nutrients 11, no. 8: 1779. https://doi.org/10.3390/nu11081779
APA StyleNakamura, A., Kitamura, N., Yokoyama, Y., Uchida, S., Kumadaki, K., Tsubota, K., & Watanabe, M. (2019). Melon GliSODin® Prevents Diet-Induced NASH Onset by Reducing Fat Synthesis and Improving Liver Function. Nutrients, 11(8), 1779. https://doi.org/10.3390/nu11081779