Interscapular and Perivascular Brown Adipose Tissue Respond Differently to a Short-Term High-Fat Diet

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
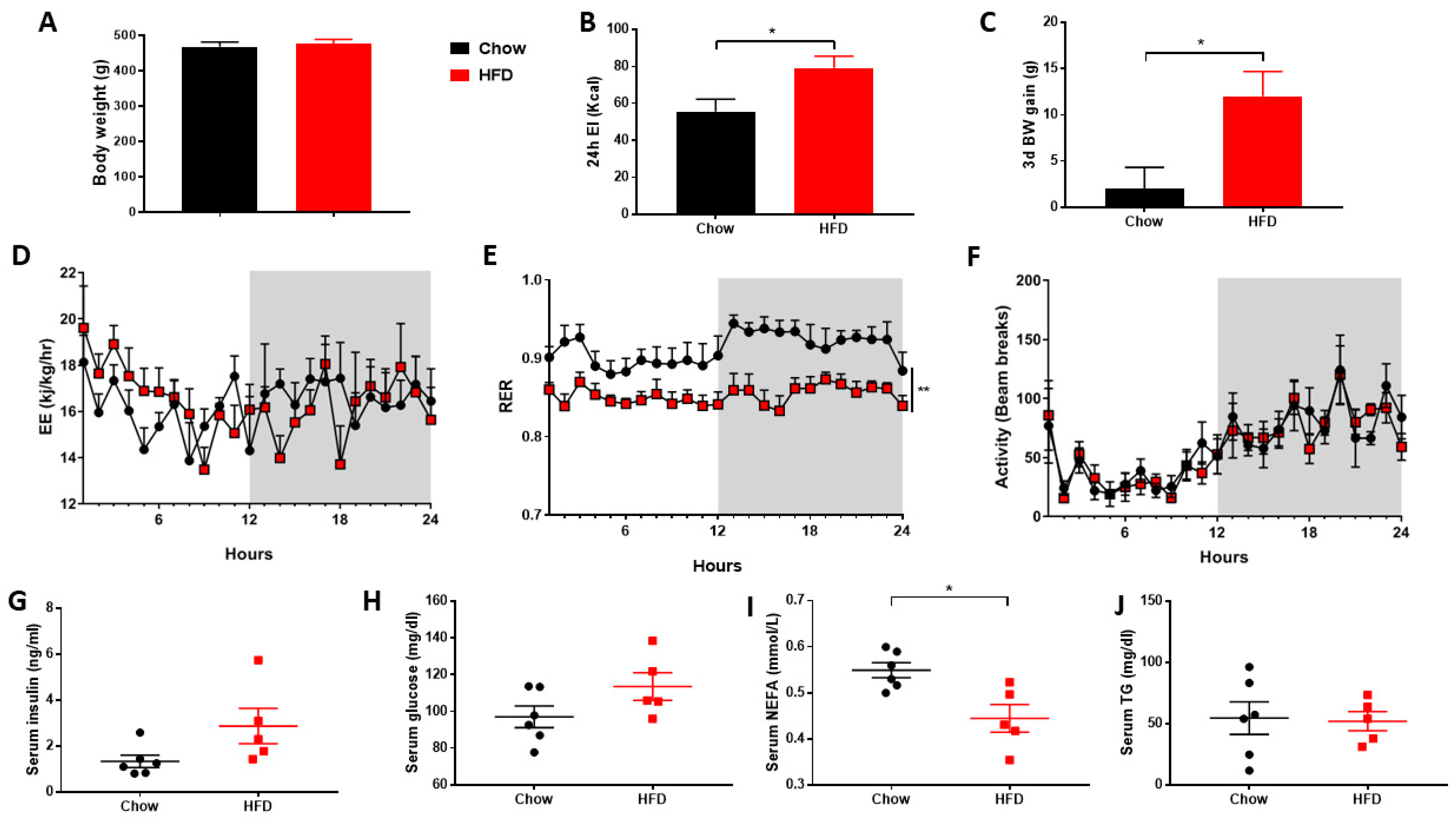
2.1. Metabolic Cages
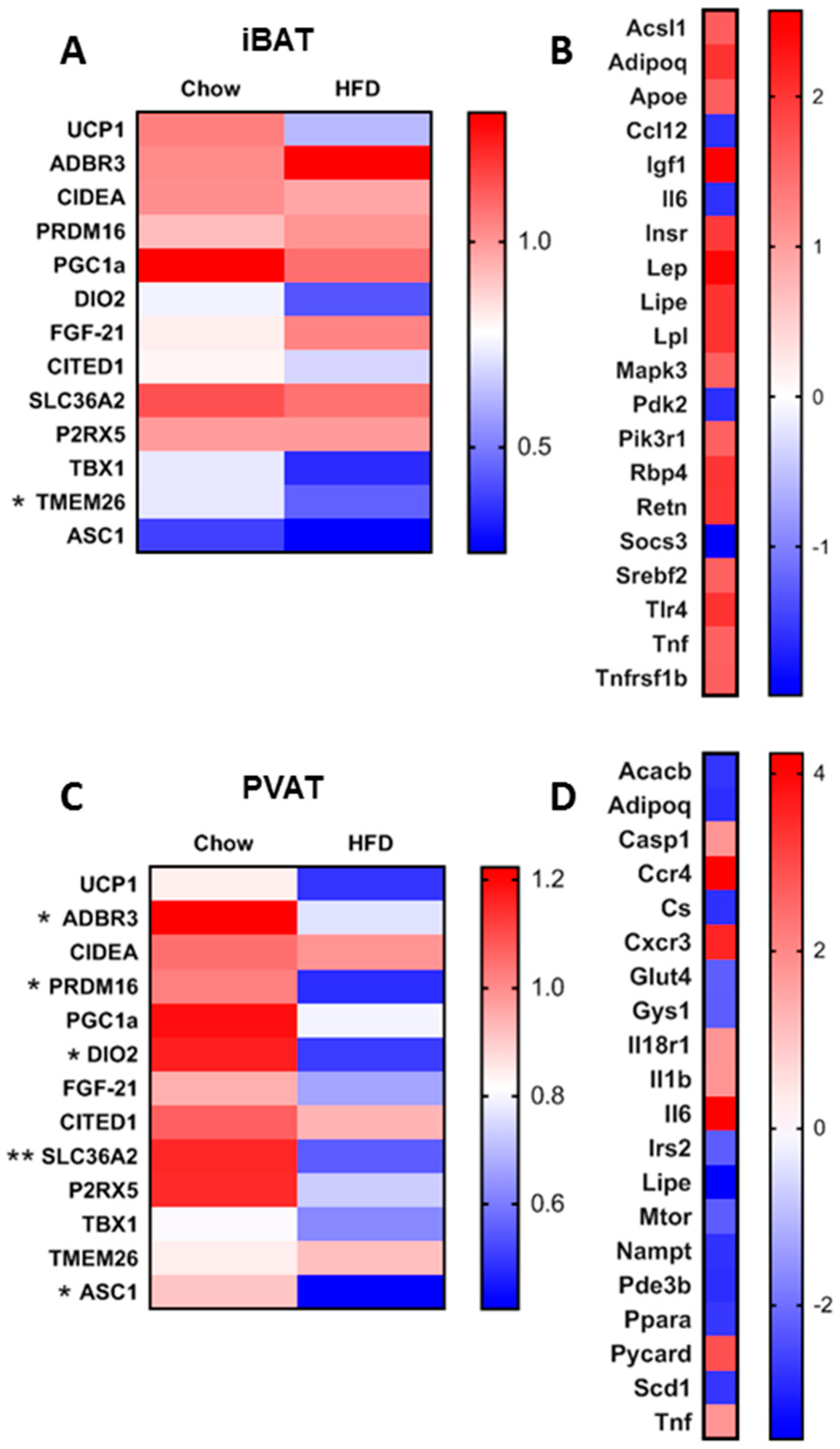
2.2. Gene Expression Analysis
2.3. Targeted Insulin Resistance PCR Arrays
2.4. Serum Analysis
2.5. Protein Extraction, Clean-Up and Trypsinization
2.6. Mass Spectrometry
2.7. Statistical Analysis
3. Results
3.1. Short-Term HFD Downregulates Genes Involved in Thermogenesis in PVAT Only
3.2. Short-Term HFD Alters Insulin Signalling Pathways in a Depot-Specific Manner
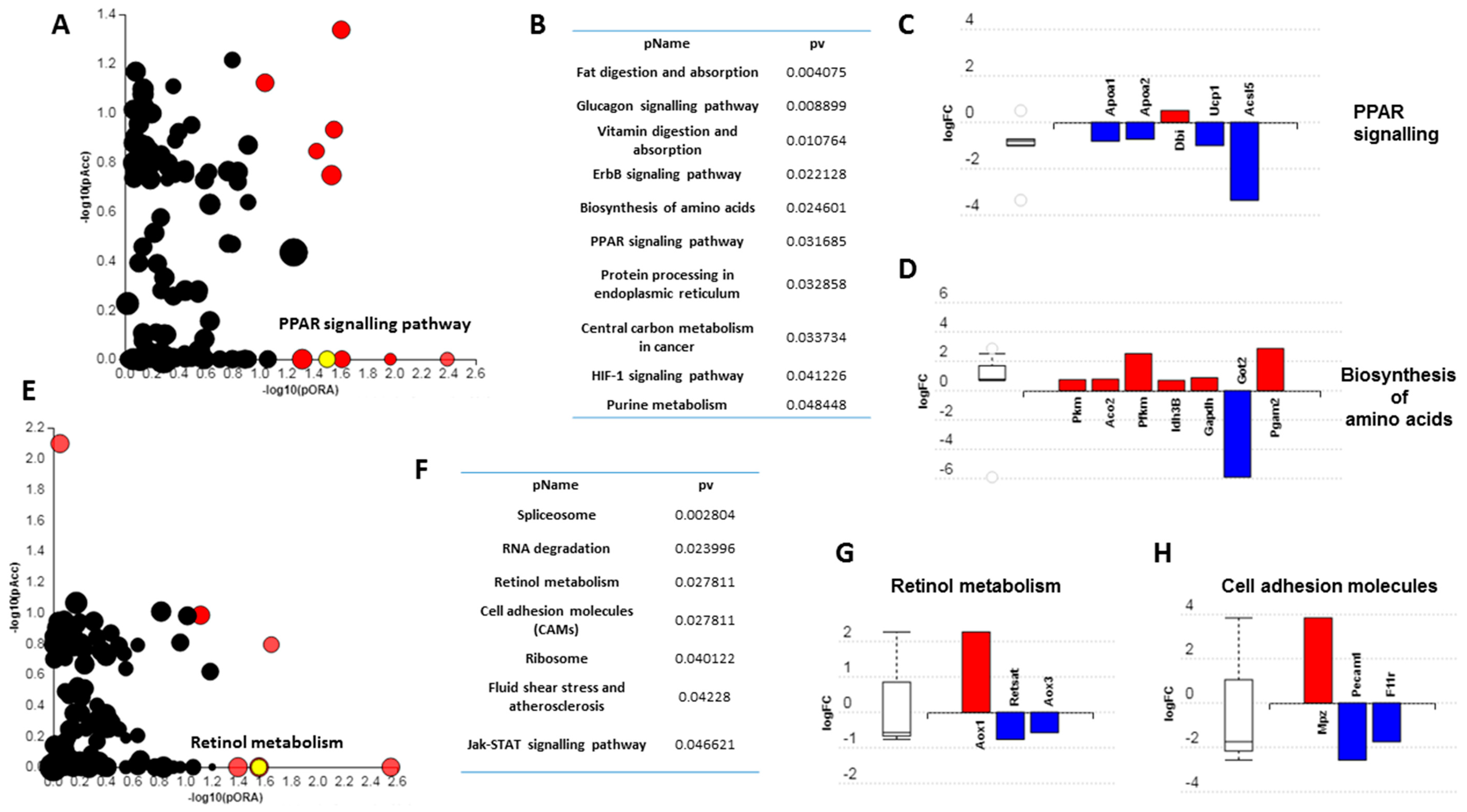
3.3. Short-Term High Fat Feeding Induces Divergent Alterations in the BAT and PVAT Proteome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Ethical Approval
Availability of Data and Material
References
- Sanchez-Gurmaches, J.; Guertin, D.A. Adipocyte lineages: Tracing back the origins of fat. Biochim. Biophys. Acta 2014, 1842, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Villacorta, L.; Li, R.; Hamblin, M.; Xu, W.; Dou, C.; Zhang, J.; Wu, J.; Zeng, R.; Chen, Y.E. Loss of perivascular adipose tissue on peroxisome proliferator-activated receptor-gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation 2012, 126, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Blondin, D.P.; Tingelstad, H.C.; Noll, C.; Frisch, F.; Phoenix, S.; Guérin, B.; Turcotte, É.E.; Richard, D.; Haman, F.; Carpentier, A.C. Dietary fatty acid metabolism of brown adipose tissue in cold-acclimated men. Nat. Commun. 2017, 8, 14146. [Google Scholar] [CrossRef]
- Lee, P.; Bova, R.; Schofield, L.; Bryant, W.; Dieckmann, W.; Slattery, A.; Govendir, M.A.; Emmet, L.; Greenfield, J.R. Brown Adipose Tissue Exhibits a Glucose-Responsive Thermogenic Biorhythm in Humans. Cell Metab. 2016, 23, 602–609. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Scheele, C.; Nielsen, S. Metabolic regulation and the anti-obesity perspectives of human brown fat. Redox. Biol. 2017, 12, 770–775. [Google Scholar] [CrossRef]
- Nosalski, R.; Guzik, T.J. Perivascular adipose tissue inflammation in vascular disease. Br. J. Pharm. 2017, 174, 3496–3513. [Google Scholar] [CrossRef]
- Marlatt, K.L.; Ravussin, E. Brown Adipose Tissue: An Update on Recent Findings. Curr. Obes. Rep. 2017, 6, 389–396. [Google Scholar] [CrossRef]
- Waise, T.M.Z.; Toshinai, K.; Naznin, F.; NamKoong, C.; Md Moin, A.S.; Sakoda, H.; Nakazato, M. One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem. Biophys. Res. Commun. 2015, 464, 1157–1162. [Google Scholar] [CrossRef]
- Boghossian, S.; Lemmon, K.; Park, M.; York, D.A. High-fat diets induce a rapid loss of the insulin anorectic response in the amygdala. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1302–R1311. [Google Scholar] [CrossRef]
- Clegg, D.J.; Gotoh, K.; Kemp, C.; Wortman, M.D.; Benoit, S.C.; Brown, L.M.; D’Alessio, D.; Tso, P.; Seeley, R.J.; Woods, S.C. Consumption of a high-fat diet induces central insulin resistance independent of adiposity. Physiol. Behav. 2011, 103, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, W.; Chen, L.; Chen, J.; Jiang, P.; Fu, X.; Nie, X.; Kwan, H.; Liu, Y.; Zhao, X. The effects of short-term high-fat feeding on exercise capacity: Multi-tissue transcriptome changes by RNA sequencing analysis. Lipids Health Dis. 2017, 16, 28. [Google Scholar] [CrossRef][Green Version]
- Ji, Y.; Sun, S.; Xia, S.; Yang, L.; Li, X.; Qi, L. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J. Biol. Chem. 2012, 287, 24378–24386. [Google Scholar] [CrossRef]
- Lee, Y.S.; Li, P.; Huh, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, M.S.; Wueest, S.; Item, F.; Schoenle, E.J.; Konrad, D. Adipose tissue inflammation contributes to short-term high-fat diet-induced hepatic insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E388–E395. [Google Scholar] [CrossRef] [PubMed]
- Mullen, K.L.; Tishinsky, J.M.; Robinson, L.E.; Dyck, D.J. Skeletal muscle inflammation is not responsible for the rapid impairment in adiponectin response with high-fat feeding in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R500–R508. [Google Scholar] [CrossRef]
- Cuthbertson, D.J.; Steele, T.; Wilding, J.P.; Halford, J.C.; Harrold, J.A.; Hamer, M.; Karpe, F. What have human experimental overfeeding studies taught us about adipose tissue expansion and susceptibility to obesity and metabolic complications? Int. J. Obes. (Lond.) 2017, 41, 853–865. [Google Scholar] [CrossRef]
- Fitzgibbons, T.P.; Kogan, S.; Aouadi, M.; Hendricks, G.M.; Straubhaar, J.; Czech, M.P. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1425–H1437. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J. The mouse thermoregulatory system: Its impact on translating biomedical data to humans. Physiol. Behav. 2017, 179, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Ganeshan, K.; Hong, C.; Nguyen, K.D.; Qiu, Y.; Kim, J.; Tangirala, R.K.; Tontonoz, P.; Chawla, A. Thermoneutral Housing Accelerates Metabolic Inflammation to Potentiate Atherosclerosis but Not Insulin Resistance. Cell Metab. 2016, 23, 165–178. [Google Scholar] [CrossRef]
- Hawkins, P.; Golledge, H.D.R. The 9 to 5 Rodent—Time for Change? Scientific and animal welfare implications of circadian and light effects on laboratory mice and rats. J. Neurosci. Methods 2018, 300, 20–25. [Google Scholar] [CrossRef]
- Warner, A.; Jethwa, P.H.; Wyse, C.A.; l’Anson, H.; Brameld, J.M.; Ebling, F.J.P. Effects of photoperiod on daily locomotor activity, energy expenditure, and feeding behavior in a seasonal mammal. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1409–R1416. [Google Scholar] [CrossRef]
- Samms, R.J.; Lewis, J.E.; Lory, A.; Fowler, M.J.; Cooper, S.; Warner, A.; Emmerson, P.; Adams, A.C.; Luckett, J.C.; Perkins, A.C.; et al. Antibody-Mediated Inhibition of the FGFR1c Isoform Induces a Catabolic Lean State in Siberian Hamsters. Curr. Biol. 2015, 25, 2997–3003. [Google Scholar] [CrossRef][Green Version]
- Frayn, K.N. Calculation of substrate oxidation rates in vivo from gaseous exchange. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 628–634. [Google Scholar] [CrossRef]
- Gil-Ortega, M.; Somoza, B.; Huang, Y.; Gollasch, M.; Fernández-Alfonso, M.S. Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol. Metab. 2015, 26, 367–375. [Google Scholar] [CrossRef]
- Lambert, J.P.; Ivosev, G.; Couzens, A.L.; Larsen, B.; Taipale, M.; Lin, Z.Y.; Zhong, Q.; Lindquist, S.; Vidal, M.; Aebersold, R.; et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 2013, 10, 1239–1245. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG database. Novartis Found. Symp. 2002, 247, 91–101, discussion 101–103, 119–128, 244–252. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef]
- Ussar, S.; Lee, K.Y.; Dankel, S.N.; Boucher, J.; Hearing, M.F.; Kleinridders, A.; Thomou, T.; Xue, R.; Macotela, Y.; Cypess, A.M.; et al. ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Sci. Transl. Med. 2014, 6, 247ra103. [Google Scholar] [CrossRef] [PubMed]
- Sakamuri, S.S.; Putcha, U.K.; Veettil, G.N.; Ayyalasomayajula, V. Transcriptome profiling of visceral adipose tissue in a novel obese rat model, WNIN/Ob & its comparison with other animal models. Indian J. Med. Res. 2016, 144, 409–423. [Google Scholar]
- Bartelt, A.; Widenmaier, S.B.; Schlein, C.; Johann, K.; Goncalves, R.L.S.; Eguchi, K.; Fischer, A.W.; Parlakgül, G.; Snyder, N.A.; Nguyen, T.B.; et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018, 24, 292–303. [Google Scholar] [CrossRef]
- Hanein, S.; Garcia, M.; Fares-Taie, L.; Serre, V.; De Keyzer, Y.; Delaveau, T.; Perrault, I.; Delphin, N.; Gerber, S.; Schmitt, A.; et al. TMEM126A is a mitochondrial located mRNA (MLR) protein of the mitochondrial inner membrane. Biochim. Biophys. Acta 2013, 1830, 3719–3733. [Google Scholar] [CrossRef]
- Hanein, S.; Perrault, I.; Roche, O.; Gerber, S.; Khadom, N.; Rio, M.; Boddaert, N.; Jean-Pierre, M.; Brahimi, N.; Serre, V.; et al. TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am. J. Hum. Genet. 2009, 84, 493–498. [Google Scholar] [CrossRef]
- Bae, J.S.; Choi, J.K.; Moon, J.H.; Kim, E.C.; Croft, M.; Lee, H.W. Novel transmembrane protein 126A (TMEM126A) couples with CD137L reverse signals in myeloid cells. Cell Signal. 2012, 24, 2227–2236. [Google Scholar] [CrossRef]
- Kim, E.C.; Moon, J.H.; Kang, S.W.; Kwon, B.; Lee, H.W. TMEM126A, a CD137 ligand binding protein, couples with the TLR4 signal transduction pathway in macrophages. Mol. Immunol. 2015, 64, 244–251. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef]
- Scuteri, A.; Sanna, S.; Chen, W.M.; Uda, M.; Albai, G.; Strait, J.; Najjar, S.; Nagaraja, R.; Orrú, M.; Usala, G.; et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007, 3, e115. [Google Scholar] [CrossRef]
- Liu, Y.J.; Liu, X.G.; Wang, L.; Dina, C.; Yan, H.; Liu, J.F.; Levy, S.; Papasian, C.J.; Drees, B.M.; Hamilton, J.J.; et al. Genome-wide association scans identified CTNNBL1 as a novel gene for obesity. Hum. Mol. Genet. 2008, 17, 1803–1813. [Google Scholar] [CrossRef]
- Basse, A.L.; Isidor, M.S.; Winther, S.; Skjoldborg, N.B.; Murholm, M.; Andersen, E.S.; Pedersen, S.B.; Wolfrum, C.; Quistorff, B.; Hansen, J.B. Regulation of glycolysis in brown adipocytes by HIF-1alpha. Sci. Rep. 2017, 7, 4052. [Google Scholar] [CrossRef]
- Oka, T.; Sayano, T.; Tamai, S.; Yokota, S.; Kato, H.; Fujii, G.; Mihara, K. Identification of a novel protein MICS1 that is involved in maintenance of mitochondrial morphology and apoptotic release of cytochrome c. Mol. Biol. Cell 2008, 19, 2597–2608. [Google Scholar] [CrossRef]
- Yang, F.H.; Pyle, W.G. Reduced cardiac CapZ protein protects hearts against acute ischemia-reperfusion injury and enhances preconditioning. J. Mol. Cell. Cardiol. 2012, 52, 761–772. [Google Scholar] [CrossRef]
- Sarkar, S.; Leaman, D.W.; Gupta, S.; Sil, P.; Young, D.; Morehead, A.; Mukherjee, D.; Ratfliff, N.; Sun, Y.; Rayborn, M. Cardiac overexpression of myotrophin triggers myocardial hypertrophy and heart failure in transgenic mice. J. Biol. Chem. 2004, 279, 20422–20434. [Google Scholar] [CrossRef]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef]
- Berbee, J.F.; Boon, M.R.; Khedoe, P.P.; Bartelt, A.; Schlein, C.; Worthmann, A.; Kooijman, S.; Hoeke, G.; Mol, I.M.; John, C.; et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat. Commun. 2015, 6, 6356. [Google Scholar] [CrossRef]
- Hoeke, G.; Kooijman, S.; Boon, M.R.; Rensen, P.C.; Berbee, J.F. Role of Brown Fat in Lipoprotein Metabolism and Atherosclerosis. Circ. Res. 2016, 118, 173–182. [Google Scholar] [CrossRef]
- Krude, T. Chromatin: Nucleosome assembly during DNA replication. Curr. Biol. 1995, 5, 1232–1234. [Google Scholar] [CrossRef]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef]
- Rhee, E.J.; Nallamshetty, S.; Plutzky, J. Retinoid metabolism and its effects on the vasculature. Biochim. Biophys. Acta 2012, 1821, 230–240. [Google Scholar] [CrossRef]
- Schupp, M.; Lefterova, M.I.; Janke, J.; Leitner, K.; Cristancho, A.G.; Mullican, S.E.; Qatanani, M.; Szwergold, N.; Steger, D.J.; Curtin, J.C.; et al. Retinol saturase promotes adipogenesis and is downregulated in obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 1105–1110. [Google Scholar] [CrossRef]
- Moise, A.R.; Lobo, G.P.; Erokwu, B.; Wilson, D.L.; Peck, D.; Alvarez, S.; Dominguez, M.; Alvarez, R.; Flask, C.A.; de Lera, A.R.; et al. Increased adiposity in the retinol saturase-knockout mouse. FASEB J. 2010, 24, 1261–1270. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Vascular adhesion molecules in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2292–2301. [Google Scholar] [CrossRef]
- Caro, C.G. Discovery of the role of wall shear in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 158–161. [Google Scholar] [CrossRef]
- Stevens, H.Y.; Melchior, B.; Bell, K.S.; Yun, S.; Yeh, J.C.; Frangos, J.A. PECAM-1 is a critical mediator of atherosclerosis. Dis. Models Mech. 2008, 1, 175–181, discussion 179. [Google Scholar] [CrossRef]
- Chen, Z.; Tzima, E. PECAM-1 is necessary for flow-induced vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1067–1073. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Symbol | Gene Name | Entrez | Logfc | Adjpv |
|---|---|---|---|---|
| Psma3l | Proteasome subunit alpha type-3 | 408248 | 0.793509 | 0.000116 |
| Tmem126a | Transmembrane protein 126A | 293113 | −1.83882 | 0.000185 |
| Ssr3 | Signal Sequence Receptor Subunit 3 | 81784 | −1.61099 | 0.0002 |
| Ccdc51 | Coiled-Coil Domain Containing 51 | 316008 | −0.693 | 0.000763 |
| Pkm | Pyruvate Kinase M1/2 | 25630 | 0.7335 | 0.00279 |
| Vps4a | Vacuolar Protein Sorting 4 Homolog A | 246772 | −0.71726 | 0.003363 |
| Apoa4 | Apolipoprotein A4 | 25080 | −1.0162 | 0.004051 |
| Prss1 | Serine Protease 1 | 24691 | 0.827668 | 0.005684 |
| Serpina3n | Serpin Family A Member 3 | 24795 | −0.58665 | 0.006792 |
| Camk2d | Calcium/Calmodulin Dependent Protein Kinase II Delta | 24246 | −2.55115 | 0.007442 |
| Symbol | Gene Name | Entrez | Logfc | Adjpv |
|---|---|---|---|---|
| Pfkp | Phosphofructokinase, Platelet | 60416 | −1.38584 | 0.000225 |
| Hdgf | Heparin Binding Growth Factor | 114499 | 0.69362 | 0.000445 |
| Rbmxrtl | RNA-binding motif protein, X chromosome retrogene-like | 307779 | 1.91652 | 0.001389 |
| Ywhaq | 14-3-3 Protein Theta | 25577 | 0.613265 | 0.001405 |
| Ghitm | Growth Hormone Inducible Transmembrane Protein | 290596 | −0.71467 | 0.001854 |
| Capza1 | Capping Actin Protein Of Muscle Z-Line Subunit Alpha 1 | 691149 | 1.194102 | 0.002081 |
| Mtpn | Myotrophin | 79215 | 0.669685 | 0.002234 |
| Ube2n | Ubiquitin Conjugating Enzyme E2 N | 116725 | 0.80585 | 0.002495 |
| B2m | Beta-2- Microglobulin | 24223 | 0.809804 | 0.002742 |
| Acox1 | Acyl-CoA Oxidase 1 | 50681 | −3.50641 | 0.004222 |
| GoId | GoName | CountDE | CountAll | Pv_elim |
| Biological Process | ||||
| GO:0039536 | negative regulation of RIG-I signaling pathway | 3 | 3 | 0.0011 |
| GO:0050996 | positive regulation of lipid catabolic process | 4 | 6 | 0.0014 |
| GO:0046470 | phosphatidylcholine metabolic process | 5 | 7 | 0.0038 |
| GO:0030300 | regulation of intestinal cholesterol absorption | 3 | 4 | 0.004 |
| GO:0034380 | high-density lipoprotein particle assembly | 3 | 4 | 0.004 |
| Molecular Function | ||||
| GO:0031210 | phosphatidylcholine binding | 3 | 3 | 0.0011 |
| GO:0060228 | phosphatidylcholine-sterol O-acyltransferase activator activity | 3 | 4 | 0.004 |
| GO:0003713 | transcription coactivator activity | 4 | 8 | 0.0054 |
| GO:0001047 | core promoter binding | 3 | 5 | 0.0091 |
| GO:0017127 | cholesterol transporter activity | 3 | 5 | 0.0091 |
| Cellular Component | ||||
| GO:0034366 | spherical high-density lipoprotein particle | 3 | 4 | 0.0041 |
| GO:0042627 | chylomicron | 3 | 4 | 0.0041 |
| GO:0005667 | transcription factor complex | 3 | 6 | 0.0174 |
| GO:0034361 | very-low-density lipoprotein particle | 3 | 6 | 0.0174 |
| GoId | GoName | CountDE | CountAll | Pv_elim |
| Biological Process | ||||
| GO:0006334 | nucleosome assembly | 8 | 10 | 0.00023 |
| GO:0017144 | drug metabolic process | 5 | 8 | 0.00287 |
| GO:0043486 | histone exchange | 3 | 3 | 0.00343 |
| GO:1901655 | cellular response to ketone | 8 | 21 | 0.00825 |
| GO:0021766 | hippocampus development | 6 | 14 | 0.0116 |
| Molecular Function | ||||
| GO:0042393 | histone binding | 7 | 8 | 0.000011 |
| GO:0003785 | actin monomer binding | 3 | 3 | 0.0033 |
| GO:0043565 | sequence-specific DNA binding | 8 | 23 | 0.0143 |
| GO:0035091 | phosphatidylinositol binding | 5 | 8 | 0.0219 |
| GO:0005506 | iron ion binding | 6 | 16 | 0.0227 |
| Cellular Component | ||||
| GO:0000788 | nuclear nucleosome | 3 | 3 | 0.0035 |
| GO:0000784 | nuclear chromosome, telomeric region | 3 | 4 | 0.0123 |
| GO:0030054 | cell junction | 38 | 183 | 0.0174 |
| GO:0071013 | catalytic step 2 spliceosome | 5 | 12 | 0.0246 |
| GO:0001931 | uropod | 3 | 5 | 0.0273 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldiss, P.; Lewis, J.E.; Boocock, D.J.; Miles, A.K.; Bloor, I.; Ebling, F.J.P.; Budge, H.; Symonds, M.E. Interscapular and Perivascular Brown Adipose Tissue Respond Differently to a Short-Term High-Fat Diet. Nutrients 2019, 11, 1065. https://doi.org/10.3390/nu11051065
Aldiss P, Lewis JE, Boocock DJ, Miles AK, Bloor I, Ebling FJP, Budge H, Symonds ME. Interscapular and Perivascular Brown Adipose Tissue Respond Differently to a Short-Term High-Fat Diet. Nutrients. 2019; 11(5):1065. https://doi.org/10.3390/nu11051065
Chicago/Turabian StyleAldiss, Peter, Jo E. Lewis, David J. Boocock, Amanda K. Miles, Ian Bloor, Francis J. P. Ebling, Helen Budge, and Michael E. Symonds. 2019. "Interscapular and Perivascular Brown Adipose Tissue Respond Differently to a Short-Term High-Fat Diet" Nutrients 11, no. 5: 1065. https://doi.org/10.3390/nu11051065
APA StyleAldiss, P., Lewis, J. E., Boocock, D. J., Miles, A. K., Bloor, I., Ebling, F. J. P., Budge, H., & Symonds, M. E. (2019). Interscapular and Perivascular Brown Adipose Tissue Respond Differently to a Short-Term High-Fat Diet. Nutrients, 11(5), 1065. https://doi.org/10.3390/nu11051065