Uroguanylin Improves Leptin Responsiveness in Diet-Induced Obese Mice

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Treatments and Surgeries
2.3. Dissection of Brain Areas
2.4. Real Time Polymerase Chain Reaction (PCR)
2.5. Western Blotting
2.6. Blood Biochemistry
2.7. Statistical Analysis
3. Results
3.1. Central UGN Administration Restores Leptin Sensitivity in Diet-Induced Obese (DIO) Mice
3.2. Central UGN Administration Restores Hypothalamic Leptin-Induced Signaling Pathways in DIO Mice
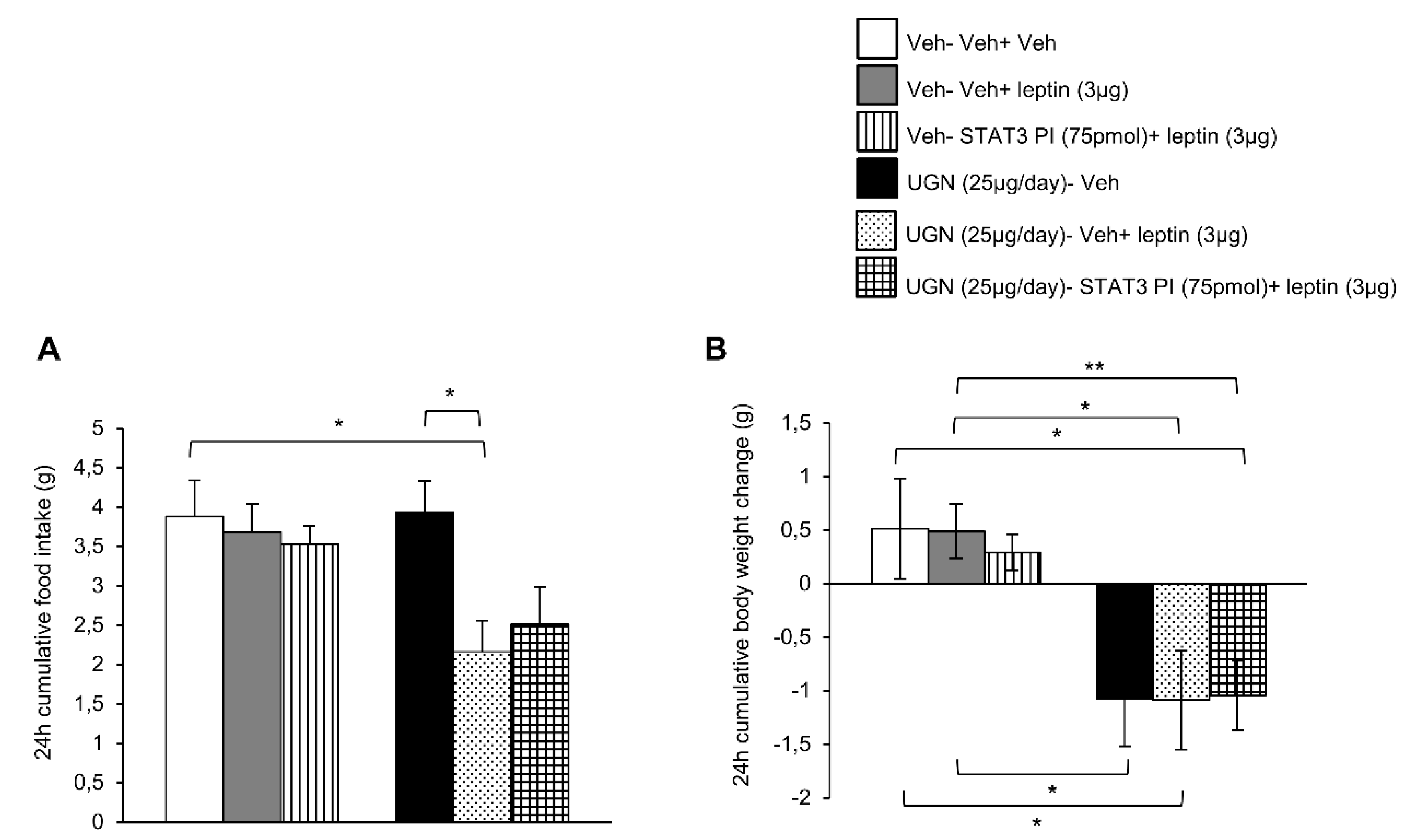
3.3. Blockade of STAT3 does not Affect UGN-Mediated Leptin Responsiveness
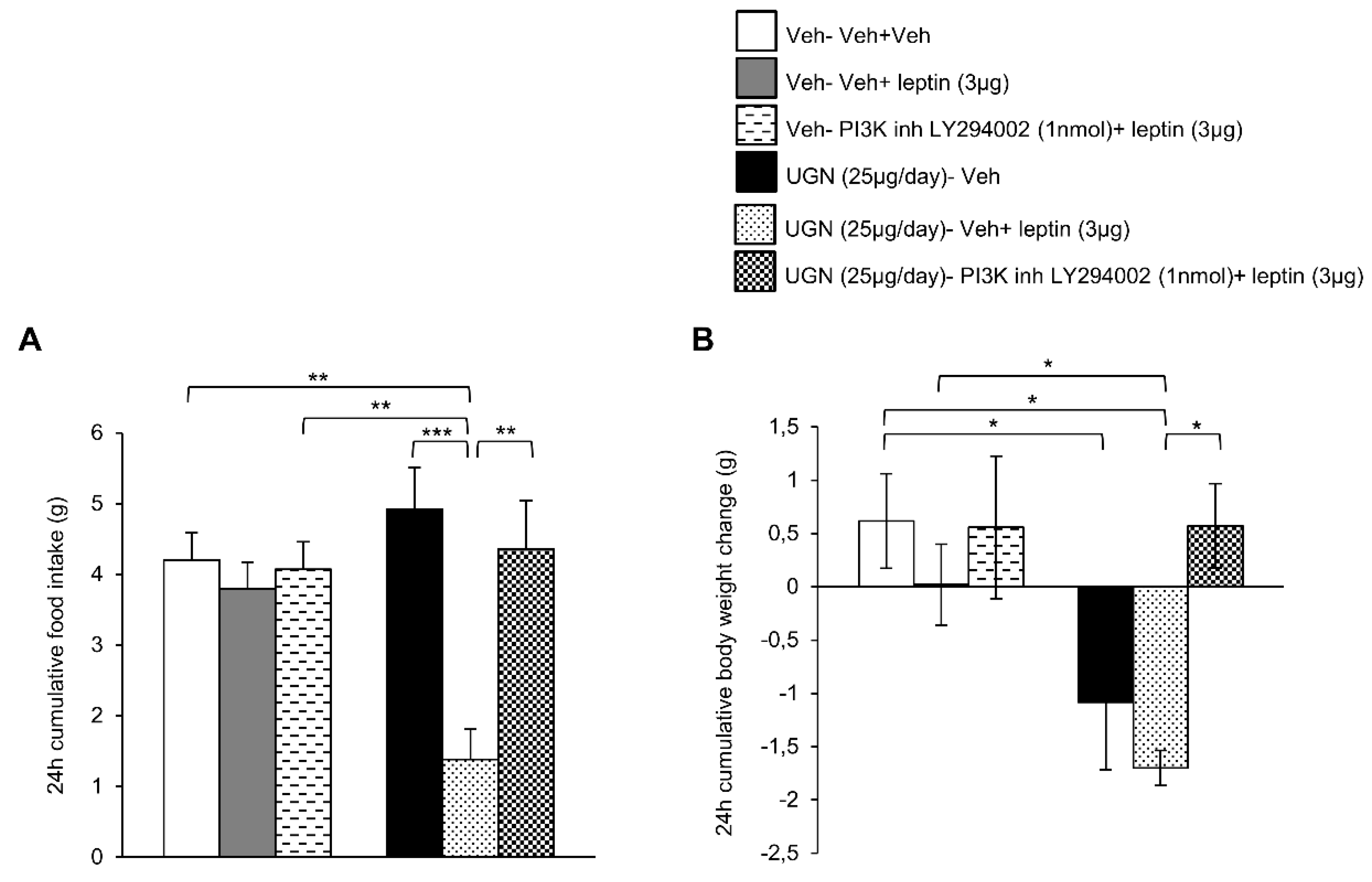
3.4. Blockade of PI3K Blunts UGN-Mediated Leptin Responsiveness
3.5. Central UGN Restores Leptin Sensitivity Independently of Body Weight Changes and Leptin Levels in DIO Mice
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Cummings, D.E.; Overduin, J. Gastrointestinal regulation of food intake. J. Clin. Investig. 2007, 117, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Lin, J.E.; Waldman, S.A. GUCY2C: At the intersection of obesity and cancer. Trends Endocrinol. Metab. 2013, 24, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Blomain, E.S.; Merlino, D.J.; Pattison, A.M.; Snook, A.E.; Waldman, S.A. Guanylyl Cyclase C Hormone Axis at the Intersection of Obesity and Colorectal Cancer. Mol. Pharmacol. 2016, 90, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeck, G. Gastrointestinal hormones: Uroguanylin-a new gut-derived weapon against obesity? Nat. Rev. Endocrinol. 2011, 8, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Folgueira, C.; Barja-Fernandez, S.; Gonzalez-Saenz, P.; Pena-Leon, V.; Castelao, C.; Ruiz-Pinon, M.; Casanueva, F.F.; Nogueiras, R.; Seoane, L.M. Uroguanylin: A new actor in the energy balance movie. J. Mol. Endocrinol. 2018, 60, R31–R38. [Google Scholar] [CrossRef] [PubMed]
- Valentino, M.A.; Lin, J.E.; Snook, A.E.; Li, P.; Kim, G.W.; Marszalowicz, G.; Magee, M.S.; Hyslop, T.; Schulz, S.; Waldman, S.A. A uroguanylin-GUCY2C endocrine axis regulates feeding in mice. J. Clin. Investig. 2011, 121, 3578–3588. [Google Scholar] [CrossRef] [PubMed]
- Hamra, F.K.; Forte, L.R.; Eber, S.L.; Pidhorodeckyj, N.V.; Krause, W.J.; Freeman, R.H.; Chin, D.T.; Tompkins, J.A.; Fok, K.F.; Smith, C.E.; et al. Uroguanylin: Structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase. Proc. Natl. Acad. Sci. USA 1993, 90, 10464–10468. [Google Scholar] [CrossRef]
- Begg, D.P.; Steinbrecher, K.A.; Mul, J.D.; Chambers, A.P.; Kohli, R.; Haller, A.; Cohen, M.B.; Woods, S.C.; Seeley, R.J. Effect of guanylate cyclase-C activity on energy and glucose homeostasis. Diabetes 2014, 63, 3798–3804. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cachon, M.L.; Pedersen, S.L.; Rigbolt, K.T.; Zhang, C.; Fabricius, K.; Hansen, H.H.; Elster, L.; Fink, L.N.; Schafer, M.; Rhee, N.A.; et al. Guanylin and uroguanylin mRNA expression is increased following Roux-en-Y gastric bypass, but guanylins do not play a significant role in body weight regulation and glycemic control. Peptides 2018, 101, 32–43. [Google Scholar] [CrossRef]
- Folgueira, C.; Beiroa, D.; Callon, A.; Al-Massadi, O.; Barja-Fernandez, S.; Senra, A.; Ferno, J.; Lopez, M.; Dieguez, C.; Casanueva, F.F.; et al. Uroguanylin action in the brain reduces weight gain in obese mice via different efferent autonomic pathways. Diabetes 2016, 65, 421–432. [Google Scholar] [CrossRef]
- Folgueira, C.; Sanchez-Rebordelo, E.; Barja-Fernandez, S.; Leis, R.; Tovar, S.; Casanueva, F.F.; Dieguez, C.; Nogueiras, R.; Seoane, L.M. Uroguanylin levels in intestine and plasma are regulated by nutritional status in a leptin-dependent manner. Eur. J. Nutr. 2016, 55, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef]
- El-Haschimi, K.; Pierroz, D.D.; Hileman, S.M.; Bjorbaek, C.; Flier, J.S. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Investig. 2000, 105, 1827–1832. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Greenberg, A.S.; Fujioka, K.; Dixon, R.M.; Kushner, R.; Hunt, T.; Lubina, J.A.; Patane, J.; Self, B.; Hunt, P.; et al. Recombinant leptin for weight loss in obese and lean adults: A randomized, controlled, dose-escalation trial. JAMA 1999, 282, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M. Hypothalamic Leptin Resistance: From BBB to BBSome. PLoS Genet. 2016, 12, e1005980. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Lopez, M.; Rahmouni, K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat. Rev. Endocrinol. 2017, 13, 338–351. [Google Scholar] [CrossRef]
- Munzberg, H.; Flier, J.S.; Bjorbaek, C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 2004, 145, 4880–4889. [Google Scholar] [CrossRef]
- Enriori, P.J.; Evans, A.E.; Sinnayah, P.; Jobst, E.E.; Tonelli-Lemos, L.; Billes, S.K.; Glavas, M.M.; Grayson, B.E.; Perello, M.; Nillni, E.A.; et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007, 5, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Simonds, S.E.; Pryor, J.T.; Ravussin, E.; Greenway, F.L.; Dileone, R.; Allen, A.M.; Bassi, J.; Elmquist, J.K.; Keogh, J.M.; Henning, E.; et al. Leptin mediates the increase in blood pressure associated with obesity. Cell 2014, 159, 1404–1416. [Google Scholar] [CrossRef]
- Roth, J.D.; Roland, B.L.; Cole, R.L.; Trevaskis, J.L.; Weyer, C.; Koda, J.E.; Anderson, C.M.; Parkes, D.G.; Baron, A.D. Leptin responsiveness restored by amylin agonism in diet-induced obesity: Evidence from nonclinical and clinical studies. Proc. Natl. Acad. Sci. USA 2008, 105, 7257–7262. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Sullivan, L.M.; Habegger, K.; Yi, C.X.; Kabra, D.; Grant, E.; Ottaway, N.; Krishna, R.; Holland, J.; Hembree, J.; et al. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. J. Pept. Sci. 2012, 18, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Clemmensen, C.; Chabenne, J.; Finan, B.; Sullivan, L.; Fischer, K.; Kuchler, D.; Sehrer, L.; Ograjsek, T.; Hofmann, S.M.; Schriever, S.C.; et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes 2014, 63, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Sadry, S.A.; Drucker, D.J. Emerging combinatorial hormone therapies for the treatment of obesity and T2DM. Nat. Rev. Endocrinol. 2013, 9, 425–433. [Google Scholar] [CrossRef]
- Imbernon, M.; Sanchez-Rebordelo, E.; Romero-Pico, A.; Kallo, I.; Chee, M.J.; Porteiro, B.; Al-Massadi, O.; Contreras, C.; Ferno, J.; Senra, A.; et al. Hypothalamic kappa opioid receptor mediates both diet-induced and melanin concentrating hormone-induced liver damage through inflammation and endoplasmic reticulum stress. Hepatology 2016, 64, 1086–1104. [Google Scholar] [CrossRef]
- Quinones, M.; Al-Massadi, O.; Folgueira, C.; Bremser, S.; Gallego, R.; Torres-Leal, L.; Haddad-Tovolli, R.; Garcia-Caceres, C.; Hernandez-Bautista, R.; Lam, B.Y.H.; et al. p53 in AgRP neurons is required for protection against diet-induced obesity via JNK1. Nat. Commun. 2018, 9, 3432. [Google Scholar] [CrossRef]
- Imbernon, M.; Beiroa, D.; Vazquez, M.J.; Morgan, D.A.; Veyrat-Durebex, C.; Porteiro, B.; Diaz-Arteaga, A.; Senra, A.; Busquets, S.; Velasquez, D.A.; et al. Central melanin-concentrating hormone influences liver and adipose metabolism via specific hypothalamic nuclei and efferent autonomic/JNK1 pathways. Gastroenterology 2013, 144, 636–649. [Google Scholar] [CrossRef]
- Imbernon, M.; Sanchez-Rebordelo, E.; Gallego, R.; Gandara, M.; Lear, P.; Lopez, M.; Dieguez, C.; Nogueiras, R. Hypothalamic KLF4 mediates leptin’s effects on food intake via AgRP. Mol. Metab. 2014, 3, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Quinones, M.; Al-Massadi, O.; Gallego, R.; Ferno, J.; Dieguez, C.; Lopez, M.; Nogueiras, R. Hypothalamic CaMKKbeta mediates glucagon anorectic effect and its diet-induced resistance. Mol. Metab. 2015, 4, 961–970. [Google Scholar] [CrossRef]
- Martinez-Sanchez, N.; Seoane-Collazo, P.; Contreras, C.; Varela, L.; Villarroya, J.; Rial-Pensado, E.; Buque, X.; Aurrekoetxea, I.; Delgado, T.C.; Vazquez-Martinez, R.; et al. Hypothalamic AMPK-ER Stress-JNK1 Axis Mediates the Central Actions of Thyroid Hormones on Energy Balance. Cell Metab. 2017, 26, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; Gonzalez-Garcia, I.; Martinez-Sanchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef]
- Seeley, R.J.; Tschop, M.H. Uroguanylin: How the gut got another satiety hormone. J. Clin. Investig. 2011, 121, 3384–3386. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Gomez-Ambrosi, J.; Catalan, V.; Ezquerro, S.; Mendez-Gimenez, L.; Becerril, S.; Ibanez, P.; Vila, N.; Margall, M.A.; Moncada, R.; et al. Guanylin and uroguanylin stimulate lipolysis in human visceral adipocytes. Int. J. Obes. 2016, 40, 1405–1415. [Google Scholar] [CrossRef]
- Di Guglielmo, M.D.; Tonb, D.; He, Z.; Adeyemi, A.; van Golen, K.L. Pilot Study Measuring the novel satiety hormone, pro-uroguanylin, in adolescents with and without obesity. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 489–495. [Google Scholar] [CrossRef]
- Caricilli, A.M.; Penteado, E.; de Abreu, L.L.; Quaresma, P.G.; Santos, A.C.; Guadagnini, D.; Razolli, D.; Mittestainer, F.C.; Carvalheira, J.B.; Velloso, L.A.; et al. Topiramate treatment improves hypothalamic insulin and leptin signaling and action and reduces obesity in mice. Endocrinology 2012, 153, 4401–4411. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Aleman, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef]
- Swartling, F.J.; Ferletta, M.; Kastemar, M.; Weiss, W.A.; Westermark, B. Cyclic GMP-dependent protein kinase II inhibits cell proliferation, Sox9 expression and Akt phosphorylation in human glioma cell lines. Oncogene 2009, 28, 3121–3131. [Google Scholar] [CrossRef]
- Hashikawa-Hobara, N.; Hashikawa, N.; Yutani, C.; Zamami, Y.; Jin, X.; Takatori, S.; Mio, M.; Kawasaki, H. The Akt-nitric oxide-cGMP pathway contributes to nerve growth factor-mediated neurite outgrowth in apolipoprotein E knockout mice. J. Pharmacol. Exp. Ther. 2011, 338, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.E.; Li, P.; Snook, A.E.; Schulz, S.; Dasgupta, A.; Hyslop, T.M.; Gibbons, A.V.; Marszlowicz, G.; Pitari, G.M.; Waldman, S.A. The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology 2010, 138, 241–254. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Direction | Primer Sequence 5’ → 3’ |
|---|---|---|
| HPRT | FWD | AGCCGACCGGTTCTGTCAT |
| REV | GGTCATAACCTGGTTCATCATCAC | |
| PB | CGACCCTCAGTCCCAGCGTCGTGAT | |
| AgRP | FWD | GCACAAGTGGCCAGGAACTC |
| REV | CAGGACACAGCTCAGCAACAT | |
| PB | CAAGCATCAACAAGCAAAGGCCATGC | |
| POMC | FWD | CGTCCTCAGAGAGCTGCCTTT |
| REV | TGTAGCAGAATCTCGGCATCTTC | |
| PB | CGGGACAGAGCCTCAGCCACC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folgueira, C.; Beiroa, D.; González-Rellán, M.J.; Porteiro, B.; Milbank, E.; Castelao, C.; García-Palacios, M.; Casanueva, F.F.; López, M.; Diéguez, C.; et al. Uroguanylin Improves Leptin Responsiveness in Diet-Induced Obese Mice. Nutrients 2019, 11, 752. https://doi.org/10.3390/nu11040752
Folgueira C, Beiroa D, González-Rellán MJ, Porteiro B, Milbank E, Castelao C, García-Palacios M, Casanueva FF, López M, Diéguez C, et al. Uroguanylin Improves Leptin Responsiveness in Diet-Induced Obese Mice. Nutrients. 2019; 11(4):752. https://doi.org/10.3390/nu11040752
Chicago/Turabian StyleFolgueira, Cintia, Daniel Beiroa, María Jesús González-Rellán, Begoña Porteiro, Edward Milbank, Cecilia Castelao, María García-Palacios, Felipe F Casanueva, Miguel López, Carlos Diéguez, and et al. 2019. "Uroguanylin Improves Leptin Responsiveness in Diet-Induced Obese Mice" Nutrients 11, no. 4: 752. https://doi.org/10.3390/nu11040752
APA StyleFolgueira, C., Beiroa, D., González-Rellán, M. J., Porteiro, B., Milbank, E., Castelao, C., García-Palacios, M., Casanueva, F. F., López, M., Diéguez, C., Seoane, L. M., & Nogueiras, R. (2019). Uroguanylin Improves Leptin Responsiveness in Diet-Induced Obese Mice. Nutrients, 11(4), 752. https://doi.org/10.3390/nu11040752